
Revisiting the Impact of Local Leptin Signaling in Folliculogenesis and Oocyte Maturation in Obese Mothers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
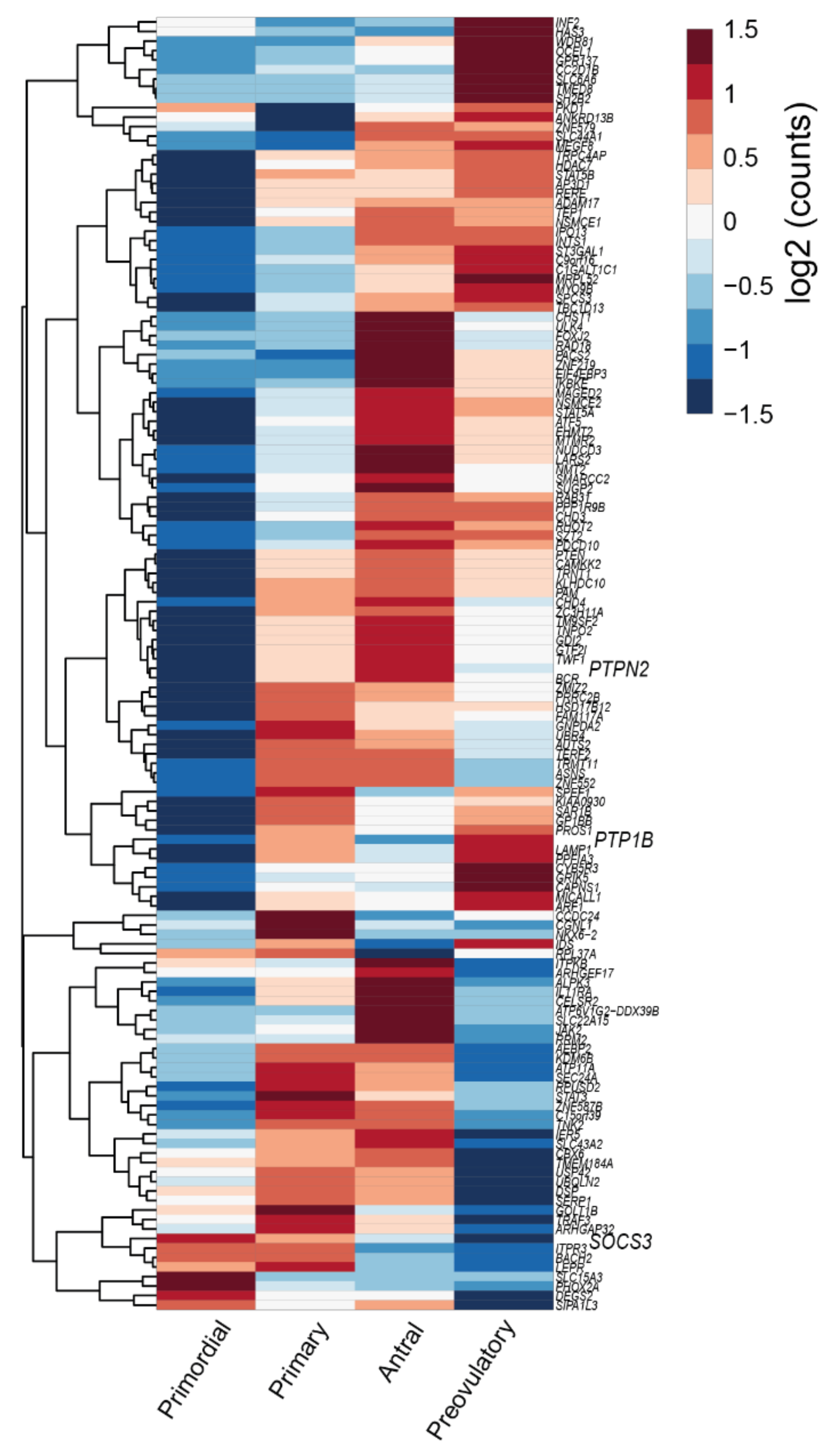{kind=link}
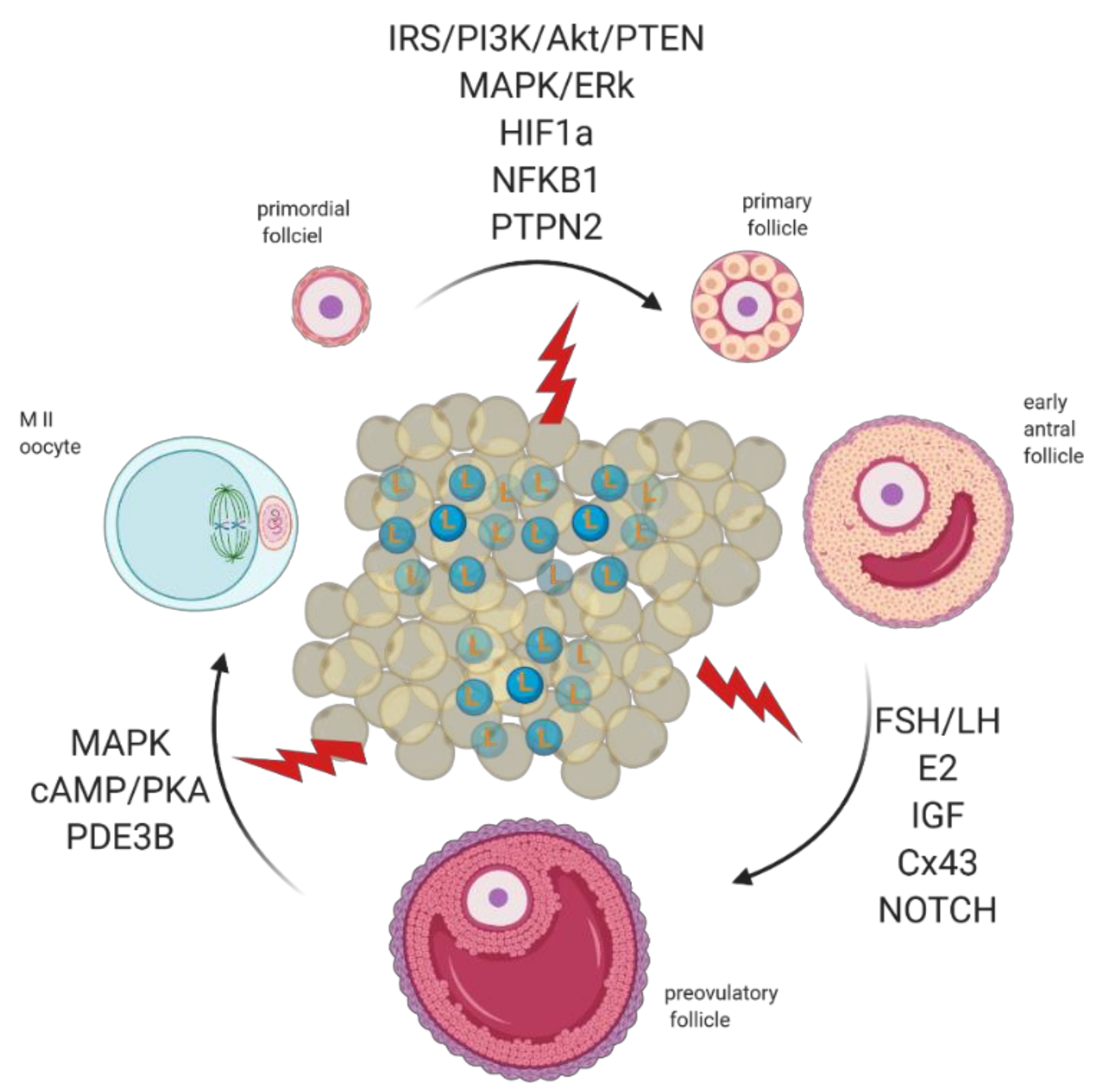{kind=link}
Abstract
:1. Introduction
2. Obesity and Ovarian Function
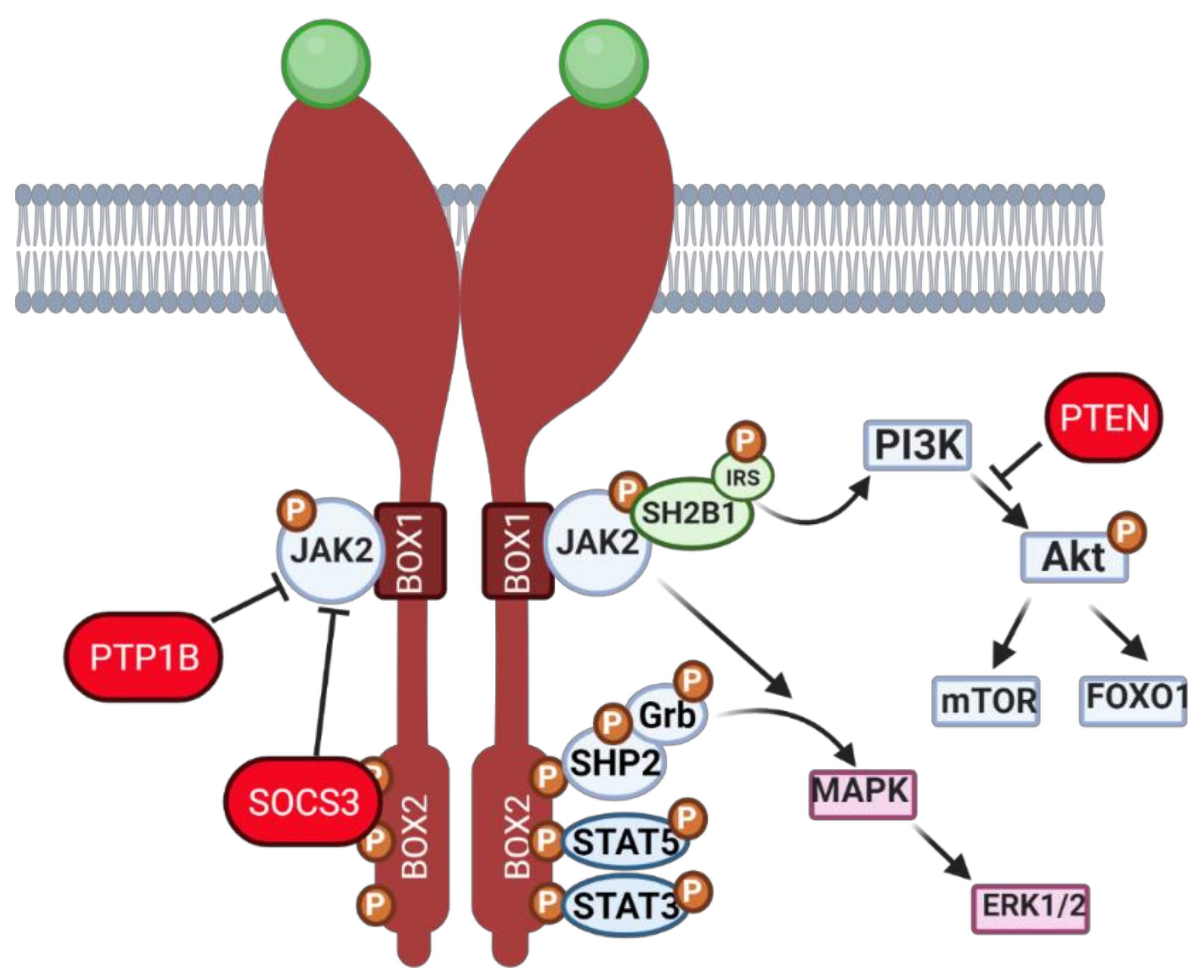
2.1. Leptin—A Common Denominator between Ovarian Function and Obesity
2.2. Other Adipokines and Ovarian Function during Obesity
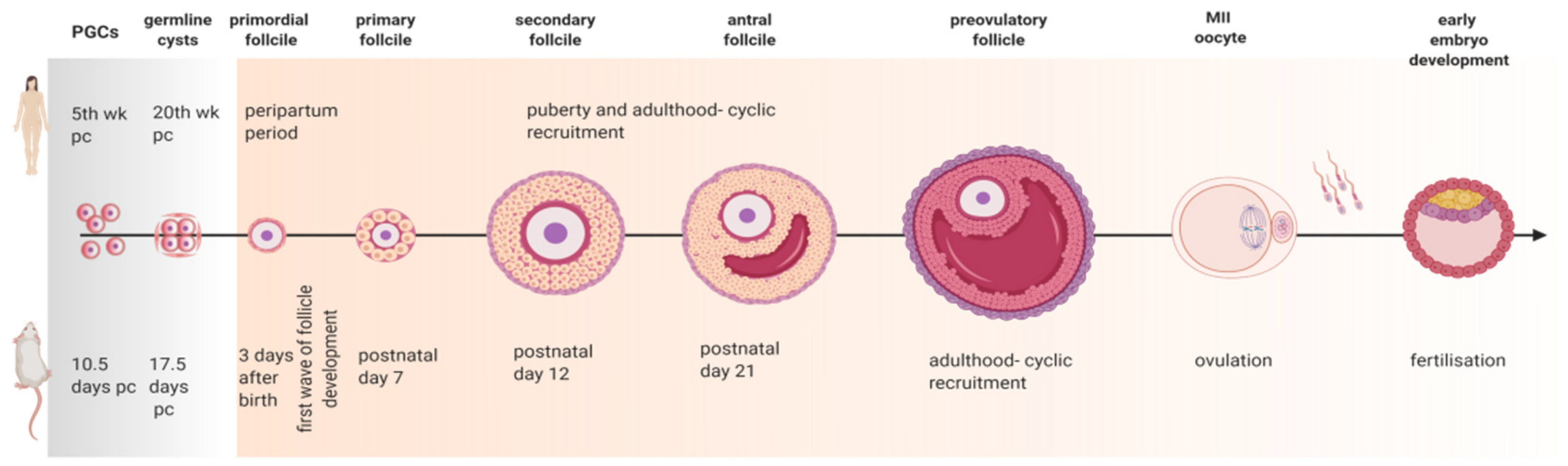
3. Revisiting Folliculogenesis in Mice and Women: A Morphofunctional Characterization
3.1. Primordial Follicle Assembly
3.2. Primary Follicle Development and Growth
3.3. The Road to Ovulation
4. Molecular Mechanisms Regulating Primordial to Primary Follicle Transition
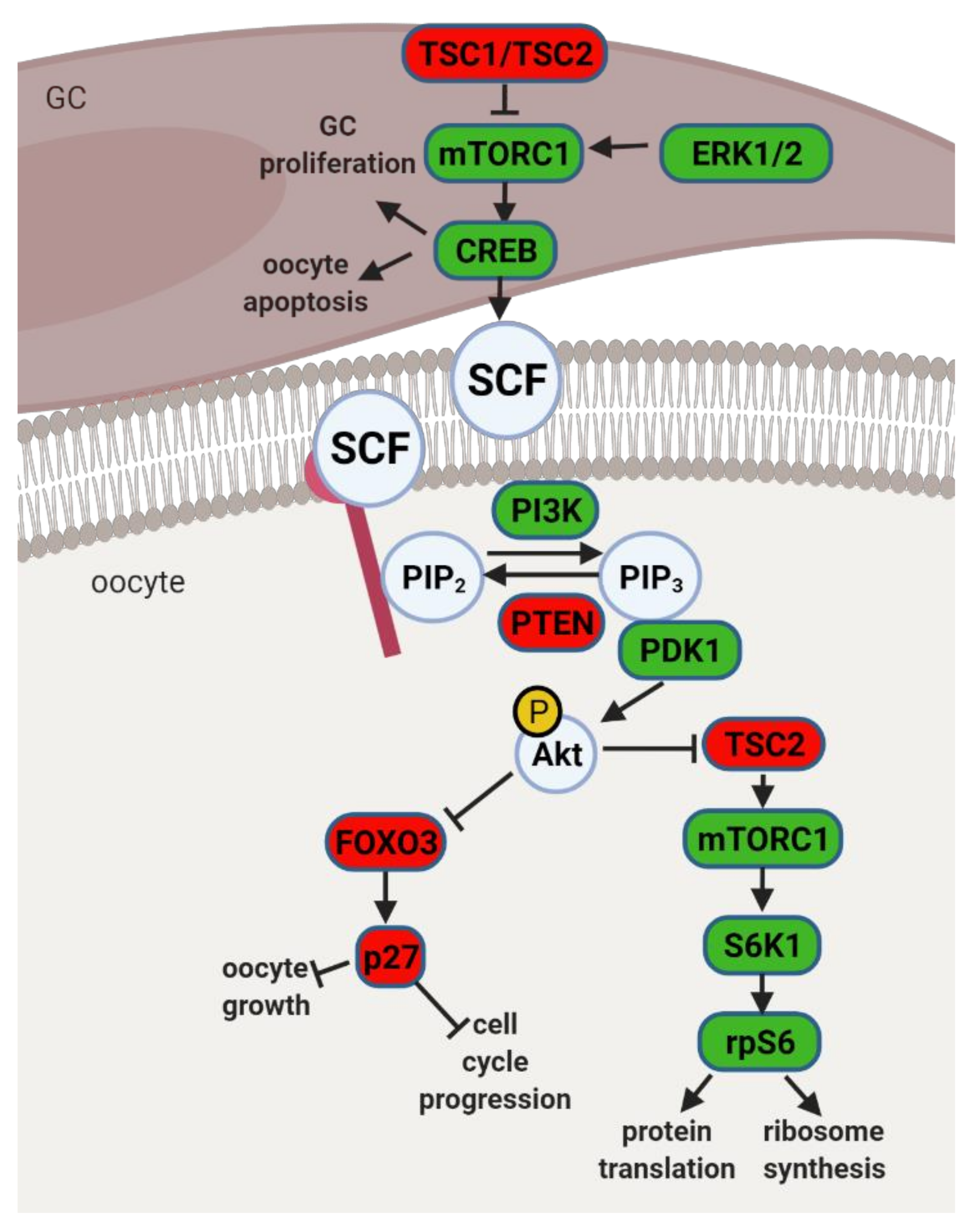
4.1. The PI3K Pathway and Primordial to Primary Follicle Transition
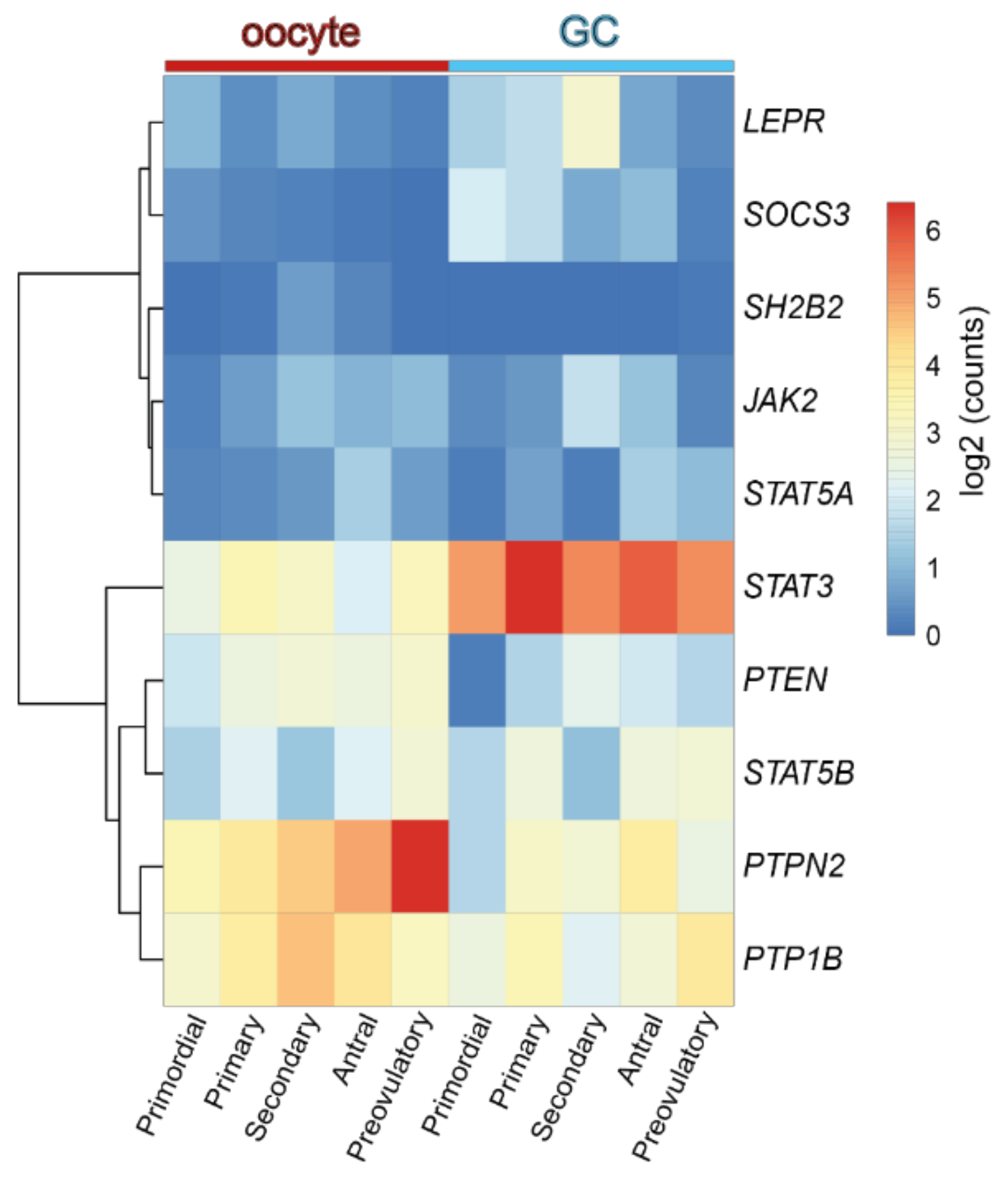
4.2. Obesity, Leptin Signaling and Primordial Follicle Activation
5. Molecular Regulation of Early Antral to Preovulatory Follicle Transition
5.1. Preovulatory Follicle Formation—The Role of Estradiol
5.2. Preovulatory Follicle Formation—The Role of Leptin
5.3. Obesity and Leptin Signaling Disruption during Preovulatory Follicle Formation
6. Oocyte Maturation: The Last Step before Fertilization
6.1. Regulation of Oocyte Maturation
6.2. Leptin Effects on Oocyte Maturation
6.3. Oocyte Maturation and Early Embryo Development in Obese Mothers
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| acyl-CoA | acetyl coenzyme A |
| Akt | Protein kinase B |
| AMH | Anti-Mullerian hormone |
| ATF | Activating transcription factor |
| ATP | Adenosine triphosphate |
| BDNF | Brain-derived nerve factor |
| BMI | Body mass index |
| BMP | Bone morphogenic protein |
| cAMP | Cyclic adenosine monophosphate |
| CART | Cocaine- and amphetamine-regulated transcript |
| CC | Cumulus cells |
| CDC25B | Cell division cycle 25 homolog B |
| cGMP | Cyclic guanosine monophosphate |
| CNS | Central nervous system |
| COCs | Cumulus–oocyte complexes |
| CREB | Activates cyclic AMP-response element binding protein |
| Cx | Connexin |
| CYP | Cytochrome P450 |
| DEGs | Differently expressed genes |
| DIO | Diet-induced obesity |
| DLL | Delta like canonical notch ligand |
| dpc | Days post coitum |
| E2 | Estradiol |
| EGF | Epidermal growth factor |
| Egfr | Epidermal growth factor receptor |
| EOMES | Eomesodermin |
| EnR | Endoplasmic reticulum |
| ER | Estradiol receptor |
| EREs | Estrogens response elements |
| ERK | Extracellular signal regulated kinase |
| FGF | Basic fibroblast growth factor |
| FIGa | Folliculogenesis-specific basic helix–loop–helix |
| FOXO | Forkhead box O |
| FSH | Follicule-stimulating hormone |
| Galnt | Polypeptide N-acetylgalactosaminyltransferase |
| GC | Granulosa cells |
| GDF | Growth differentiation factor |
| GnRH | Gonadotropin releasing hormone |
| GPER | G protein coupled Estrogen receptor |
| GPR | G protein coupled receptor |
| GVBD | Germinal vesicle breakdown |
| HES | Hes family BHLH transcription factor |
| HFD | High-fat diet |
| HSD | Hydroxysteroid dehydrogenase |
| IGF | Insulin-like growth factor |
| IRS | Insulin receptor substrate |
| JAG2 | Jagged canonical notch ligand |
| JAK | Janus kinase |
| Jaml | Junction adhesion molecule like |
| Kit | Stem cell factor receptor |
| LH | Luteinizing hormone |
| Lhx8 | LIM homeobox protein |
| MAPK | Mitogen-activated protein kinase |
| Mater | Maternal antigen that embryos require |
| MEK | Mitogen-activated protein kinas |
| MII | Metaphase II oocyte |
| miR | Micro RNA |
| MPF | Maturation promoting factor |
| MTOCs | Microtubule organizing centers |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NFKB | Nuclear factor kappa B |
| NPP | Natriuretic peptides |
| NPR | Natriuretic peptide receptors |
| NSN | Non-surrounded nucleolus oocyte |
| NT | Neurotrophin |
| ObR | Leptin receptor |
| ObRb | Leptin receptor isoform b |
| p27 | Cyclin-dependent kinase inhibitor 1B |
| P4 | Progesterone |
| P450scc | Cytochrome P450 side chain cleavage |
| PADI | Peptidyl arginine deiminase |
| PB | Polar body |
| PCOS | Polycystic ovary syndrome |
| PDE3A | Phosphodiesterase 3A |
| PDK | Phosphoinositide-dependent kinase |
| PGC | Primordial germ cells |
| PI3K | Phosphatidylinositol 3 kinase |
| PIP2 | Phosphatidylinositol-4,5-biphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-triphosphate |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| POF | Premature ovarian failure |
| PTEN | Phosphatase and tensin homolog |
| PTP1B | Protein tyrosine phosphatase |
| PTPN2 | Protein tyrosine phosphatase non-receptor type 2 |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| rpS6 | Ribosomal protein S6 |
| S1P | Sphingosine 1-phosphate |
| S6K1 | Ribosomal protein S6 kinase beta-1 |
| SCF | Stem cell factor |
| SHP-2 | SH2-domain containing protein tyrosine phosphatase |
| SIRT | Sirtuin |
| SN | Surrounded nucleolus oocyte |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SPC | Sphingosylphosphorylcholine |
| STAT | Signal transducer and activator of transcription |
| TC | Theca cells |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| TSC | Tuberous sclerosis complex |
| TZPs | Transzonal projections |
| wk | Weeks |
References
- Gambineri, A.; Laudisio, D.; Marocco, C.; Radellini, S.; Colao, A.; Savastano, S. Female infertility: Which role for obesity? Int. J. Obes. Suppl. 2019, 9, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Igosheva, N.; Abramov, A.Y.; Poston, L.; Eckert, J.J.; Fleming, T.P.; Duchen, M.R.; McConnell, J. Maternal Diet-Induced Obesity Alters Mitochondrial Activity and Redox Status in Mouse Oocytes and Zygotes. PLoS ONE 2010, 5, e10074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robker, R.L.; Wu, L.L.-Y.; Yang, X. Inflammatory pathways linking obesity and ovarian dysfunction. J. Reprod. Immunol. 2011, 88, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, L.L.; Chura, L.R.; Liang, X.; Lane, M.; Norman, R.J.; Robker, R.L. Exposure to lipid-rich follicular fluid is associated with endoplasmic reticulum stress and impaired oocyte maturation in cumulus-oocyte complexes. Fertil. Steril. 2012, 97, 1438–1443. [Google Scholar] [CrossRef]
- Han, L.; Ren, C.; Li, L.; Li, X.; Ge, J.; Wang, H.; Miao, Y.-L.; Guo, X.; Moley, K.H.; Shu, W.; et al. Embryonic defects induced by maternal obesity in mice derive from Stella insufficiency in oocytes. Nat. Genet. 2018, 50, 432–442. [Google Scholar] [CrossRef]
- Wołodko, K.; Walewska, E.; Adamowski, M.; Castillo-Fernandez, J.; Kelsey, G.; Galvão, A. Leptin Resistance in the Ovary of Obese Mice is Associated with Profound Changes in the Transcriptome of Cumulus Cells. Cell. Physiol. Biochem. 2020, 54, 417–437. [Google Scholar] [CrossRef]
- Dag, Z.O.; Dilbaz, B. Impact of obesity on infertility in women. J. Turk. Gynecol. Assoc. 2015, 16, 111–117. [Google Scholar] [CrossRef]
- Cree-Green, M.; Newcomer, B.R.; Coe, G.; Newnes, L.; Baumgartner, A.; Brown, M.S.; Pyle, L.; Reusch, J.E.; Nadeau, K.J. Peripheral insulin resistance in obese girls with hyperandrogenism is related to oxidative phosphor-ylation and elevated serum free fatty acids. Am. J. Physiol. Metab. 2015, 308, E726–E733. [Google Scholar]
- Chang, A.S.; Dale, A.N.; Moley, K.H. Maternal Diabetes Adversely Affects Preovulatory Oocyte Maturation, Development, and Granulosa Cell Apoptosis. Endocrinology 2005, 146, 2445–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaznik-Wikiel, M.E.; Swindle, D.C.; Allshouse, A.A.; Polotsky, A.J.; McManaman, J.L. High-Fat Diet Causes Subfertility and Compromised Ovarian Function Independent of Obesity in Mice1. Biol. Reprod. 2016, 94, 108. [Google Scholar] [CrossRef]
- Wu, L.L.-Y.; Dunning, K.R.; Yang, X.; Russell, D.L.; Lane, M.; Norman, R.J.; Robker, R.L. High-Fat Diet Causes Lipotoxicity Responses in Cumulus–Oocyte Complexes and Decreased Fertilization Rates. Endocrinology 2010, 151, 5438–5445. [Google Scholar] [CrossRef] [PubMed]
- Boudoures, A.L.; Saben, J.; Drury, A.; Scheaffer, S.; Modi, Z.; Zhang, W.; Moley, K.H. Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy. Dev. Biol. 2017, 426, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-J.; Zhu, C.-C.; Duan, X.; Liu, H.-L.; Wang, Q.; Sun, S.-C. Both diet and gene mutation induced obesity affect oocyte quality in mice. Sci. Rep. 2016, 6, srep18858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robker, R.L.; Akison, L.K.; Bennett, B.D.; Thrupp, P.N.; Chura, L.R.; Russell, D.L.; Lane, M.; Norman, R.J. Obese Women Exhibit Differences in Ovarian Metabolites, Hormones, and Gene Expression Compared with Moderate-Weight Women. J. Clin. Endocrinol. Metab. 2009, 94, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Vaisse, C.; Halaas, J.L.; Horvath, C.M.; Darnell, J.E.; Stoffel, M.; Friedman, J.M. Leptin activation of Stat3 in the hypothalamus of wild–type and ob/ob mice but not db/db mice. Nat. Genet. 1996, 14, 95–97. [Google Scholar] [CrossRef]
- Odle, A.K.; Akhter, N.; Syed, M.M.; Allensworth-James, M.L.; Beneš, H.; Castillo, A.I.M.; MacNicol, M.C.; MacNicol, A.M.; Childs, G.V. Leptin Regulation of Gonadotrope Gonadotropin-Releasing Hormone Receptors as a Metabolic Checkpoint and Gateway to Reproductive Competence. Front. Endocrinol. 2018, 8, 367. [Google Scholar] [CrossRef] [Green Version]
- Ryan, N.K.; Woodhouse, C.M.; Van Der Hoek, K.H.; Gilchrist, R.B.; Armstrong, D.T.; Norman, R.J. Expression of leptin and its receptor in the murine ovary: Possible role in the regulation of oocyte maturation. Biol. Reprod. 2002, 66, 1548–1554. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, C.; Lindell, K.; Svensson, E.; Bergh, C.; Lind, P.; Billig, H.; Carlsson, L.M.; Carlsson, B. Expression of Functional Leptin Receptors in the Human Ovary 1. J. Clin. Endocrinol. Metab. 1997, 82, 4144–4148. [Google Scholar] [CrossRef]
- Welt, C.K.; Schneyer, A.L.; Heist, K.; Mantzoros, C.S. Leptin and Soluble Leptin Receptor in Follicular Fluid. J. Assist. Reprod. Genet. 2003, 20, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Maffei, M.; Halaas, J.L.; Ravussin, E.; Pratley, R.E.; Lee, G.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef]
- Hegyi, K.; Fülöp, K.; Kovacs, K.; Tóth, S.; Falus, A. Leptin-induced signal transduction pathways. Cell Biol. Int. 2004, 28, 159–169. [Google Scholar] [CrossRef]
- Bjørbæk, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent Signaling Capacities of the Long and Short Isoforms of the Leptin Receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef] [Green Version]
- Bjørbæk, C.; Buchholz, R.M.; Davis, S.M.; Bates, S.H.; Pierroz, D.D.; Gu, H.; Neel, B.G.; Myers, M.G., Jr.; Flier, J.S. Divergent Roles of SHP-2 in ERK Activation by Leptin Receptors. J. Biol. Chem. 2001, 276, 4747–4755. [Google Scholar] [CrossRef] [Green Version]
- Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.-M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nat. Cell Biol. 1998, 392, 398–401. [Google Scholar] [CrossRef]
- Tu, X.; Kuang, Z.; Gong, X.; Shi, Y.; Yu, L.; Shi, H.; Wang, J.; Sun, Z. The Influence of LepR Tyrosine Site Mutations on Mouse Ovary Development and Related Gene Expression Changes. PLoS ONE 2015, 10, e0141800. [Google Scholar] [CrossRef] [Green Version]
- Bilbao, M.G.; Di Yorio, M.P.; Faletti, A.G. Different levels of leptin regulate different target enzymes involved in proges-terone synthesis. Fertil. Steril. 2013, 99, 1460–1466. [Google Scholar] [CrossRef]
- Galvão, A.; Tramontano, A.; Rebordão, M.R.; Amaral, A.; Bravo, P.P.; Szóstek, A.; Skarzynski, D.; Mollo, A.; Ferreira-Dias, G. Opposing Roles of Leptin and Ghrelin in the Equine Corpus Luteum Regulation: An In Vitro Study. Mediat. Inflamm. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Panwar, S.; Herrid, M.; Kauter, K.G.; McFarlane, J.R. Effect of passive immunization against leptin on ovarian follicular development in prepubertal mice. J. Reprod. Immunol. 2012, 96, 19–24. [Google Scholar] [CrossRef]
- Swain, J.E.; Dunn, R.L.; McConnell, D.; Gonzalez-Martinez, J.; Smith, G.D. Direct Effects of Leptin on Mouse Reproductive Function: Regulation of Follicular, Oocyte, and Embryo Development. Biol. Reprod. 2004, 71, 1446–1452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yan, Z.; Qin, Q.; Nisenblat, V.; Chang, H.-M.; Yu, Y.; Wang, T.; Lu, C.; Yang, M.; Yang, S.; et al. Transcriptome Landscape of Human Folliculogenesis Reveals Oocyte and Granulosa Cell Interactions. Mol. Cell 2018, 72, 1021–1034.e4. [Google Scholar] [CrossRef] [Green Version]
- Campos, D.B.; Palin, M.F.; Bordignon, V.; Murphy, B.D. The ‘beneficial’ adipokines in reproduction and fertility. Ternational J. Obes. 2008, 32, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Jia, X.; Qiao, J.; Guan, Y.; Kang, J. Adipokines in reproductive function: A link between obesity and polycystic ovary syndrome. J. Mol. Endocrinol. 2013, 50, R21–R37. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Ho-meostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Di Carlo, C.; Tommaselli, G.A.; De Filippo, E.; Pisano, G.; Nasti, A.; Bifulco, G.; Contaldo, F.; Nappi, C. Menstrual status and serum leptin levels in anorectic and in menstruating women with low body mass indexes. Fertil. Steril. 2002, 78, 376–382. [Google Scholar] [CrossRef]
- Richards, J.S.; Liu, Z.; Kawai, T.; Tabata, K.; Watanabe, H.; Suresh, D.; Kuo, F.-T.; Pisarska, M.D.; Shimada, M. Adiponectin and its receptors modulate granulosa cell and cumulus cell functions, fertility, and early embryo development in the mouse and human. Fertil. Steril. 2012, 98, 471–479.e1. [Google Scholar] [CrossRef] [Green Version]
- Maillard, V.; Uzbekova, S.; Guignot, F.; Perreau, C.; Ramé, C.; Coyral-Castel, S.; Dupont, J. Effect of adiponectin on bovine granulosa cell steroidogenesis, oocyte maturation and embryo development. Reprod. Biol. Endocrinol. 2010, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, Y.; Hua, L.; Feng, B.; Jiang, X.; Li, J.; Jiang, D.; Huang, X.; Zhu, Y.; Li, Z.; Yan, L.; et al. Fibroblast growth factor 21 coordinates adiponectin to mediate the beneficial effects of low-protein diet on primordial follicle reserve. EBioMedicine 2019, 41, 623–635. [Google Scholar] [CrossRef] [Green Version]
- Zieba, D.; Biernat, W.; Barć, J. Roles of leptin and resistin in metabolism, reproduction, and leptin resistance. Domest. Anim. Endocrinol. 2020, 73, 106472. [Google Scholar] [CrossRef]
- Biernat, W.; Kirsz, K.; Szczesna, M.; Zieba, D.A. Resistin regulates reproductive hormone secretion from the ovine adenohypophysis depending on season. Domest. Anim. Endocrinol. 2018, 65, 95–100. [Google Scholar] [CrossRef]
- Estienne, A.; Bongrani, A.; Reverchon, M.; Ramé, C.; Ducluzeau, P.-H.; Froment, P.; Dupont, J. Involvement of Novel Adipokines, Chemerin, Visfatin, Resistin and Apelin in Reproductive Functions in Normal and Pathological Conditions in Humans and Animal Models. Int. J. Mol. Sci. 2019, 20, 4431. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Tian, J.; Blizzard, L.; Oddy, W.H.; Dwyer, T.; Bazzano, L.A.; Hickey, M.; Harville, E.W.; Venn, A.J. Associations of childhood adiposity with menstrual irregularity and polycystic ovary syndrome in adulthood: The Childhood Determinants of Adult Health Study and the Bogalusa Heart Study. Hum. Reprod. 2020, 35, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Monk, M.; McLaren, A. X-chromosome activity in foetal germ cells of the mouse. J. Embryol. Exp. Morphol. 1981, 63, 75–84. [Google Scholar] [PubMed]
- Borum, K. Oogenesis in the mouse. A study of the origin of the mature ova. Exp. Cell Res. 1967, 45, 39–47. [Google Scholar] [CrossRef]
- Pepling, M.E.; Spradling, A.C. Mouse Ovarian Germ Cell Cysts Undergo Programmed Breakdown to Form Primordial Follicles. Dev. Biol. 2001, 234, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepling, M.E.; Sundman, E.A.; Patterson, N.L.; Gephardt, G.W.; Medico, L.; Wilson, K.I. Differences in oocyte development and estradiol sensitivity among mouse strains. Reproduction 2010, 139, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Baerwald, A.R.; Adams, G.P.; Pierson, R.A. Ovarian antral folliculogenesis during the human menstrual cycle: A review. Hum. Reprod. Update 2011, 18, 73–91. [Google Scholar] [CrossRef] [Green Version]
- Peters, H.; Byskov, A.; Grinsted, J. Follicular growth in fetal and prepubertal ovaries of humans and other primates. Clin. Endocrinol. Metab. 1978, 7, 469–485. [Google Scholar] [CrossRef]
- Skinner, M.K. Regulation of primordial follicle assembly and development. Hum. Reprod. Update 2005, 11, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Jamnongjit, M.; Hammes, S.R. Oocyte Maturation: The Coming of Age of a Germ Cell. Semin. Reprod. Med. 2005, 23, 234–241. [Google Scholar] [CrossRef] [Green Version]
- McGee, E.A.; Hsueh, A.J.W. Initial and Cyclic Recruitment of Ovarian Follicles. Endocr. Rev. 2000, 21, 200–214. [Google Scholar] [CrossRef]
- Kaipia, A.; Hsueh, A.J.W. Regulation of ovarian follicle atresia. Annu. Rev. Physiol. 1997, 59, 349–363. [Google Scholar] [CrossRef]
- Faddy, M.J.; Gosden, R.G.; Edwards, R.G. Ovarian follicle dynamics in mice: A comparative study of three inbred strains and an F1 hybrid. J. Endocrinol. 1983, 96, 23–33. [Google Scholar] [CrossRef]
- Findlay, J.K.; Hutt, K.J.; Hickey, M.; Anderson, R.A. How Is the Number of Primordial Follicles in the Ovarian Reserve Established? Biol. Reprod. 2015, 93, 111. [Google Scholar] [CrossRef]
- Tsafriri, A.; Reich, R.; Abisogun, A.O. The Ovarian Egg and Ovulation. Marshall’s Physiol. Reprod. 1994, 3, 1–91. [Google Scholar]
- Guzeloglu-Kayisli, O.; Basar, M.; Arici, A. Basic aspects of implantation. Reprod. Biomed. Online 2007, 15, 728–739. [Google Scholar] [CrossRef]
- Fortune, J.; Cushman, R.; Wahl, C.; Kito, S. The primordial to primary follicle transition. Mol. Cell. Endocrinol. 2000, 163, 53–60. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Zheng, N.; Li, B.; Yang, J.; Zhang, C.; Xia, G.; Zhang, M. CREB activity is required for mTORC1 signaling-induced primordial follicle activation in mice. Histochem. Cell Biol. 2020, 154, 287–299. [Google Scholar] [CrossRef]
- Castrillon, D.H.; Miao, L.; Kollipara, R.; Horner, J.W.; Depinho, R.A. Suppression of Ovarian Follicle Activation in Mice by the Transcription Factor Foxo3a. Science 2003, 301, 215–218. [Google Scholar] [CrossRef]
- Reddy, P.; Adhikari, D.; Zheng, W.; Liang, S.; Hämäläinen, T.; Tohonen, V.; Ogawa, W.; Noda, T.; Volarevic, S.; Huhtaniemi, I.; et al. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum. Mol. Genet. 2009, 18, 2813–2824. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.; Liu, L.; Adhikari, D.; Jagarlamudi, K.; Rajareddy, S.; Shen, Y.; Du, C.; Tang, W.; La Inen, T.H.M.; Peng, S.L.; et al. Oocyte-Specific Deletion of Pten Causes Premature Activation of the Primordial Follicle Pool. Science 2008, 319, 611–613. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, D.; Zheng, W.; Shen, Y.; Gorre, N.; Hämäläinen, T.; Cooney, A.J.; Huhtaniemi, I.; Lan, Z.-J.; Liu, K. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum. Mol. Genet. 2009, 19, 397–410. [Google Scholar] [CrossRef]
- Li, J.; Kawamura, K.; Cheng, Y.; Liu, S.; Klein, C.; Duan, E.-K.; Hsueh, A.J.W. Activation of dormant ovarian follicles to generate mature eggs. Proc. Natl. Acad. Sci. USA 2010, 107, 10280–10284. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Wang, W.; Zhou, B.; Zhang, W.; Huang, P.; Shi, F.; Taya, K. Formation of Primordial Follicles and Immunolocalization of PTEN, PKB and FOXO3A Proteins in the Ovaries of Fetal and Neonatal Pigs. J. Reprod. Dev. 2010, 56, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Albertini, D.F.; Nishimori, K.; Kumar, T.R.; Lu, N.; Matzuk, M.M. Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nat. Cell Biol. 1996, 383, 531–535. [Google Scholar] [CrossRef]
- Yoshida, H.; Takakura, N.; Kataoka, H.; Kunisada, T.; Okamura, H.; Nishikawa, S.I. Stepwise requirement of c-kit tyrosine kinase in mouse ovarian follicle development. Dev. Biol. 1997, 184, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Kezele, P.R.; Nilsson, E.E.; Skinner, M.K. Insulin but not insulin-like growth factor-1 promotes the primordial to primary follicle transition. Mol. Cell. Endocrinol. 2002, 192, 37–43. [Google Scholar] [CrossRef]
- Wang, N.; Luo, L.-L.; Xu, J.-J.; Xu, M.-Y.; Zhang, X.-M.; Zhou, X.-L.; Liu, W.-J.; Fu, Y.-C. Obesity accelerates ovarian follicle development and follicle loss in rats. Metabolism 2014, 63, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Nteeba, J.; Ross, J.; Ii, J.P.; Keating, A. High fat diet induced obesity alters ovarian phosphatidylinositol-3 kinase signaling gene expression. Reprod. Toxicol. 2013, 42, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Zhang, R.; Guan, H.; Zhang, W. Follicle loss and PTEN/PI3K/mTOR signaling pathway activated in LepR-mutated mice. Gynecol. Endocrinol. 2019, 35, 44–48. [Google Scholar] [CrossRef]
- Loh, K.; Fukushima, A.; Zhang, X.; Galic, S.; Briggs, D.; Enriori, P.J.; Simonds, S.; Wiede, F.; Reichenbach, A.; Hauser, C.; et al. Elevated Hypothalamic TCPTP in Obesity Contributes to Cellular Leptin Resistance. Cell Metab. 2011, 14, 684–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durlinger, A.L.L.; Kramer, P.; Karels, B.; De Jong, F.H.; Uilenbroek, J.T.J.; Grootegoed, J.A.; Themmen, A.P.N. Control of Primordial Follicle Recruitment by Anti-Müllerian Hormone in the Mouse Ovary1. Endocrinology 1999, 140, 5789–5796. [Google Scholar] [CrossRef]
- Ren, Y.; Suzuki, H.; Jagarlamudi, K.; Golnoski, K.; McGuire, M.; Lopes, R.; Pachnis, V.; Rajkovic, A. Lhx8 regulates primordial follicle activation and postnatal folliculogenesis. BMC Biol. 2015, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-P.; Yang, J.-L.; Zhang, J.; Li, L.; Huang, L.; Ji, S.-Y.; Hu, Z.-Y.; Gao, F.; Liu, Y.-X. Notch Signaling Is Involved in Ovarian Follicle Development by Regulating Granulosa Cell Proliferation. Endocrinology 2011, 152, 2437–2447. [Google Scholar] [CrossRef]
- Yu, J.; Yaba, A.; Kasiman, C.; Thomson, T.; Johnson, J. mTOR Controls Ovarian Follicle Growth by Regulating Granulosa Cell Proliferation. PLoS ONE 2011, 6, e21415. [Google Scholar] [CrossRef]
- Hsueh, A.J.W.; Billig, H.; Tsafriri, A. Ovarian Follicle Atresia: A Hormonally Controlled Apoptotic Process. Endocr. Rev. 1994, 15, 707–724. [Google Scholar] [CrossRef]
- Lin, F.; Li, R.; Pan, Z.X.; Zhou, B.; Yu, D.B.; Wang, X.G.; Ma, X.S.; Han, J.; Shen, M.; Liu, H.L. miR-26b Promotes Granulosa Cell Apoptosis by Targeting ATM during Follicular Atresia in Porcine Ovary. PLoS ONE 2012, 7, e38640. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Du, X.; Liu, L.; Liu, H.; Pan, Z. Upregulation of miR-146b promotes porcine ovarian granulosa cell apoptosis by attenuating CYP19A1. Domest. Anim. Endocrinol. 2021, 74, 106509. [Google Scholar] [CrossRef]
- Yuan, S.; Wen, J.; Cheng, J.; Shen, W.; Zhou, S.; Yan, W.; Shen, L.; Luo, A.; Wang, S. Age-associated up-regulation of EGR1 promotes granulosa cell apoptosis during follicle atresia in mice through the NF-κB pathway. Cell Cycle 2016, 15, 2895–2905. [Google Scholar] [CrossRef] [Green Version]
- Braw, R.H.; Tsafriri, A. Effect of PMSG on follicular atresia in the immature rat ovary. Reproduction 1980, 59, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Franks, S.; Hardy, K. Androgen Action in the Ovary. Front. Endocrinol. 2018, 9, 452. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.T.; Dorrington, J.H. Estrogen biosynthesis in the ovaries and testes. Adv. Sex Horm. Res. 1977, 3, 217–258. [Google Scholar] [PubMed]
- Kristensen, S.G.; Mamsen, L.S.; Jeppesen, J.V.; Bøtkjær, J.A.; Pors, S.E.; Borgbo, T.; Ernst, E.; Macklon, K.T.; Andersen, C.Y. Hallmarks of Human Small Antral Follicle Development: Implications for Regulation of Ovarian Steroidogenesis and Selection of the Dominant Follicle. Front. Endocrinol. 2018, 8, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Xin, Q.; Wang, X.; Wang, S.; Wang, H.; Zhang, W.; Yang, Y.; Zhang, Y.; Zhang, Z.; Wang, C.; et al. Estrogen receptors in granulosa cells govern meiotic resumption of pre-ovulatory oocytes in mammals. Cell Death Dis. 2017, 8, e2662. [Google Scholar] [CrossRef] [Green Version]
- Sar, M.; Welsch, F. Differential Expression of Estrogen Receptor-β and Estrogen Receptor-α in the Rat Ovary. Endocrinology 1999, 140, 963–971. [Google Scholar] [CrossRef]
- Dupont, S.; Krust, A.; Gansmuller, A.; Dierich, A.; Chambon, P.; Mark, M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 2000, 127, 4277–4291. [Google Scholar]
- Chakravarthi, V.P.; Ratri, A.; Masumi, S.; Borosha, S.; Ghosh, S.; Christenson, L.K.; Roby, K.F.; Wolfe, M.W.; Rumi, M.K. Granulosa cell genes that regulate ovarian follicle development beyond the antral stage: The role of estrogen receptor β. Mol. Cell. Endocrinol. 2021, 528, 111212. [Google Scholar] [CrossRef]
- Kovács, T.; Szabó-Meleg, E.; Ábrahám, I.M. Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression. Int. J. Mol. Sci. 2020, 21, 3177. [Google Scholar] [CrossRef]
- Simon, A.M.; Goodenough, D.A.; Li, E.; Paul, D.L. Female infertility in mice lacking connexin 37. Nat. Cell Biol. 1997, 385, 525–529. [Google Scholar] [CrossRef]
- Granot, I.; Dekel, N. The ovarian gap junction protein connexin43: Regulation by gonadotropins. Trends Endocrinol. Metab. 2002, 13, 310–313. [Google Scholar] [CrossRef]
- Wiesen, J.F.; Midgley, A.R. Changes in expression of connexin 43 gap junction messenger ribonucleic acid and protein during ovarian follicular growth. Endocrinology 1993, 133, 741–746. [Google Scholar] [CrossRef]
- Albertini, D.F.; Combelles, C.M.; Benecchi, E.; Carabatsos, M.J. Cellular basis for paracrine regulation of ovarian follicle development. Reproduction 2001, 121, 647–653. [Google Scholar] [CrossRef]
- Combelles, C.M.; Carabatsos, M.J.; Kumar, T.R.; Matzuk, M.M.; Albertini, D.F. Hormonal control of somatic cell oocyte interactions during ovarian follicle development. Mol. Reprod. Dev. 2004, 69, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Makita, M.; Miyano, T. Steroid hormones promote bovine oocyte growth and connection with granulosa cells. Theriogenology 2014, 82, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Zachow, R.J.; Magoffin, D.A. Direct Intraovarian Effects of Leptin: Impairment of the Synergistic Action of Insulin-Like Growth Factor-I on Follicle-Stimulating Hormone-Dependent Estradiol-17β Production by Rat Ovarian Granulosa Cells. Endocrinology 1997, 138, 847–850. [Google Scholar] [CrossRef]
- Guo, X.; Chen, S.; Xing, F. Effects of leptin on estradiol and progesterone production by human luteinized granulosa cells in vitro. Zhonghua Fu Chan Ke Za Zhi 2001, 36, 95–97. [Google Scholar]
- Gregoraszczuk, E.Ł.; Wójtowicz, A.K.; Ptak, A.; Nowak, K. In vitro effect of leptin on steroids’ secretion by FSH- and LH-treated porcine small, medium and large preovulatory follicles. Reprod. Biol. 2003, 3, 227–239. [Google Scholar]
- Agarwal, S.K.; Vogel, K.; Weitsman, S.R.; Magoffin, D.A. Leptin Antagonizes the Insulin-Like Growth Factor-I Aug-mentation of Steroidogenesis in Granulosa and Theca Cells of the Human Ovary 1. J. Clin. Endocrinol. Metab. 1999, 84, 1072–1076. [Google Scholar] [PubMed] [Green Version]
- Ma, X.; Hayes, E.; Prizant, H.; Srivastava, R.K.; Hammes, S.R.; Sen, A. Leptin-induced CART (cocaine- and amphetamine-regulated transcript) is a novel intraovarian mediator of obesity-related infertility in females. Endocrinology 2016, 157, 1248–1257. [Google Scholar] [CrossRef]
- Mirshamsi, S.; Laidlaw, H.A.; Ning, K.; Anderson, E.; Burgess, L.A.; Gray, A.; Sutherland, C.; Ashford, M.L.J. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci. 2004, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liang, J.; Wu, W.K.K.; Yu, X.; Yu, J.; Weng, X.; Shen, J. Leptin Activates RhoA/ROCK Pathway to Induce Cytoskeleton Remodeling in Nucleus Pulposus Cells. Int. J. Mol. Sci. 2014, 15, 1176–1188. [Google Scholar] [CrossRef]
- Antczak, M.; Blerkom, V.J. Oocyte influences on early development: The regulatory proteins leptin and STAT3 are polarized in mouse and human oocytes and differentially distributed within the cells of the preimplantation stage embryo. Mol. Hum. Reprod. 1997, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.-H.; Liao, J.; Zhang, J.-Y.; Liang, C.; Song, C.-H.; Han, M.; Wang, L.-H.; Xue, H.; Zhang, K.; Zabeau, L.; et al. Inhibition of the Connexin 43 Elevation May be Involved in the Neuroprotective Activity of Leptin Against Brain Ischemic Injury. Cell. Mol. Neurobiol. 2014, 34, 871–879. [Google Scholar] [CrossRef]
- Panza, S.; Russo, U.; Giordano, F.; Leggio, A.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Malivindi, R.; Conforti, F.L.; Naimo, G.D.; et al. Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression. Biomolecules 2020, 10, 886. [Google Scholar] [CrossRef]
- Quinkler, M.; Sinha, B.; Tomlinson, J.W.; Bujalska, I.J.; Stewart, P.M.; Arlt, W. Androgen generation in adipose tissue in women with simple obesity—A site-specific role for 17β-hydroxysteroid dehydrogenase type 5. J. Endocrinol. 2004, 183, 331–342. [Google Scholar] [CrossRef]
- Wu, S.; Divall, S.; Nwaopara, A.; Radovick, S.; Wondisford, F.; Ko, C.; Wolfe, A. Obesity-Induced Infertility and Hyperandrogenism Are Corrected by Deletion of the Insulin Receptor in the Ovarian Theca Cell. Diabetes 2013, 63, 1270–1282. [Google Scholar] [CrossRef] [Green Version]
- Souter, I.; Baltagi, L.M.; Kuleta, D.; Meeker, J.D.; Petrozza, J.C. Women, weight, and fertility: The effect of body mass index on the outcome of superovulation/intrauterine insemination cycles. Fertil. Steril. 2011, 95, 1042–1047. [Google Scholar] [CrossRef]
- Sagae, S.C.; Menezes, E.F.; Bonfleur, M.L.; Vanzela, E.C.; Zacharias, P.; Lubaczeuski, C.; Franci, C.R.; Sanvitto, G.L. Early onset of obesity induces reproductive deficits in female rats. Physiol. Behav. 2012, 105, 1104–1111. [Google Scholar] [CrossRef]
- Arnold, S.F.; Melamed, M.; Vorojeikina, D.P.; Notides, A.C.; Sasson, S. Estradiol-Binding Mechanism and Binding Capacity of the Human Estrogen Receptor Is Regulated by Tyrosine Phosphorylation. Mol. Endocrinol. 1997, 11, 48–53. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Malivindi, R.; Lanzino, M.; Rizza, P.; Casaburi, I.; Bonofiglio, D.; Catalano, S.; Andò, S. Estrogens and PTP1B Function in a Novel Pathway to Regulate Aromatase Enzymatic Activity in Breast Cancer Cells. Endocrinology 2012, 153, 5157–5166. [Google Scholar] [CrossRef] [Green Version]
- White, C.L.; Whittington, A.; Barnes, M.J.; Wang, Z.; Bray, G.A.; Morrison, C.D. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am. J. Physiol. Metab. 2009, 296, E291–E299. [Google Scholar] [CrossRef]
- Fan, H.Y.; Liu, Z.; Shimada, M.; Sterneck, E.; Johnson, P.F.; Hedrick, S.M.; Richards, J.S. MAPK3/1 (ERK1/2) in ovarian granulosa cells are essential for female fertility. Science 2009, 324, 938–941. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.-Y.; Li, M.; Luo, Y.-B.; Song, S.; Tian, D.; Yang, J.; Zhang, B.; Hou, Y.; Schatten, H.; Liu, Z.; et al. Maternal factors required for oocyte developmental competence in mice: Transcriptome analysis of non-surrounded nucleolus (NSN) and surrounded nucleolus (SN) oocytes. Cell Cycle 2013, 12, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Hinckley, M.; Vaccari, S.; Horner, K.; Chen, R.; Conti, M. The G-protein-coupled receptors GPR3 and GPR12 are involved in cAMP signaling and maintenance of meiotic arrest in rodent oocytes. Dev. Biol. 2005, 287, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.S.; Han, S.J.; Conti, M. Wee1B, Myt1, and Cdc25 function in distinct compartments of the mouse oocyte to control meiotic resumption. J. Cell Biol. 2010, 188, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Li, J. The art of oocyte meiotic arrest regulation 11 Medical and Health Sciences 1114 Paediatrics and Reproductive Medicine. Reprod. Biol. Endocrinol. 2019, 17, 1–12. [Google Scholar]
- Vaccari, S.; Weeks, J.L.; Hsieh, M.; Menniti, F.S.; Conti, M. Cyclic GMP signaling is involved in the luteinizing hormone-dependent meiotic maturation of mouse oocytes. Biol. Reprod. 2009, 81, 595–604. [Google Scholar] [CrossRef]
- Nogueira, D.; Ron-El, R.; Friedler, S.; Schachter, M.; Raziel, A.; Cortvrindt, R.; Smitz, J. Meiotic Arrest In Vitro by Phosphodiesterase 3-Inhibitor Enhances Maturation Capacity of Human Oocytes and Allows Subsequent Embryonic Development. Biol. Reprod. 2006, 74, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y.; Xu, X.; Li, J.; Yuan, F.; Bo, S.; Qiao, J.; Xia, G.; Su, Y.; Zhang, M. Transforming growth factor-β is involved in maintaining oocyte meiotic arrest by promoting natriuretic peptide type C expression in mouse granulosa cells. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.; Jamnongjit, M.; Hammes, S.R. Androgens Promote Maturation and Signaling in Mouse Oocytes Independent of Transcription: A Release of Inhibition Model for Mammalian Oocyte Meiosis. Mol. Endocrinol. 2004, 18, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holubcová, Z.; Blayney, M.; Elder, K.; Schuh, M. Error-prone chromosome-mediated spindle assembly favors chromosome segregation defects in human oocytes. Science 2015, 348, 1143–1147. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.J. Oocyte cytoplasmic maturation: A key mediator of oocyte and embryo developmental competence1. J. Anim. Sci. 2007, 85, E1–E3. [Google Scholar] [CrossRef] [Green Version]
- Schisa, J.A. New Insights into the Regulation of RNP Granule Assembly in Oocytes. Int. Rev. Cell Mol. Biol. 2012, 295, 233–289. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Vitale, A.M.; Leijten, F.P.J.; Strik, A.M.; Koonen-Reemst, A.M.C.B.; Yurttas, P.; Robben, T.J.A.A.; Coonrod, S.; Gossen, J.A. Peptidylarginine deiminase (PAD) 6 is essential for oocyte cytoskeletal sheet formation and female fertility. Mol. Cell. Endocrinol. 2007, 273, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Kan, R.; Anguish, L.; Nelson, L.M.; Coonrod, S.A. Potential Role for MATER in Cytoplasmic Lattice Formation in Murine Oocytes. PLoS ONE 2010, 5, e12587. [Google Scholar] [CrossRef]
- Steeves, T.E.; Gardner, D.K.; Zuelke, K.A.; Squires, T.S.; Fry, R.C. In vitro development and nutrient uptake by embryos derived from oocytes of pre-pubertal and adult cows. Mol. Reprod. Dev. 1999, 54, 49–56. [Google Scholar] [CrossRef]
- Cui, X.-S.; Li, X.-Y.; Yin, X.-J.; Kong, I.K.; Kang, J.-J.; Kim, N.-H. Maternal Gene Transcription in Mouse Oocytes: Genes Implicated in Oocyte Maturation and Fertilization. J. Reprod. Dev. 2007, 53, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Katz-Jaffe, M.; McCallie, B.; Preis, K.; Filipovits, J.; Gardner, D. Transcriptome analysis of in vivo and in vitro matured bovine MII oocytes. Theriogenology 2009, 71, 939–946. [Google Scholar] [CrossRef]
- Downs, S.M.; Hudson, E.D. Energy substrates and the completion of spontaneous meiotic maturation. Zygote 2000, 8, 339–351. [Google Scholar] [CrossRef]
- Sanfins, A.; Rodrigues, P.; Albertini, D.F. GDF-9 and BMP-15 direct the follicle symphony. J. Assist. Reprod. Genet. 2018, 35, 1741–1750. [Google Scholar] [CrossRef]
- Wilding, M.; Coppola, G.; Dale, B.; Di Matteo, L. Mitochondria and human preimplantation embryo development. Reproduction 2009, 137, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Dunning, K.R.; Anastasi, M.R.; Zhang, V.J.; Russell, D.L.; Robker, R.L. Regulation of Fatty Acid Oxidation in Mouse Cumulus-Oocyte Complexes during Maturation and Modulation by PPAR Agonists. PLoS ONE 2014, 9, e87327. [Google Scholar] [CrossRef] [Green Version]
- Reidy, S.P.; Weber, J.-M. Leptin: An essential regulator of lipid metabolism. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2000, 125, 285–298. [Google Scholar] [CrossRef]
- Paula-Lopes, F.F.; Boelhauve, M.; Habermann, F.A.; Sinowatz, F.; Wolf, E. Leptin Promotes Meiotic Progression and Developmental Capacity of Bovine Oocytes Via Cumulus Cell-Independent and -Dependent Mechanisms1. Biol. Reprod. 2007, 76, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Craig, J.; Zhu, H.; Dyce, P.W.; Petrik, J.; Li, J. Leptin enhances oocyte nuclear and cytoplasmic maturation via the mitogen-activated protein kinase pathway. Endocrinology 2004, 145, 5355–5363. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Kawamura, K.; Sasaki, M.; Kawamura, N.; Groenen, P.; Gelpke, M.D.S.; Kumagai, J.; Fukuda, J.; Tanaka, T. Leptin and ObRa/MEK signalling in mouse oocyte maturation and preimplantation embryo development. Reprod. Biomed. Online 2009, 19, 181–190. [Google Scholar] [CrossRef]
- Ahrén, B.; Havel, P.J. Leptin inhibits insulin secretion induced by cellular cAMP in a pancreatic B cell line (INS-1 cells). Am. J. Physiol. Content 1999, 277, R959–R966. [Google Scholar] [CrossRef]
- Zhao, A.Z.; Bornfeldt, K.E.; Beavo, J.A. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J. Clin. Investig. 1998, 102, 869–873. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Hou, M.; Xia, M.; Wang, Q.; Zhu, H.; Xiao, Y.; Tang, Z.; Ma, J.; Ling, W. Globular adiponectin decreases leptin-induced tumor necrosis factor-α expression by murine macrophages: Involvement of cAMP-PKA and MAPK pathways. Cell. Immunol. 2005, 238, 19–30. [Google Scholar] [CrossRef]
- Morton, G.J.; Schwartz, M.W. Leptin and the Central Nervous System Control of Glucose Metabolism. Physiol. Rev. 2011, 91, 389–411. [Google Scholar] [CrossRef] [Green Version]
- Silva, E.; Paczkowski, M.; Krisher, R.L. The effect of leptin on maturing porcine oocytes is dependent on glucose concentration. Mol. Reprod. Dev. 2012, 79, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.H.; Moley, K.H. The impact of obesity on egg quality. J. Assist. Reprod. Genet. 2011, 28, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.H.; Chi, M.M.; Moley, K.H. Insulin-Stimulated Glucose Uptake Occurs in Specialized Cells within the Cumulus Oocyte Complex. Endocrinology 2012, 153, 2444–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton-McDowall, M.L.; Gilchrist, R.B.; Thompson, J.G. The pivotal role of glucose metabolism in determining oocyte developmental competence. Reproduction 2010, 139, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Robker, R.L. Evidence that obesity alters the quality of oocytes and embryos. Pathophysiology 2008, 15, 115–121. [Google Scholar] [CrossRef]
- Huang, X.; Hao, C.; Shen, X.; Liu, X.; Shan, Y.; Zhang, Y.; Chen, L. Differences in the transcriptional profiles of human cumulus cells isolated from MI and MII oocytes of patients with polycystic ovary syndrome. Reproduction 2013, 145, 597–608. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wołodko, K.; Castillo-Fernandez, J.; Kelsey, G.; Galvão, A. Revisiting the Impact of Local Leptin Signaling in Folliculogenesis and Oocyte Maturation in Obese Mothers. Int. J. Mol. Sci. 2021, 22, 4270. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084270
Wołodko K, Castillo-Fernandez J, Kelsey G, Galvão A. Revisiting the Impact of Local Leptin Signaling in Folliculogenesis and Oocyte Maturation in Obese Mothers. International Journal of Molecular Sciences. 2021; 22(8):4270. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084270
Chicago/Turabian StyleWołodko, Karolina, Juan Castillo-Fernandez, Gavin Kelsey, and António Galvão. 2021. "Revisiting the Impact of Local Leptin Signaling in Folliculogenesis and Oocyte Maturation in Obese Mothers" International Journal of Molecular Sciences 22, no. 8: 4270. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084270