
Leucine Reconstitutes Phagocytosis-Induced Cell Death in E. coli-Infected Neonatal Monocytes—Effects on Energy Metabolism and mTOR Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
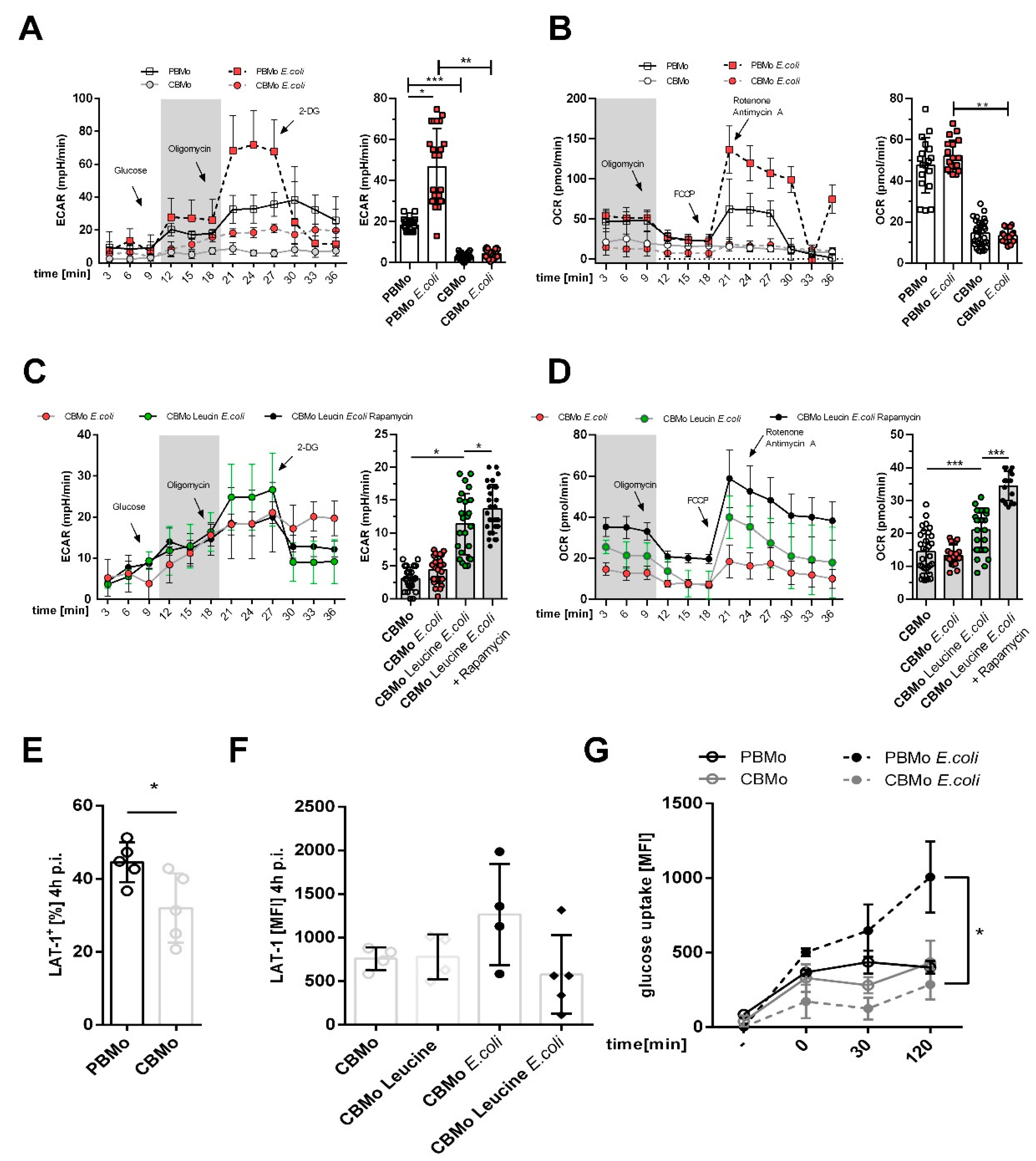
2.1. Glucose-Dependent Energy Metabolism Differs between PBMo and CBMo
2.2. Leucine Supplementation Does Not Interfere with Peri-Phagocytic Reactions
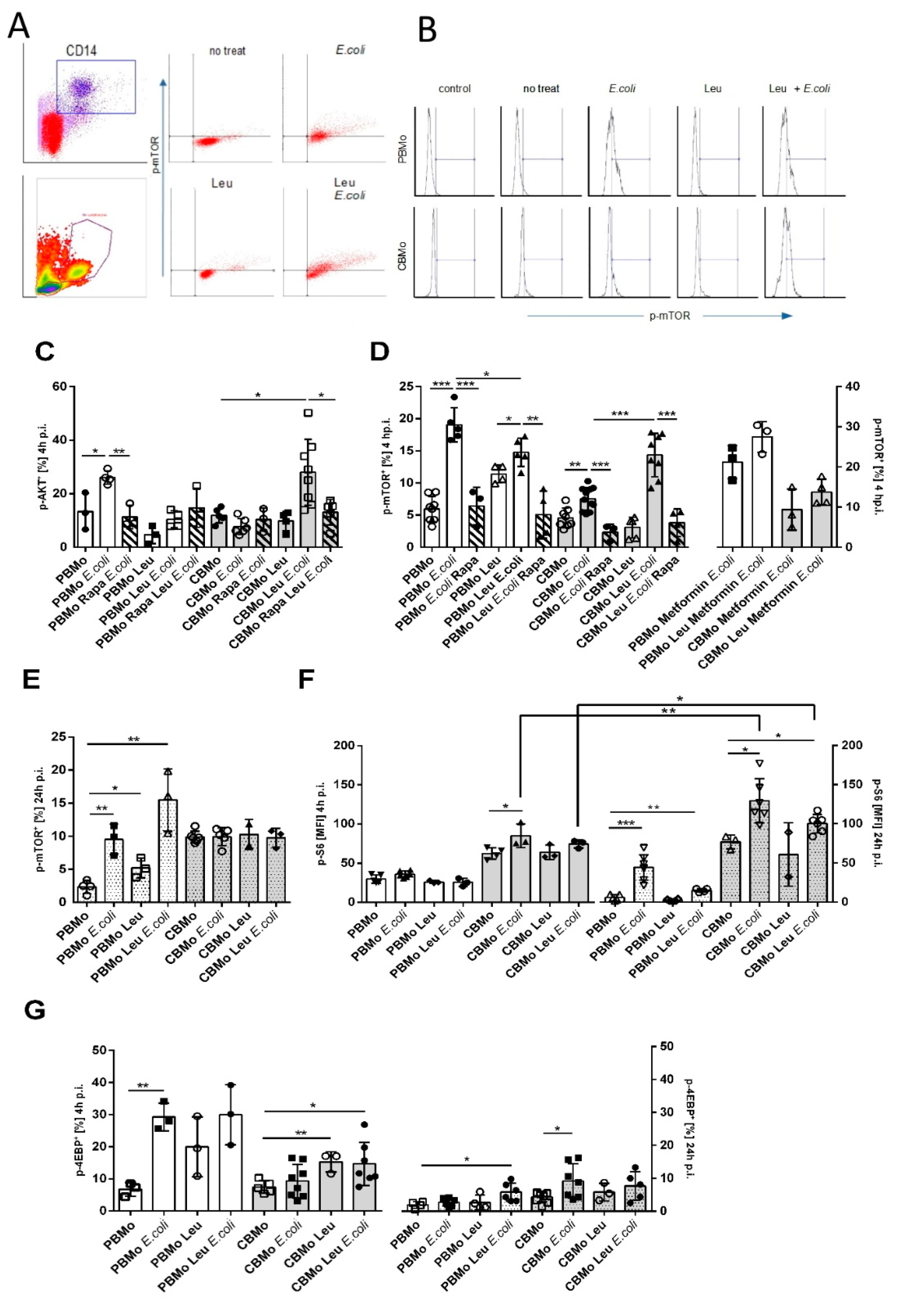
2.3. Leucine Increases AKT and mTOR Activation in CBMo
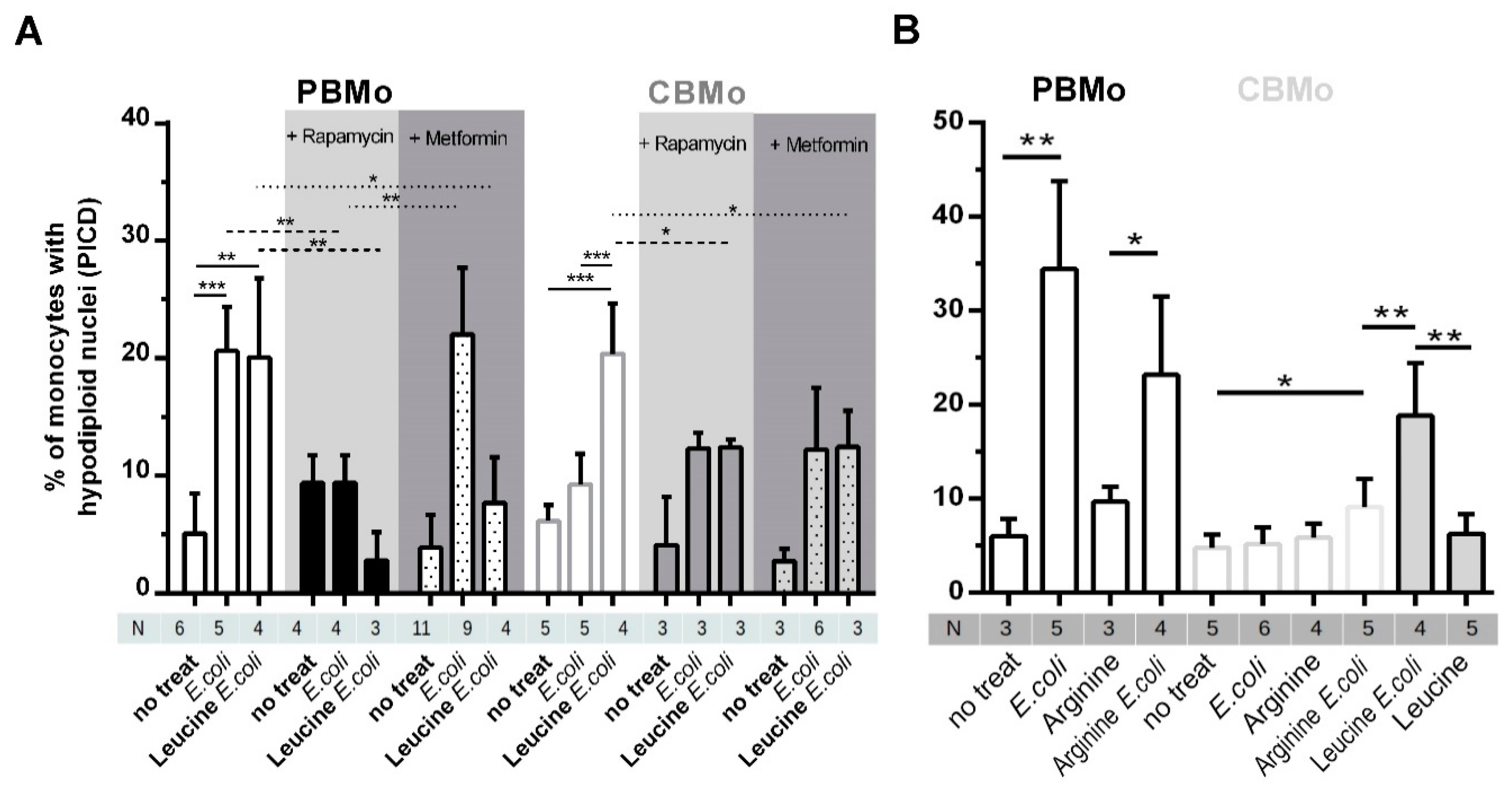
2.4. Leucine Increases PICD in CBMo in an mTOR-Dependent Manner
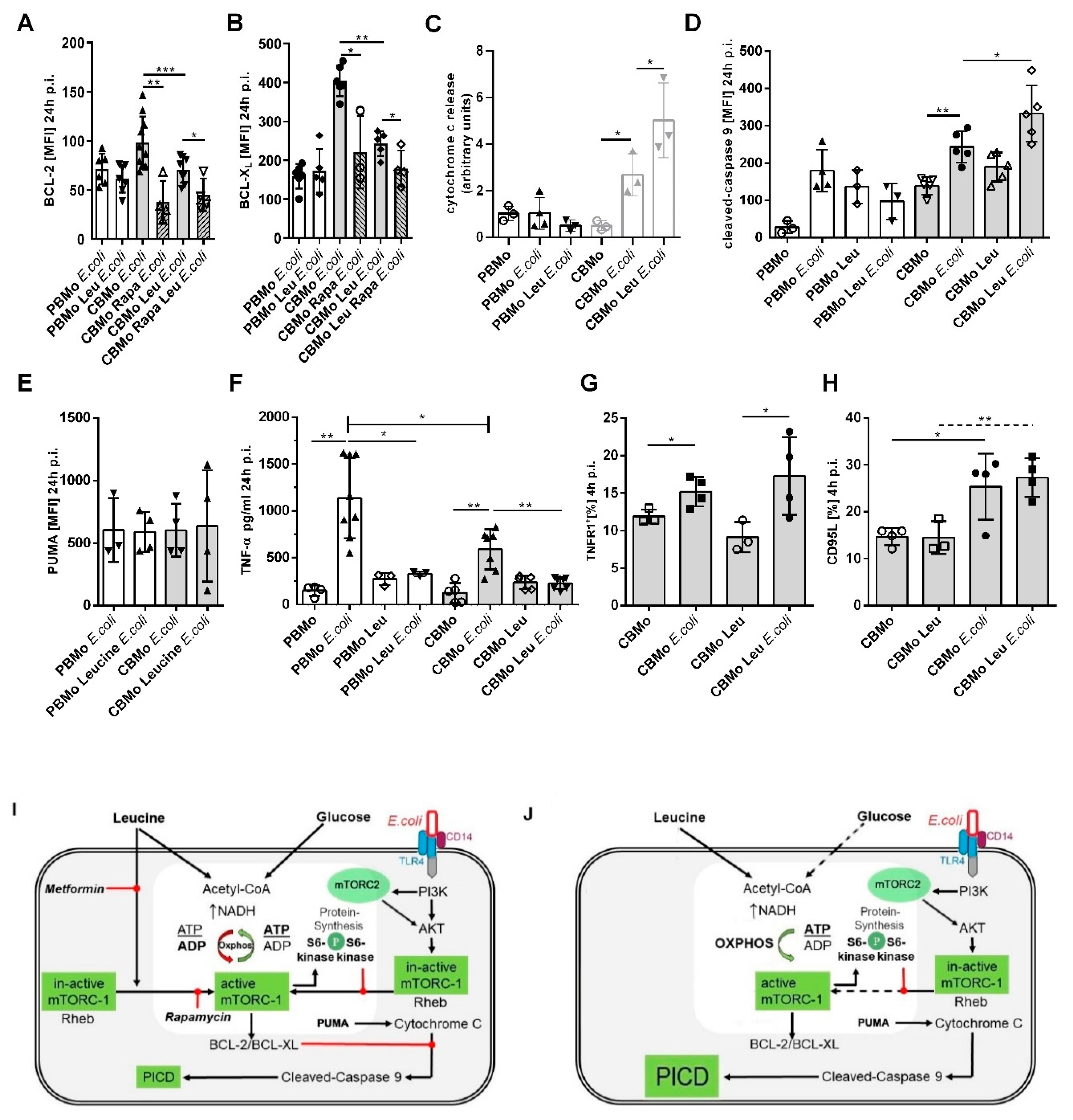
2.5. The Intrinsic Apoptotic Pathway is Engaged in the Leucine-Induced PICD
3. Discussion
4. Material and Methods
4.1. Patients
4.2. Reagents
4.3. Isolation of Mo and Stimulation
4.4. Flow Cytometry
4.5. Seahorse Assay
4.6. ELISA
4.7. Western Blot
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carbonea, F.; Montecucco, F.; Sahebkard, A. Current and emerging treatments for neonatal sepsis. Expert Opin. Pharmacother. 2020, 21, 549–556. [Google Scholar] [CrossRef]
- Humberg, A.; Fortmann, I.; Siller, B.; Kopp, M.V.; Herting, E.; Göpel, W.; Härtel, C. Preterm birth and sustained inflammation: Consequences for the neonate. Semin Immunopathol. 2020, 21, 549–556. [Google Scholar] [CrossRef]
- Dammann, O.; Leviton, A. Intermittent or sustained systemic inflammation and the preterm brain. Pediatr. Res. 2014, 75, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Dreschers, S.; Ohl, K.; Lehrke, M.; Möllmann, J.; Denecke, B.; Costa, I.; Vogl, T.; Viemann, D.; Roth, J.; Orlikowsky, T.; et al. Impaired cellular energy metabolism in cord blood macrophages contributes to abortive response toward inflammatory threats. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Lee, D.S.; Madrenas, J. Evolving Bacterial Envelopes and Plasticity of TLR2-Dependent Responses: Basic Research and Translational Opportunities. Front Immunol. 2013, 4, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; Bi, D.; Zheng, R.; Cai, N.; Xu, H.; Zhou, R.; Lu, J.; Wan, M.; Xu, X. Identification and activation of TLR4-mediated signalling pathways by alginate-derived guluronate oligosaccharide in RAW264.7 macrophages. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ulas, T.; Pirr, S.; Fehlhaber, B.; Bickes, M.S.; Loof, T.G.; Vogl, T.; Mellinger, L.; Heinemann, A.S.; Burgmann, J.; Schöning, J.; et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat. Immunol. 2017, 18, 622–632. [Google Scholar] [CrossRef]
- Grace, Y.L.; David, M.S. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 4, 183–203. [Google Scholar]
- Castedo, M.; Ferri, K.F.; Kroemer, G. Mammalian target of rapamycin (mTOR): Pro- and anti-apoptotic. Cell Death Differ. 2002, 9, 99–100. [Google Scholar] [CrossRef]
- Lang, C.H.; Frost, R.A. Glucocorticoids and TNFalpha interact cooperatively to mediate sepsis-induced leucine resistance in skeletal muscle. Mol. Med. 2006, 12, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Frost, R.A.; Vary, T.C. Regulation of muscle protein synthesis during sepsis and inflammation. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E453–E459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Columbus, D.A.; Fiorotto, M.L.; Davis, T.A. Leucine is a major regulator of muscle protein synthesis in neonates. Amino Acids 2015, 47, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-García, A.D.; Columbus, D.A.; Manjarín, R.; Nguyen, H.V.; Suryawan, A.; Orellana, R.A.; Davis, T.A. Leucine supplementation stimulates protein synthesis and reduces degradation signal activation in muscle of newborn pigs during acute endotoxemia. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E791–E801. [Google Scholar] [CrossRef]
- Escobar, J.; Frank, J.W.; Suryawan, A.; Nguyen, H.V.; Kimball, S.R.; Jefferson, L.S.; Davis, T.A. Regulation of cardiac and skeletal muscle protein synthesis by individual branched-chain amino acids in neonatal pigs. Am. J. Physiol. Metab. 2006, 290, E612–E621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibrian, D.; Castillo-González, R.; Fernández-Gallego, N.; de la Fuente, H.; Jorge, I.; Saiz, M.L.; Punzón, C.; Ramírez-Huesca, M.; Vicente-Manzanares, M.; Fresno, M.; et al. Targeting L-type amino acid transporter 1 in innate and adaptive T cells efficiently controls skin inflammation. J. Allergy Clin. Immunol. 2020, 145, 199–214.e11. [Google Scholar] [CrossRef] [Green Version]
- Dreschers, S.; Gille, C.; Haas, M.; Seubert, F.; Platen, C.; Orlikowsky, T.W. Reduced internalization of TNF-ɑ/TNFR1 down-regulates caspase dependent phagocytosis induced cell death (PICD) in neonatal monocytes. PLoS ONE 2017, 12, e0182415. [Google Scholar] [CrossRef] [Green Version]
- Dreschers, S.; Saupp, P.; Hornef, M.; Prehn, A.; Platen, C.; Morschhäuser, J.; Orlikowsky, T.W. Reduced PICD in Monocytes Mounts Altered Neonate Immune Response to Candida albicans. PLoS ONE 2016, 11, e0166648. [Google Scholar] [CrossRef] [Green Version]
- Gille, C.; Dreschers, S.; Leiber, A.; Lepiorz, F.; Krusch, M.; Grosse-Opphoff, J.; Spring, B.; Haas, M.; Urschitz, M.; Poets, C.F.; et al. The CD95/CD95L pathway is involved in phagocytosis-induced cell death of monocytes and may account for sustained inflammation in neonates. Pediatr. Res. 2012, 73, 402–408. [Google Scholar] [CrossRef]
- Leiber, A.; Graf, B.; Spring, B.; Rudner, J.; Köstlin, N.; Orlikowsky, T.W.; Poets, C.F.; Gille, C. Neonatal monocytes express antiapoptotic pattern of Bcl-2 proteins and show diminished apoptosis upon infection with Escherichia coli. Pediatr. Res. 2014, 76, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Platen, C.; Dreschers, S.; Wappler, J.; Ludwig, A.; Düsterhöft, S.; Reiss, L.K.; Orlikowsky, T.W. Amphiregulin Regulates Phagocytosis-Induced Cell Death in Monocytes via EGFR and the Bcl-2 Protein Family. Mediat. Inflamm. 2019, 2019, 1603131. [Google Scholar] [CrossRef]
- Gille, C.; Steffen, F.; Lauber, K.; Keppeler, H.; Leiber, A.; Spring, B.; Poets, C.F.; Orlikowsky, T.W. Clearance of Apoptotic Neutrophils Is Diminished in Cord Blood Monocytes and Does Not Lead to Reduced IL-8 Production. Pediatr. Res. 2009, 66, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Kan, B.; Michalski, C.; Fu, H.; Au, H.H.T.; Lee, K.; Marchant, E.A.; Cheng, M.F.; Anderson-Baucum, E.; Aharoni-Simon, M.; Tilley, P.; et al. Cellular metabolism constrains innate immune responses in early human ontogeny. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gille, C.; Leiber, A.; Mundle, I.; Spring, B.; Abele, H.; Spellerberg, B.; Hartmann, H. Poets ChF, Orlikowsky TW. Phagocytosis and postphagocytic reaction of cord blood and adult blood monocyte after infection with green fluorescent protein-labeled Escherichia coli and group B Streptococci. Cytom. B Clin. Cytom. 2009, 76, 271–284. [Google Scholar] [CrossRef]
- Bonvini, A.; Coqueiro, A.Y.; Tirapegui, J.; Calder, P.C.; Rogero, M.M. Immunomodulatory role of branched-chain amino acids. Nutr. Rev. 2018, 76, 840–856. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Zhang, W.; Ruvolo, V.R.; Gao, C.; Zhou, L.; Bornmann, W.; Tsao, T.; Schober, W.D.; Smith, P.; Guichard, S.; Konopleva, M.; et al. Evaluation of Apoptosis Induction by Concomitant Inhibition of MEK, mTOR, and Bcl-2 in Human Acute Myelogenous Leukemia Cells. Mol. Cancer Ther. 2014, 13, 1848–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.C.; Peruchetti, D.B.; Silva, L.S.; Silva-Filho, J.L.; Souza, M.C.; Henriques, M.D.G.; Caruso-Neves, C.; Pinheiro, A.A.S. LPS Induces mTORC1 and mTORC2 Activation During Monocyte Adhesion. Front. Mol. Biosci. 2018, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Woltman, A.M.; de Fijter, J.W.; Kamerling, S.W.; van Der Kooij, S.W.; Paul, L.C.; Daha, M.R.; van Kooten, C. Rapamycin induces apoptosis in monocyte- and CD34-derived dendritic cells but not in monocytes and macrophages. Blood 2001, 98, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Perl, A. Mechanistic Target of Rapamycin Complex 1 Expands Th17 and IL-4+ CD4−CD8− Double-Negative T Cells and Contracts Regulatory T Cells in Systemic Lupus Erythematosus. J. Immunol. 2014, 192, 4134–4144. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Mannam, P. Warburg revisited: Lessons for innate immunity and sepsis. Front. Physiol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, R.J.W.; Gresnigt, M.S.; Joosten, L.A.B.; Netea, M.G. Cellular metabolism of myeloid cells in sepsis. J. Leukoc. Biol. 2016, 101, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papathanassiu, A.E.; Ko, J.H.; Imprialou, M.; Bagnati, M.; Srivastava, P.K.; Vu, H.A.; Cucchi, D.; McAdoo, S.P.; Ananieva, E.A.; Mauro, C.; et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Erg. Pharma. 2017, 8, 16040. [Google Scholar] [CrossRef] [PubMed]
- Lachmandas, E.; Boutens, L.; Ratter, J.M.; Hijmans, A.; Hooiveld, G.J.; Joosten, L.A.B.; Rodenburg, R.J.; Fransen, J.A.M.; Houtkooper, R.H.; Van Crevel, R.; et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol. 2017, 2, 16246. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Ji, L.; Kanga, L.; Hua, X. Metabolic regulation of innate immunity. Adv. Immunol. 2020, 145, 129–157. [Google Scholar]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Wischmeyer, P.E. Glutamine. World Rev. Nut.r Diet. 2013, 105, 90–96. [Google Scholar]
- Wischmeyer, P.E. Nutrition Therapy in Sepsis. Crit. Care Clin. 2018, 34, 107–125. [Google Scholar] [CrossRef]
- Yogaratnam, D.; Miller, M.A.; Ross, B.; DiNapoli, M. Nutrition as medical therapy. Crit. Care Nurs. Clin. North Am. 2014, 26, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Gille, C.; Dreschers, S.; Spring, B.; Tárnok, A.; Bocsi, J.; Poets, C.F.; Orlikowsky, T.W. Differential modulation of cord blood and peripheral blood monocytes by intravenous immunoglobulin. Cytom. Part B Clin. Cytom. 2011, 82, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Deutz, N.E.; Matheson, E.M.; Matarese, L.E.; Luo, M.; Baggs, G.E.; Nelson, J.L.; Hegazi, R.A.; Tappenden, K.A.; Ziegler, T.R. Readmission and mortality in malnourished, older, hospitalized adults treated with a specialized oral nutritional supplement: A randomized clinical trial. Clin. Nutr. 2016, 35, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Coloff, J.L.; MacIntyre, A.N.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-Dependent Glucose Metabolism Promotes Mcl-1 Synthesis to Maintain Cell Survival and Resistance to Bcl-2 Inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef] [Green Version]
- Huang, N.; Perl, A. Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol. 2018, 39, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Häcker, G. Apoptosis induced by direct triggering of mitochondrial apoptosis proceeds in the near-absence of some apoptotic markers. Apoptosis 2007, 12, 2003–2011. [Google Scholar] [CrossRef]
- Dockrell, D.H.; Lee, M.; Lynch, D.H.; Read, R.C. Immune-Mediated Phagocytosis and Killing ofStreptococcus pneumoniaeAre Associated with Direct and Bystander Macrophage Apoptosis. J. Infect. Dis. 2001, 184, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Ananieva, E.A.; Powell, J.D.; Hutson, S.M. Leucine Metabolism in T Cell Activation: mTOR Signaling and Beyond. Adv. Nutr. 2016, 7, 798S–805S. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dreschers, S.; Ohl, K.; Möllmann, J.; Tenbrock, K.; Orlikowsky, T.W. Leucine Reconstitutes Phagocytosis-Induced Cell Death in E. coli-Infected Neonatal Monocytes—Effects on Energy Metabolism and mTOR Signaling. Int. J. Mol. Sci. 2021, 22, 4271. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084271
Dreschers S, Ohl K, Möllmann J, Tenbrock K, Orlikowsky TW. Leucine Reconstitutes Phagocytosis-Induced Cell Death in E. coli-Infected Neonatal Monocytes—Effects on Energy Metabolism and mTOR Signaling. International Journal of Molecular Sciences. 2021; 22(8):4271. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084271
Chicago/Turabian StyleDreschers, Stephan, Kim Ohl, Julia Möllmann, Klaus Tenbrock, and Thorsten W. Orlikowsky. 2021. "Leucine Reconstitutes Phagocytosis-Induced Cell Death in E. coli-Infected Neonatal Monocytes—Effects on Energy Metabolism and mTOR Signaling" International Journal of Molecular Sciences 22, no. 8: 4271. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084271