
Pepsin Promotes Activation of Epidermal Growth Factor Receptor and Downstream Oncogenic Pathways, at Slightly Acidic and Neutral pH, in Exposed Hypopharyngeal Cells


Abstract
:1. Introduction
2. Results
2.1. Slightly Acidic and Neutral Pepsin Preserves Cell Viability in HCs
2.2. Slightly Acidic and Neutral Pepsin Results in EGFR Activation and bcl-2 Overexpression in HCs
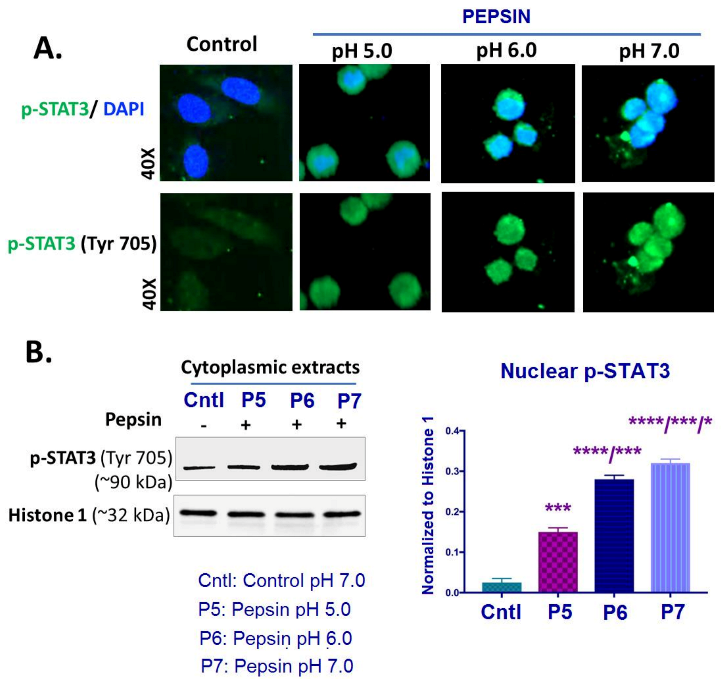
2.3. Slightly Acidic and Neutral Pepsin Enhances STAT3 Activation in HCs
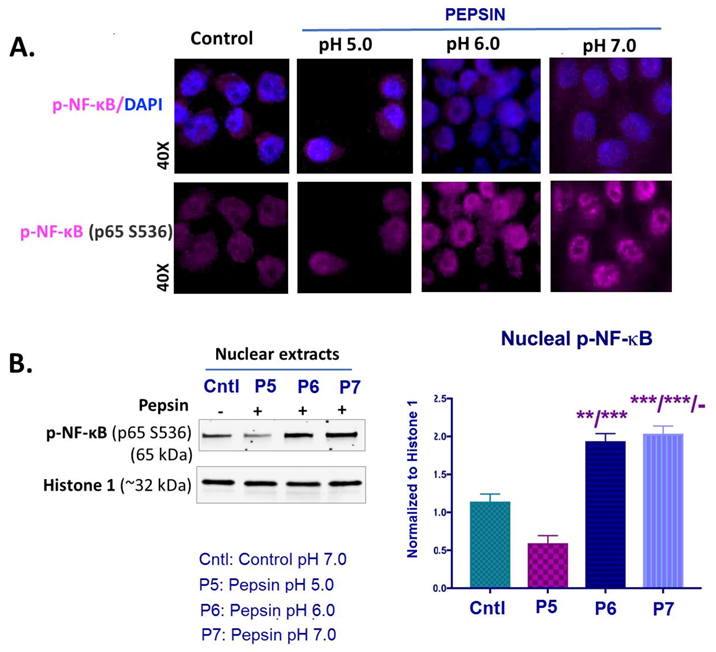
2.4. Slightly Acidic and Neutral Pepsin Promotes NF-κB (p65) Activation in treated HCs
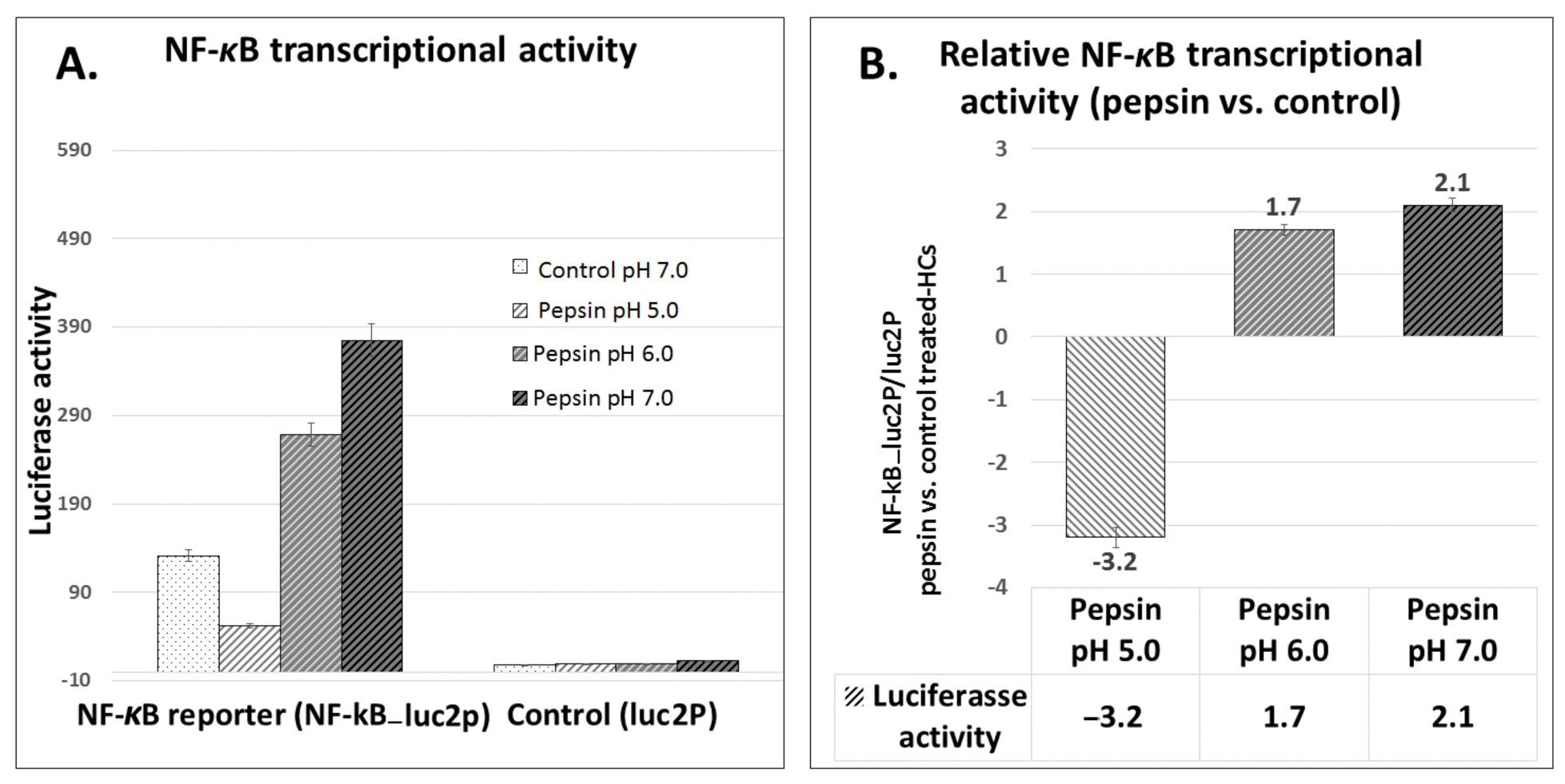
2.5. Pepsin Induces Elevated NF-κB Transcriptional Activity in Treated HCs, at Slightly Acidic pH 6.0 and Neutral pH 7.0
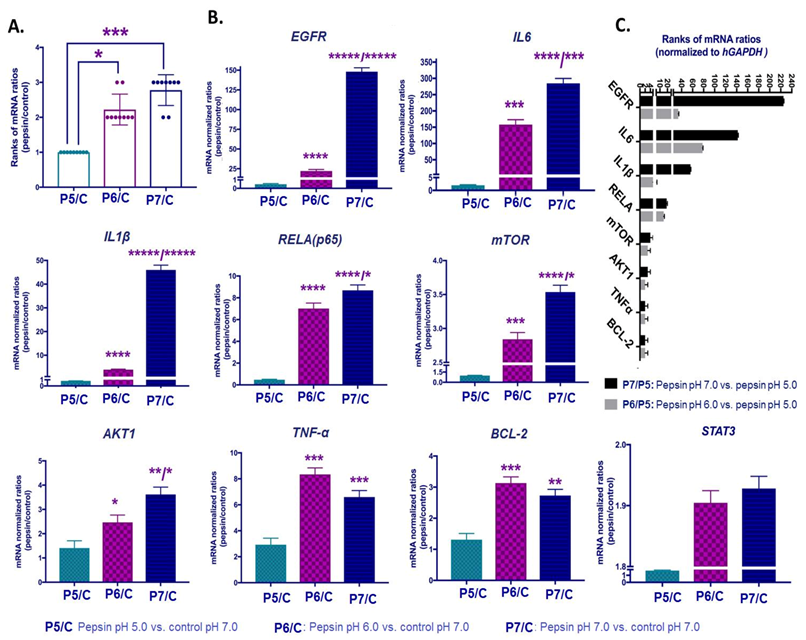
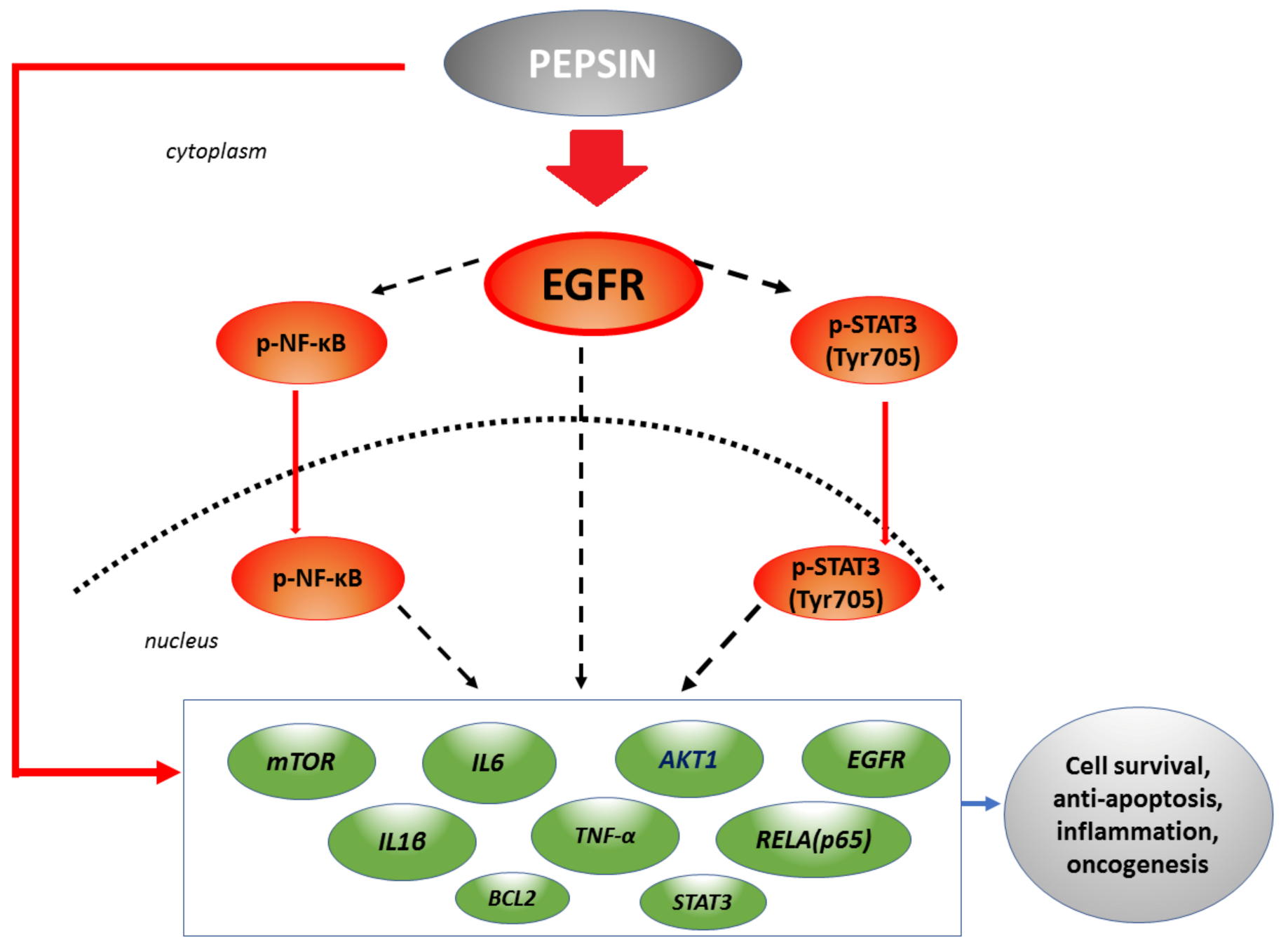
2.6. Slightly Acidic and Neutral Pepsin Promotes the Transcriptional Activation of EGFR-Related Oncogenic Factors in HCs
2.7. Correlations among Pepsin-Induced Protein and Gene Expression Levels
3. Discussion
4. Materials and Methods
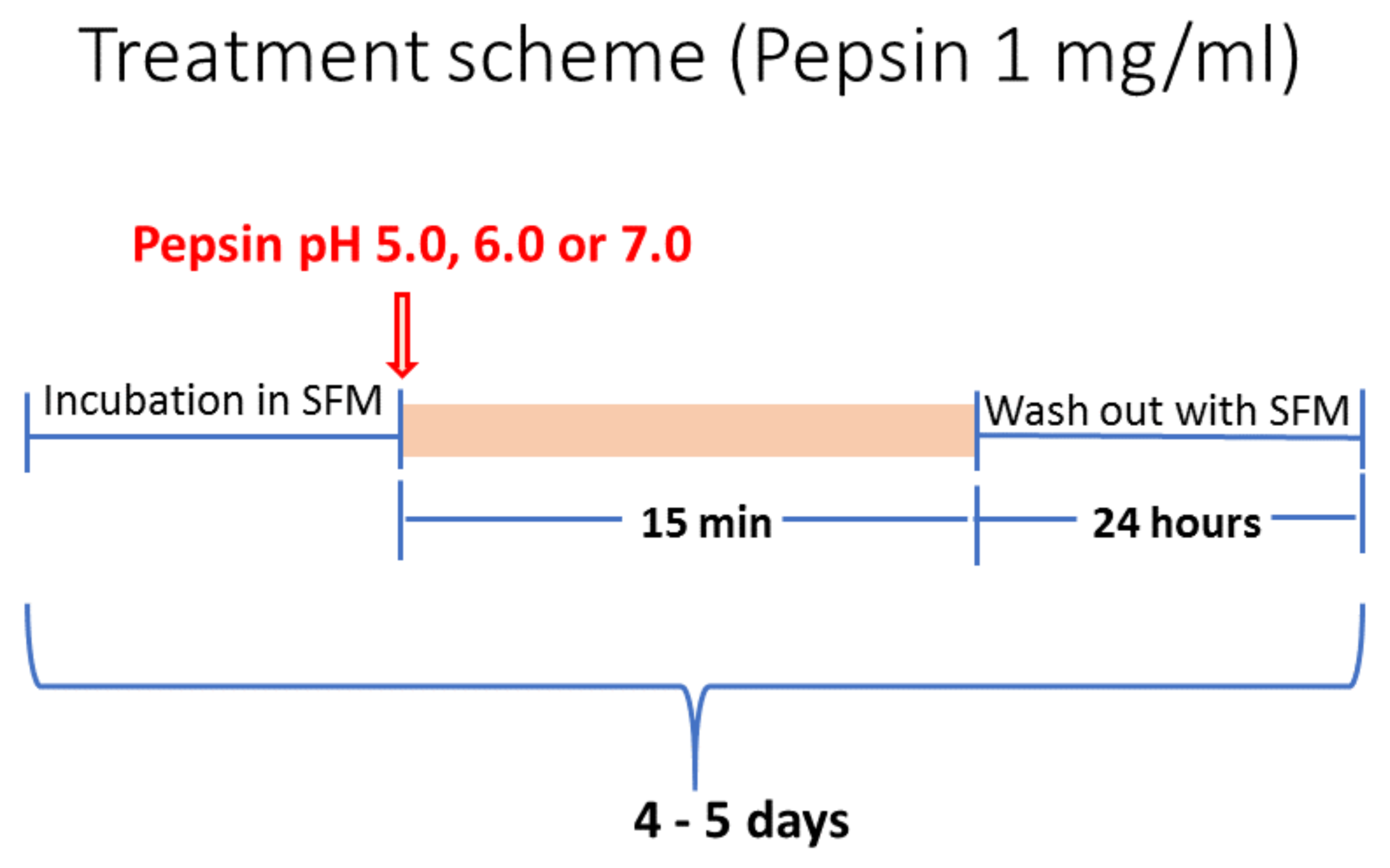
4.1. Cell Culture and Treatment Conditions
4.2. Cell Viability Assay
4.3. Immunofluorescence Cell Staining
4.4. Luciferase Assay
4.5. Protein Expression Analysis
4.5.1. Enzyme-Linked Immunosorbent Assay Quantification
4.5.2. Western Blot Analysis
4.6. Quantitative Real-Time Polymerase Chain Reaction
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, D.A.; Blanco, M.; Vaezi, M.F. Laryngopharyngeal Reflux and Functional Laryngeal Disorder: Perspective and Common Practice of the General Gastroenterologist. Gastroenterol. Hepatol. 2018, 14, 512–520. [Google Scholar]
- Lechien, J.R.; Bobin, F.; Muls, V.; Eisendrath, P.; Horoi, M.; Thill, M.; Dequanter, D.; Durdurez, J.; Rodriguez, A.; Saussez, S. Gastroesophageal reflux in laryngopharyngeal reflux patients: Clinical features and therapeutic response. Laryngoscope 2020, 130. [Google Scholar] [CrossRef]
- Galli, J.; Cammarota, G.; Volante, M.; De Corso, E.; Almadori, G.; Paludetti, G. Laryngeal carcinoma and laryngo-pharyngeal reflux disease. Acta Otorhinolaryngol. Ital. 2006, 26, 260–263. [Google Scholar] [PubMed]
- Copper, M.P.; Smit, C.F.; Stanojcic, L.D.; Devriese, P.P.; Schouwenburg, P.F.; Mathus-Vliegen, L.M. High Incidence of Laryngopharyngeal Reflux in Patients with Head and Neck Cancer. Laryngoscope 2000, 110, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, K.; Krzeski, A. The role of pepsin in the laryngopharyngeal reflux. Otolaryngol. Polska 2017, 71, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.; Yan, J.C.; Bs, C.R.H.; Ms, T.L.S.; Stoner, G.D.; Blumin, J.H.; Bock, J.M. Pepsin promotes proliferation of laryngeal and pharyngeal epithelial cells. Laryngoscope 2012, 122, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Sereg-Bahar, M.; Jerin, A.; Jansa, R.; Stabuc, B.; Hocevar-Boltezar, I. Pepsin and bile acids in saliva in patients with laryngopharyngeal reflux—A prospective comparative study. Clin. Otolaryngol. 2015, 40, 234–239. [Google Scholar] [CrossRef]
- Yuksel, E.S.; Hong, S.-K.S.; Strugala, V.; Slaughter, J.C.; Goutte, M.; Garrett, C.G.; Dettmar, P.W.; Vaezi, M.F. Rapid salivary pepsin test: Blinded assessment of test performance in gastroesophageal reflux disease. Laryngoscope 2012, 122, 1312–1316. [Google Scholar] [CrossRef]
- Hayat, J.O.; Gabieta-Somnez, S.; Yazaki, E.; Kang, J.-Y.; Woodcock, A.; Dettmar, P.; Mabary, J.; Knowles, C.H.; Sifrim, D. Pepsin in saliva for the diagnosis of gastro-oesophageal reflux disease. Gut 2014, 64, 373–380. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Liu, Y.-L.; Zeng, F.-F.; Wu, T.; Yang, C.-L.; Shen, H.-Y.; Li, X.-P. Correlation of pepsin-measured laryngopharyngeal reflux disease with symptoms and signs. Otolaryngol. Neck Surg. 2010, 143, 765–771. [Google Scholar] [CrossRef]
- Knight, J.; Lively, M.O.; Johnston, N.; Dettmar, P.W.; Koufman, J.A. Sensitive Pepsin Immunoassay for Detection of Laryngopharyngeal Reflux. Laryngoscope 2005, 115, 1473–1478. [Google Scholar] [CrossRef]
- Kawamura, O.; Aslam, M.; Rittmann, T.; Hofmann, C.; Shaker, R. Physical and pH Properties of Gastroesophagopharyngeal Refluxate: A 24-Hour Simultaneous Ambulatory Impedance and pH Monitoring Study. Am. J. Gastroenterol. 2004, 99, 1000–1010. [Google Scholar] [CrossRef]
- Oelschlager, B.K.; Quiroga, E.; Isch, J.A.; Cuenca-Abente, F. Gastroesophageal and Pharyngeal Reflux Detection Using Impedance and 24-Hour pH Monitoring in Asymptomatic Subjects: Defining the Normal Environment. J. Gastrointest. Surg. 2006, 10, 54–62. [Google Scholar] [CrossRef]
- Formánek, M.; Jančatová, D.; Komínek, P.; Tomanová, R.; Zeleník, K. Comparison of Impedance and Pepsin Detection in the Laryngeal Mucosa to Determine Impedance Values that Indicate Pathological Laryngopharyngeal Reflux. Clin. Transl. Gastroenterol. 2017, 8, e123. [Google Scholar] [CrossRef] [PubMed]
- Mainie, I.; Tutuian, R.; Shay, S.; Vela, M.; Zhang, X.; Sifrim, D.; Castell, D.O. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: A multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006, 55, 1398–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredenoord, A.J.; Dent, J. Proton pump inhibitor-therapy refractory gastro-oesophageal reflux disease patients, who are they? Gut 2007, 56, 593–594. [Google Scholar] [CrossRef] [Green Version]
- Tutuian, R.; Vela, M.F.; Hill, E.G.; Mainie, I.; Agrawal, A.; Castell, D.O. Characteristics of Symptomatic Reflux Episodes on Acid Suppressive Therapy. Am. J. Gastroenterol. 2008, 103, 1090–1096. [Google Scholar] [CrossRef]
- Sifrim, D.; Mittal, R.; Fass, R.; Smout, A.; Castell, D.; Tack, J.; Gregersen, H. Review article: Acidity and volume of the refluxate in the genesis of gastro-oesophageal reflux disease symptoms. Aliment. Pharmacol. Ther. 2007, 25, 1003–1017. [Google Scholar] [CrossRef]
- Johnston, N.; Dettmar, P.W.; Bishwokarma, B.; Lively, M.O.; Koufman, J.A. Activity/Stability of Human Pepsin: Implications for Reflux Attributed Laryngeal Disease. Laryngoscope 2007, 117, 1036–1039. [Google Scholar] [CrossRef] [Green Version]
- Hammer, H.F. Reflux-Associated Laryngitis and Laryngopharyngeal Reflux: A Gastroenterologist’s Point of View. Dig. Dis. 2009, 27, 14–17. [Google Scholar] [CrossRef]
- Johnston, N.; Wells, C.W.; Samuels, T.L.; Blumin, J.H. Pepsin in Nonacidic Refluxate Can Damage Hypopharyngeal Epithelial Cells. Ann. Otol. Rhinol. Laryngol. 2009, 118, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.T.; Toman, J.; Vageli, D. The In Vitro Effect of Acidic-Pepsin on Nuclear Factor KappaB Activation and Its Related Oncogenic Effect on Normal Human Hypopharyngeal Cells. PLoS ONE 2016, 11, e0168269. [Google Scholar] [CrossRef] [PubMed]
- Block, B.B.; Kuo, E.; Zalzal, H.G.; Escobar, H.; Rose, M.; Preciado, D. In Vitro Effects of Acid and Pepsin on Mouse Middle Ear Epithelial Cell Viability and MUC5B Gene Expression. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, N.; Wells, C.W.; Samuels, T.L.; Blumin, J.H. Rationale for Targeting Pepsin in the Treatment of Reflux Disease. Ann. Otol. Rhinol. Laryngol. 2010, 119, 547–558. [Google Scholar] [CrossRef]
- Samuels, T.L.; Johnston, N. Pepsin as a causal agent of inflammation during nonacidic reflux. Otolaryngol. Neck Surg. 2009, 141, 559–563. [Google Scholar] [CrossRef]
- Johnston, N.; Wells, C.W.; Blumin, J.H.; Toohill, R.J.; Merati, A.L. Receptor-mediated uptake of pepsin by laryngeal epithelial cells. Ann. Otol. Rhinol. Laryngol. 2007, 116, 934–938. [Google Scholar] [CrossRef]
- Ms, T.L.S.; Zimmermann, M.T.; Ms, A.Z.; Demos, W.; Southwood, J.E.; Blumin, J.H.; Bock, J.M.; Johnston, N. RNA Sequencing Reveals Cancer-Associated Changes in Laryngeal Cells Exposed to Non-Acid Pepsin. Laryngoscope 2021, 131, 121–129. [Google Scholar] [CrossRef]
- Klein, J.D.; Grandis, J.R. The Molecular Pathogenesis of Head and Neck Cancer. Cancer Biol. Ther. 2010, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Broek, R.V.; Snow, G.E.; Chen, Z.; Van Waes, C. Chemoprevention of head and neck squamous cell carcinoma through inhibition of NF-κB signaling. Oral Oncol. 2014, 50, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Molinolo, A.A.; Amornphimoltham, P.; Squarize, C.H.; Castilho, R.M.; Patel, V.; Gutkind, J.S. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009, 45, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.E.; Patel, M.R.; Couch, M.E.; Hayes, D.N. Molecular Biology of Head and Neck Cancer: Risks and Pathways. Hematol. Clin. N. Am. 2008, 22, 1099–1124. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Bernitsas, D.G.; Ichikawa, H.; Takada, Y.; Myers, J.N.; Lin, X.L.; Darnay, B.G.; Chaturvedi, M.M.; Aggarwal, B.B. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-κB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene 2006, 26, 1385–1397. [Google Scholar] [CrossRef] [Green Version]
- Guyer, R.A.; Macara, I.G. Loss of the Polarity Protein PAR3 Activates STAT3 Signaling via an Atypical Protein Kinase C (aPKC)/NF-κB/Interleukin-6 (IL-6) Axis in Mouse Mammary Cells. J. Biol. Chem. 2015, 290, 8457–8468. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, C.; Huang, Y.; Yu, Y.; Li, R.; Li, M.; Liu, N.; Liu, P.; Qiao, J. Up-Regulated Expression of WNT5a Increases Inflammation and Oxidative Stress via PI3K/AKT/NF-κB Signaling in the Granulosa Cells of PCOS Patients. J. Clin. Endocrinol. Metab. 2015, 100, 201–211. [Google Scholar] [CrossRef]
- Choudhary, M.M.; France, T.J.; Teknos, T.N.; Kumar, P. Interleukin-6 role in head and neck squamous cell carcinoma progression. World J. Otorhinolaryngol. Head Neck Surg. 2016, 2, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Rébé, C.; Ghiringhelli, F. Interleukin-1β and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef]
- Khaznadar, S.S.; Khan, M.; Schmid, E.; Gebhart, S.; Becker, E.-T.; Krahn, T.; Von Ahsen, O. EGFR overexpression is not common in patients with head and neck cancer. Cell lines are not representative for the clinical situation in this indication. Oncotarget 2018, 9, 28965–28975. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 1–22. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Guo, Q. EGFR signaling pathway occupies an important position in cancer-related downstream signaling pathways of Pyk2. Cell Biol. Int. 2019, 44, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-L.; Zhang, X.-T.; Meng, J.; Zhang, H.-F.; Zhao, Y.; Li, C.; Sun, Y.; Mei, Q.-B.; Zhang, F.; Zhang, T. ING5 knockdown enhances migration and invasion of lung cancer cells by inducing EMT via EGFR/PI3K/Akt and IL-6/STAT3 signaling pathways. Oncotarget 2017, 8, 54265–54276. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.; Ujvari, B.; Ramana, V.; Donald, J. Cross-talk between EGFR and IL-6 drives oncogenic signaling and offers therapeutic opportunities in cancer. Cytokine Growth Factor Rev. 2018, 41, 18–27. [Google Scholar] [CrossRef]
- Sriuranpong, V.; Park, J.I.; Amornphimoltham, P.; Patel, V.; Nelkin, B.D.; Gutkind, J.S. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003, 63, 2948–2956. [Google Scholar]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Bancroft, C.C.; Chen, Z.; Yeh, J.; Sunwoo, J.B.; Yeh, N.T.; Jackson, S.; Jackson, C.; Van Waes, C. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-?B and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int. J. Cancer 2002, 99, 538–548. [Google Scholar] [CrossRef]
- Squarize, C.H.; Castilho, R.M.; Sriuranpong, V.; Pinto, D.S.; Gutkind, J.S. Molecular Cross-Talk between the NFκB and STAT3 Signaling Pathways in Head and Neck Squamous Cell Carcinoma. Neoplasia 2006, 8, 733–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A positive feedback loop involving EGFR/Akt/mTORC1 and IKK/NF-κB regulates head and neck squamous cell carcinoma proliferation. Oncotarget 2016, 7, 31892–31906. [Google Scholar] [CrossRef]
- Broek, R.V.; Mohan, S.; Eytan, D.F.; Chen, Z.; Van Waes, C. The PI3K/Akt/mTOR axis in head and neck cancer: Functions, aberrations, cross-talk, and therapies. Oral Dis. 2013, 21, 815–825. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D. NF-κB Signaling Pathway and Its Therapeutic Implications in Human Diseases. Int. Rev. Immunol. 2008, 27, 293–319. [Google Scholar] [CrossRef]
- Nottingham, L.K.; Yan, C.H.; Yang, X.; Si, H.; Coupar, J.; Bian, Y.; Cheng, T.-F.; Allen, C.; Arun, P.; Gius, D.; et al. Aberrant IKKα and IKKβ cooperatively activate NF-κB and induce EGFR/AP1 signaling to promote survival and migration of head and neck cancer. Oncogene 2013, 33, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, R.; Catzavelos, G.; Barrett, A.; Speight, P. Differential expression of bcl-2 and bax in squamous cell carcinomas of the oral cavity. Oral Oncol. 1996, 32, 394–400. [Google Scholar] [CrossRef]
- Shkeir, O.; Athanassiou-Papaefthymiou, M.; Lapadatescu, M.; Papagerakis, P.; Czerwinski, M.J.; Bradford, C.R.; Carey, T.E.; Prince, M.E.P.; Wolf, G.T.; Papagerakis, S.; et al. In vitro cytokine release profile: Predictive value for metastatic potential in head and neck squamous cell carcinomas. Head Neck 2013, 35, 1542–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vageli, D.P.; Doukas, S.G.; Sasaki, C.T. Inhibition of NF-κB prevents the acidic bile-induced oncogenic mRNA phenotype, in human hypopharyngeal cells. Oncotarget 2017, 9, 5876–5891. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, C.T.; Doukas, S.G.; Vageli, D.P. In Vivo Short-Term Topical Application of BAY 11-7082 Prevents the Acidic Bile–Induced mRNA and miRNA Oncogenic Phenotypes in Exposed Murine Hypopharyngeal Mucosa. Neoplasia 2018, 20, 374–386. [Google Scholar] [CrossRef]
- Vageli, D.P.; Doukas, S.G.; Spock, T.; Sasaki, C.T. Curcumin prevents the bile reflux-induced NF-κB-related mRNA oncogenic phenotype, in human hypopharyngeal cells. J. Cell. Mol. Med. 2018, 22, 4209–4220. [Google Scholar] [CrossRef]
- Doukas, S.G.; Vageli, D.P.; Sasaki, C.T. NF-κB inhibition reverses acidic bile-induced miR-21, miR-155, miR-192, miR-34a, miR-375 and miR-451a deregulations in human hypopharyngeal cells. J. Cell. Mol. Med. 2018, 22, 2922–2934. [Google Scholar] [CrossRef] [Green Version]
- Doukas, P.G.; Vageli, D.P.; Doukas, S.G.; Sasaki, C.T. Temporal characteristics of NF-κB inhibition in blocking bile-induced oncogenic molecular events in hypopharyngeal cells. Oncotarget 2019, 10, 3339–3351. [Google Scholar] [CrossRef] [Green Version]
- Vageli, D.P.; Kasle, D.; Doukas, S.G.; Doukas, P.G.; Sasaki, C.T. The temporal effects of topical NF-κB inhibition, in the in vivo prevention of bile-related oncogenic mRNA and miRNA phenotypes in murine hypopharyngeal mucosa: A preclinical model. Oncotarget 2020, 11, 3303–3314. [Google Scholar] [CrossRef]
- Doukas, S.G.; Doukas, P.G.; Sasaki, C.T.; Vageli, D. The in vivo preventive and therapeutic properties of curcumin in bile reflux-related oncogenesis of the hypopharynx. J. Cell. Mol. Med. 2020, 24, 10311–10321. [Google Scholar] [CrossRef]
- Niu, K.; Guo, C.; Teng, S.; Zhou, D.; Yu, S.; Yin, W.; Wang, P.; Zhu, W.; Duan, M. Pepsin promotes laryngopharyngeal neoplasia by modulating signaling pathways to induce cell proliferation. PLoS ONE 2020, 15, e0227408. [Google Scholar] [CrossRef]
- Langevin, S.M.; Michaud, D.S.; Marsit, C.J.; Nelson, H.H.; Birnbaum, A.E.; Eliot, M.; Christensen, B.C.; McClean, M.D.; Kelsey, K.T. Gastric Reflux Is an Independent Risk Factor for Laryngopharyngeal Carcinoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Papagerakis, S.; Bellile, E.; Peterson, L.A.; Pliakas, M.; Balaskas, K.; Selman, S.; Hanauer, D.; Taylor, J.M.; Duffy, S.; Wolf, G. Proton Pump Inhibitors and Histamine 2 Blockers Are Associated with Improved Overall Survival in Patients with Head and Neck Squamous Carcinoma. Cancer Prev. Res. 2014, 7, 1258–1269. [Google Scholar] [CrossRef] [Green Version]
- Vageli, D.P.; Prasad, M.L.; Sasaki, C.T. Gastro-duodenal fluid induced Nuclear Factor-κappaB activation and early pre-malignant alterations in murine hypopharyngeal mucosa. Oncotarget 2016, 7, 5892–5908. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, C.T.; Doukas, S.G.; Costa, J.; Vageli, D.P. The Progressive Mutagenic Effects of Acidic Bile Refluxate in Hypopharyngeal Squamous Cell Carcinogenesis: New Insights. Cancers 2020, 12, 1064. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.; Doukas, S.; Doukas, P.; Vageli, D. Weakly Acidic Bile Is a Risk Factor for Hypopharyngeal Carcinogenesis Evidenced by DNA Damage, Antiapoptotic Function, and Premalignant Dysplastic Lesions In Vivo. Cancers 2021, 13, 852. [Google Scholar] [CrossRef]
- Walker, V.; Taylor, W.H. Pepsin 5 in gastric juice: Determination and relationship to the alkali-stable peptic activity. Gut 1979, 20, 977–982. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.-J.; Wang, L.; Mo, T.-T.; Wang, J.; Wang, M.-G.; Li, X.-P. Pepsin promotes IL-8 signaling-induced epithelial–mesenchymal transition in laryngeal carcinoma. Cancer Cell Int. 2019, 19, 64. [Google Scholar] [CrossRef] [Green Version]
- Bossi, P.; Resteghini, C.; Paielli, N.; Licitra, L.; Pilotti, S.; Perrone, F. Prognostic and predictive value of EGFR in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 74362–74379. [Google Scholar] [CrossRef] [Green Version]
- Zibelman, M.; Mehra, R. Overview of Current Treatment Options and Investigational Targeted Therapies for Locally Advanced Squamous Cell Carcinoma of the Head and Neck. Am. J. Clin. Oncol. 2016, 39, 396–406. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Burnett, J.R.; Friedman, J.A.; Kannabiran, V.R.; Chen, Z.; Van Waes, C. EGFR–PI3K–AKT–mTOR signaling in head and neck squamous cell carcinomas: Attractive targets for molecular-oriented therapy. Expert Opin. Ther. Targets 2010, 15, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monisha, J.; Roy, N.K.; Bordoloi, D.; Kumar, A.; Golla, R.; Kotoky, J.; Padmavathi, G.; Kunnumakkara, A.B. Nuclear Factor Kappa B: A Potential Target to Persecute Head and Neck Cancer. Curr. Drug Targets 2016, 18, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yan, B.; Van Waes, C. Role of the NF-κB transcriptome and proteome as biomarkers in human head and neck squamous cell carcinomas. Biomark. Med. 2008, 2, 409–426. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.E.; Bear, M.D.; Rosol, T.J.; Premanandan, C.; Kisseberth, W.C.; London, C.A. Characterization of STAT3 expression, signaling and inhibition in feline oral squamous cell carcinoma. BMC Vet. Res. 2015, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Van Boxel-Dezaire, A.H.H.; Cheon, H.; Yang, J.; Stark, G.R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 16975–16980. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Syu, S.-H.; Liu, K.-J.; Chu, P.-Y.; Yang, W.-C.; Lin, P.; Shieh, W.-Y. Interleukin-1 beta transactivates epidermal growth factor receptor via the CXCL1-CXCR2 axis in oral cancer. Oncotarget 2015, 6, 38866–38880. [Google Scholar] [CrossRef] [Green Version]
- Ulualp, S.O.; Roland, P.S.; Toohill, R.J.; Shaker, R. Prevalence of gastroesophagopharyngeal acid reflux events: An evidence-based systematic review. Am. J. Otolaryngol. 2005, 26, 239–244. [Google Scholar] [CrossRef]
- Hemmink, G.J.M.; Bredenoord, A.J.; Weusten, B.L.A.M.; Monkelbaan, J.F.; Timmer, R.; Smout, A.J.P.M. Esophageal pH-Impedance Monitoring in Patients with Therapy-Resistant Reflux Symptoms: ‘On’ or ‘Off’ Proton Pump Inhibitor? Am. J. Gastroenterol. 2008, 103, 2446–2453. [Google Scholar] [CrossRef]
- Sandner, A.; Illert, J.; Koitzsch, S.; Unverzagt, S.; Schön, I. Reflux induces DNA strand breaks and expression changes of MMP1+9+14 in a human miniorgan culture model. Exp. Cell Res. 2013, 319, 2905–2915. [Google Scholar] [CrossRef]
- Dai, Y.-F.; Tan, J.-J.; Deng, C.-Q.; Liu, X.; Lv, Z.-H.; Li, X.-P. Association of pepsin and DNA damage in laryngopharyngeal reflux-related vocal fold polyps. Am. J. Otolaryngol. 2020, 41, 102681. [Google Scholar] [CrossRef]
- Kelly, E.A.; Samuels, T.L.; Johnston, N. Chronic Pepsin Exposure Promotes Anchorage-Independent Growth and Migration of a Hypopharyngeal Squamous Cell Line. Otolaryngol. Neck Surg. 2014, 150, 618–624. [Google Scholar] [CrossRef]
- Bs, A.J.M.; Ms, T.L.S.; Blumin, J.H.; Johnston, N. The role of pepsin in epithelia-mesenchymal transition in idiopathic subglottic stenosis. Laryngoscope 2020, 130, 154–158. [Google Scholar] [CrossRef]
- Ayazi, S.; Hagen, J.A.; Zehetner, J.; Oezcelik, A.; Abate, E.; Kohn, G.P.; Sohn, H.J.; Lipham, J.C.; Demeester, S.R.; Demeester, T.R. Proximal Esophageal pH Monitoring: Improved Definition of Normal Values and Determination of a Composite pH Score. J. Am. Coll. Surg. 2010, 210, 345–350. [Google Scholar] [CrossRef]
- Furukawa, O.; Nakamura, E.; Okabe, S. Characterization of a novel cell damage model induced by acid and pepsin using rat gastric epithelial cells: Protective effect of sucralfate. J. Gastroenterol. Hepatol. 1997, 12, 115–121. [Google Scholar] [CrossRef]
- Pearson, J.P.; Ward, R.; Allen, A.; Roberts, N.B.; Taylor, W.H. Mucus degradation by pepsin: Comparison of mucolytic activity of human pepsin 1 and pepsin 3: Implications in peptic ulceration. Gut 1986, 27, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Ryle, A.P.; Auffret, C.A. The specificity of some pig and human pepsins towards synthetic peptide substrates. Biochem. J. 1979, 179, 247–249. [Google Scholar] [CrossRef] [Green Version]
- Doukas, S.G.; Cardoso, B.; Tower, J.I.; Vageli, D.P.; Sasaki, C.T. Biliary tumorigenic effect on hypopharyngeal cells is significantly enhanced by pH reduction. Cancer Med. 2019, 8, 4417–4427. [Google Scholar] [CrossRef] [Green Version]
- Doukas, S.G.; Vageli, D.P.; Lazopoulos, G.; Spandidos, D.A.; Sasaki, C.T.; Tsatsakis, A. The Effect of NNK, A Tobacco Smoke Carcinogen, on the miRNA and Mismatch DNA Repair Expression Profiles in Lung and Head and Neck Squamous Cancer Cells. Cells 2020, 9, 1031. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| * Pepsin-Induced mRNA Phenotype in HCs | |||
|---|---|---|---|
| pH 5.0 | pH 6.0 | pH 7.0 | |
| IL6 | ↑2 | ↑158 | ↑285 |
| EGFR | ↓1.5 | ↑22 | ↑148 |
| IL1β | ↓1.2 | ↑4 | ↑46 |
| RELA | ↓2.1 | ↑7 | ↑8.6 |
| TNF-α | ↑2.9 | ↑8.3 | ↑6.6 |
| AKT1 | ↑1.4 | ↑2.5 | ↑3.6 |
| mTOR | 1 | ↑2.8 | ↑3.5 |
| BCL-2 | ↑1.3 | ↑3.1 | ↑2.7 |
| STAT3 | ↑1.7 | ↑2 | ↑2 |
| Wnt5a | ↑1.8 | ↑1.5 | ↑1.2 |
| PTGS2 | 1 | 1 | ↑1.2 |
| PKI3CA | 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doukas, P.G.; Vageli, D.P.; Sasaki, C.T.; Judson, B.L. Pepsin Promotes Activation of Epidermal Growth Factor Receptor and Downstream Oncogenic Pathways, at Slightly Acidic and Neutral pH, in Exposed Hypopharyngeal Cells. Int. J. Mol. Sci. 2021, 22, 4275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084275
Doukas PG, Vageli DP, Sasaki CT, Judson BL. Pepsin Promotes Activation of Epidermal Growth Factor Receptor and Downstream Oncogenic Pathways, at Slightly Acidic and Neutral pH, in Exposed Hypopharyngeal Cells. International Journal of Molecular Sciences. 2021; 22(8):4275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084275
Chicago/Turabian StyleDoukas, Panagiotis G., Dimitra P. Vageli, Clarence T. Sasaki, and Benjamin L. Judson. 2021. "Pepsin Promotes Activation of Epidermal Growth Factor Receptor and Downstream Oncogenic Pathways, at Slightly Acidic and Neutral pH, in Exposed Hypopharyngeal Cells" International Journal of Molecular Sciences 22, no. 8: 4275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084275