
Molecular Mechanisms of Apoptosis Induction and Its Regulation by Fatty Acids in Pancreatic β-Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
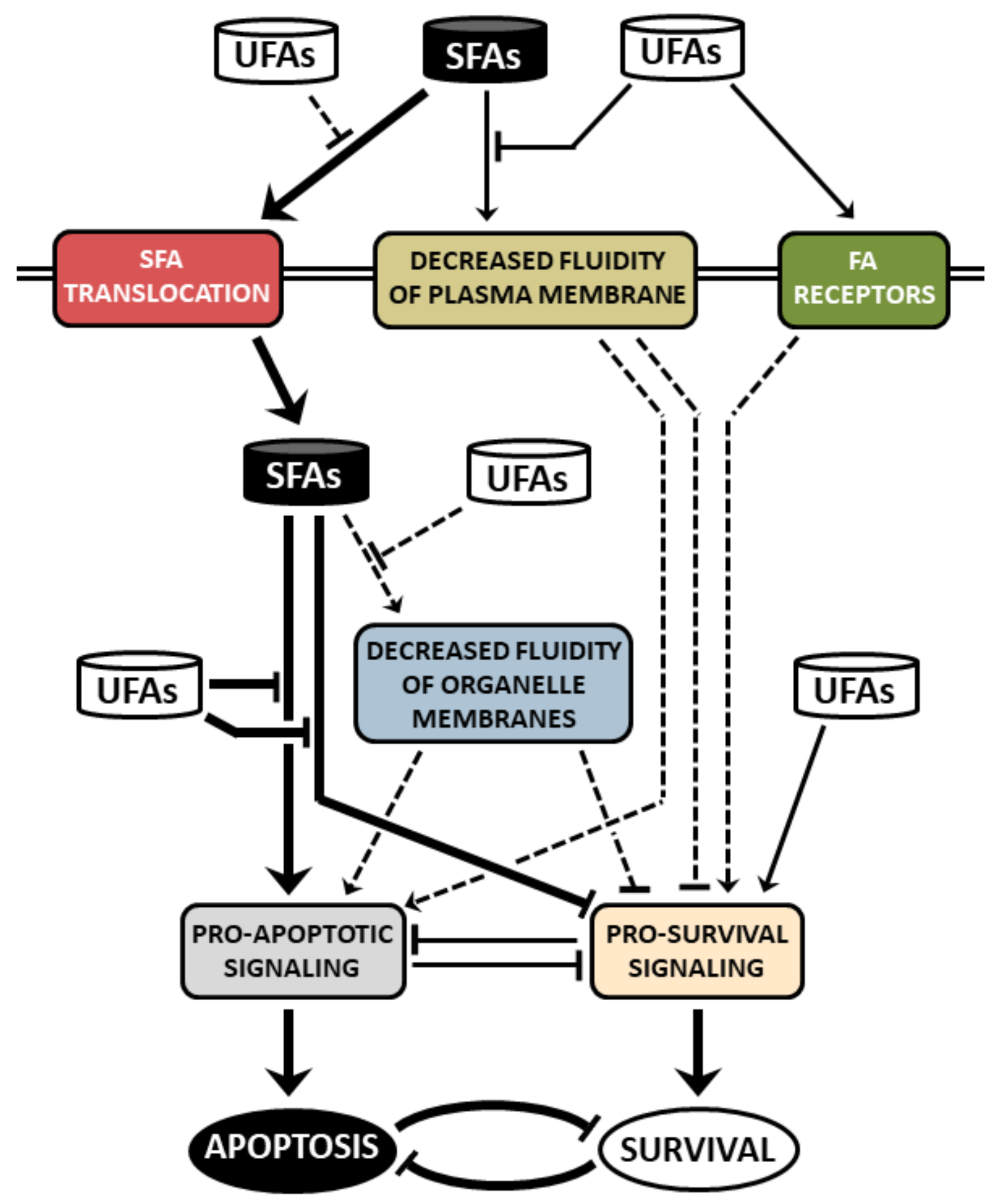
:1. Introduction
2. Effects of Fatty Acids on β-Cell Viability
2.1. Pro-Apoptotic Effect of Fatty Acids
2.2. Inhibitory Effect of Unsaturated Fatty Acids on Pro-Apoptotic Effect of Saturated Fatty Acids
2.3. The Role of Fatty Acid Metabolism and Its Intermediates
2.3.1. Fatty Acid Metabolism
2.3.2. Triacylglycerols
2.3.3. Ceramides
3. Initiation of Pro-Apoptotic Signalling by Fatty Acids
4. Mechanisms Mediating Pro-Apoptotic Effects of Saturated Fatty Acids
4.1. Pro-Survival and Pro-Proliferative Signalling
4.1.1. Akt Pathway
4.1.2. ERK Pathway
4.2. Stress Kinase Signalling
4.2.1. p38 MAPK Pathway
4.2.2. PKCδ Pathway
4.3. Endoplasmic Reticulum Stress Signalling
4.3.1. PERK Pathway
4.3.2. IRE1α Pathway
4.3.3. ATF6 Pathway
4.3.4. Mediator and Effector Molecules of Endoplasmic Reticulum Stress-Induced Signalling
4.4. Autophagy Signalling
4.5. Other Mechanisms Involved
5. Execution of Apoptosis Induction by Fatty Acids
5.1. Mitochondrial Pathway of Apoptosis Induction
5.2. Death Receptor Pathway of Apoptosis Induction
5.3. Executioner Caspases
6. Summarized Mechanisms of Apoptosis Induction by Fatty Acids
7. Mechanisms Mediating Anti-Apoptotic Effects of Unsaturated Fatty Acids
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AMPK | adenosine monophosphate-activated protein kinase |
| ATF6 | activating transcription factor 6 |
| Bcl-xL | B-cell lymphoma-extra large |
| Bcl-2 | B-cell lymphoma 2 |
| BiP | immunoglobulin heavy chain-binding protein |
| Bad | Bcl-2-associated death promoter |
| Bax | Bcl-2 associated X protein |
| CHOP | CCAAT-enhancer-binding protein C/EBP homologous protein |
| CoA | coenzyme A |
| DISC | death-inducing signalling complex |
| DP5 | death protein 5 |
| EGFR | epithelial growth factor receptor |
| eIF2α | eukaryotic initiation factor 2α |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FA | fatty acid |
| FAT/CD36 | fatty acid translocase/cluster of differentiation 36 |
| FoxO | forkhead box |
| GPR | G protein-coupled receptor |
| HFD | high-fat diet |
| iNOS | inducible form of NOS |
| IRE1α | inositol-requiring protein 1α |
| JNK | c-Jun N-terminal kinase |
| LOA | linoleic acid |
| MAPK | mitogen-activated protein kinase |
| Mcl-1 | myeloid cell leukemia-1 |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| PA | palmitic acid |
| PERK | protein kinase RNA (PKR)-like ER kinase |
| PIDD | p53-induced protein with a death domain |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| POA | palmitoil acid |
| PUMA | p53 upregulated modulator of apoptosis |
| SA | stearic acid |
| TAG | triacylglycerol |
| TLR4 | toll-like receptor 4 |
| RTK | receptor tyrosine kinase |
| ROS | reactive oxygen species |
| OA | oleic acid |
| T2DM | type 2 diabetes mellitus |
| UPR | unfolded protein response |
| XBP1 | X-box binding protein 1 |
References
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- El Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Kharroubi, I.; Ladriere, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: Role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welters, H.J.; Tadayyon, M.; Scarpello, J.H.; Smith, S.A.; Morgan, N.G. Mono-unsaturated fatty acids protect against beta-cell apoptosis induced by saturated fatty acids, serum withdrawal or cytokine exposure. FEBS Lett. 2004, 560, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Higa, M.; Shimabukuro, M.; Shimajiri, Y.; Takasu, N.; Shinjyo, T.; Inaba, T. Protein kinase B/Akt signalling is required for palmitate-induced beta-cell lipotoxicity. Diabetes Obes. Metab. 2006, 8, 228–233. [Google Scholar] [CrossRef]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [Green Version]
- Hennige, A.M.; Ranta, F.; Heinzelmann, I.; Dufer, M.; Michael, D.; Braumuller, H.; Lutz, S.Z.; Lammers, R.; Drews, G.; Bosch, F.; et al. Overexpression of kinase-negative protein kinase Cdelta in pancreatic beta-cells protects mice from diet-induced glucose intolerance and beta-cell dysfunction. Diabetes 2010, 59, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Eitel, K.; Staiger, H.; Brendel, M.D.; Brandhorst, D.; Bretzel, R.G.; Haring, H.U.; Kellerer, M. Different role of saturated and unsaturated fatty acids in beta-cell apoptosis. Biochem. Biophys. Res. Commun. 2002, 299, 853–856. [Google Scholar] [CrossRef]
- Welters, H.J.; Diakogiannaki, E.; Mordue, J.M.; Tadayyon, M.; Smith, S.A.; Morgan, N.G. Differential protective effects of palmitoleic acid and cAMP on caspase activation and cell viability in pancreatic beta-cells exposed to palmitate. Apoptosis 2006, 11, 1231–1238. [Google Scholar] [CrossRef]
- Sone, H.; Kagawa, Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005, 48, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Diakogiannaki, E.; Welters, H.J.; Morgan, N.G. Differential regulation of the endoplasmic reticulum stress response in pancreatic beta-cells exposed to long-chain saturated and monounsaturated fatty acids. J. Endocrinol. 2008, 197, 553–563. [Google Scholar] [CrossRef]
- Furstova, V.; Kopska, T.; James, R.F.; Kovar, J. Comparison of the effect of individual saturated and unsaturated fatty acids on cell growth and death induction in the human pancreatic beta-cell line NES2Y. Life Sci. 2008, 82, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Nemcova-Furstova, V.; James, R.F.L.; Kovar, J. Inhibitory Effect of Unsaturated Fatty Acids on Saturated Fatty Acid-Induced Apoptosis in Human Pancreatic beta-Cells: Activation of Caspases and ER Stress Induction. Cell. Physiol. Biochem. 2011, 27, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.W.; Koshkin, V.; Zhang, C.Y.; Wang, J.; Lowell, B.B.; Chan, C.B.; Wheeler, M.B. Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes 2002, 51, 3211–3219. [Google Scholar] [CrossRef]
- Sauter, N.S.; Schulthess, F.T.; Galasso, R.; Castellani, L.W.; Maedler, K. The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology 2008, 149, 2208–2218. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yao, D.D.; Yang, H.Y.; Wei, Y.J.; Peng, Y.R.; Ding, Y.F.; Shu, L. Puerarin Protects Pancreatic beta-Cells in Obese Diabetic Mice via Activation of GLP-1R Signaling. Mol. Endocrinol. 2016, 30, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, S.S.; Liu, Y.Q.; Yan, Z.; Zhang, F.; Lv, Q.G.; Tong, N.W. Activated PPAR beta/delta Protects Pancreatic beta Cells in Type 2 Diabetic Goto-Kakizaki Rats from Lipoapoptosis via GPR40. Lipids 2019, 54, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Morgan, N.G. Fatty acids and beta-cell toxicity. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 117–122. [Google Scholar] [CrossRef]
- Diakogiannaki, E.; Dhayal, S.; Childs, C.E.; Calder, P.C.; Welters, H.J.; Morgan, N.G. Mechanisms involved in the cytotoxic and cytoprotective actions of saturated versus monounsaturated long-chain fatty acids in pancreatic beta-cells. J. Endocrinol. 2007, 194, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Keane, D.; Welters, H.J.; Morgan, N.G. Life and death decisions of the pancreatic beta-cell: The role of fatty acids. Clin. Sci. 2007, 112, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Dhayal, S.; Welters, H.J.; Morgan, N.G. Structural requirements for the cytoprotective actions of mono-unsaturated fatty acids in the pancreatic beta-cell line, BRIN-BD11. Br. J. Pharm. 2008, 153, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Plotz, T.; von Hanstein, A.S.; Krummel, B.; Laporte, A.; Mehmeti, I.; Lenzen, S. Structure-toxicity relationships of saturated and unsaturated free fatty acids for elucidating the lipotoxic effects in human EndoC-beta H1 beta-cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165525. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, S.T.; Xu, Y.; Hoppel, C.L. Determination of plasma non-esterified fatty acids and triglyceride fatty acids by gas chromatography of their methyl esters after isolation by column chromatography on silica gel. J. Chromatogr. B Biomed. Appl. 1995, 666, 1–12. [Google Scholar] [CrossRef]
- Lagerstedt, S.A.; Hinrichs, D.R.; Batt, S.M.; Magera, M.J.; Rinaldo, P.; McConnell, J.P. Quantitative determination of plasma c8-c26 total fatty acids for the biochemical diagnosis of nutritional and metabolic disorders. Mol. Genet. Metab. 2001, 73, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Ladriere, L.; Hekerman, P.; Ortis, F.; Cardozo, A.K.; Dogusan, Z.; Flamez, D.; Boyce, M.; Yuan, J.; Eizirik, D.L. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J. Biol. Chem. 2007, 282, 3989–3997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zeng, X.; Chen, X.; Luo, R.; Li, L.; Wang, C.; Liu, J.; Cheng, J.; Lu, Y.; Chen, Y. Oleic acid protects insulin-secreting INS-1E cells against palmitic acid-induced lipotoxicity along with an amelioration of ER stress. Endocrine 2019, 64, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.; Bikopoulos, G.; Wheeler, M.B.; Rozakis-Adcock, M.; Volchuk, A. Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E540–E550. [Google Scholar] [CrossRef] [Green Version]
- Ladriere, L.; Igoillo-Esteve, M.; Cunha, D.A.; Brion, J.P.; Bugliani, M.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Enhanced signaling downstream of ribonucleic Acid-activated protein kinase-like endoplasmic reticulum kinase potentiates lipotoxic endoplasmic reticulum stress in human islets. J. Clin. Endocrinol. Metab. 2010, 95, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, K.; Hovsepyan, M.; Bergsten, P. Reduced levels of SCD1 accentuate palmitate-induced stress in insulin-producing beta-cells. Lipids Health Dis. 2010, 9, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, N.; Otabe, S.; Nakayama, H.; Yuan, X.; Yamada, K. Sequential activation of caspases and synergistic beta-cell cytotoxicity by palmitate and anti-Fas antibodies. Life Sci. 2006, 79, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.C.; Green, C.D.; Olson, L.K.; Moxley, M.A.; Corbett, J.A. A role for aberrant protein palmitoylation in FFA-induced ER stress and beta cell death. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1390–E1398. [Google Scholar] [CrossRef] [Green Version]
- Tuei, V.C.; Ha, J.S.; Ha, C.E. Effects of human serum albumin complexed with free fatty acids on cell viability and insulin secretion in the hamster pancreatic beta-cell line HIT-T15. Life Sci. 2011, 88, 810–818. [Google Scholar] [CrossRef]
- Busch, A.K.; Gurisik, E.; Cordery, D.V.; Sudlow, M.; Denyer, G.S.; Laybutt, D.R.; Hughes, W.E.; Biden, T.J. Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic beta-cells from lipoapoptosis. Diabetes 2005, 54, 2917–2924. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.E.; Kim, H.E.; Shin, H.C.; Jang, H.J.; Lee, K.W.; Kim, Y.; Kang, S.S.; Chun, J.; Kang, Y. Involvement of Ca2+-mediated apoptotic signals in palmitate-induced MIN6N8a beta cell death. Mol. Cell. Endocrinol. 2007, 272, 50–62. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Jeffrey, K.D.; Alejandro, E.U.; Luciani, D.S.; Kalynyak, T.B.; Hu, X.; Li, H.; Lin, Y.; Townsend, R.R.; Polonsky, K.S.; Johnson, J.D. Carboxypeptidase E mediates palmitate-induced beta-cell ER stress and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 8452–8457. [Google Scholar] [CrossRef] [Green Version]
- Eitel, K.; Staiger, H.; Rieger, J.; Mischak, H.; Brandhorst, H.; Brendel, M.D.; Bretzel, R.G.; Haring, H.U.; Kellerer, M. Protein kinase C delta activation and translocation to the nucleus are required for fatty acid-induced apoptosis of insulin-secreting cells. Diabetes 2003, 52, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Gehrmann, W.; Wurdemann, W.; Plotz, T.; Jorns, A.; Lenzen, S.; Elsner, M. Antagonism Between Saturated and Unsaturated Fatty Acids in ROS Mediated Lipotoxicity in Rat Insulin-Producing Cells. Cell. Physiol. Biochem. 2015, 36, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Sarnyai, F.; Donko, M.B.; Matyasi, J.; Gor-Nagy, Z.; Marczi, I.; Simon-Szabo, L.; Zambo, V.; Somogyi, A.; Csizmadia, T.; Low, P.; et al. Cellular toxicity of dietary trans fatty acids and its correlation with ceramide and diglyceride accumulation. Food Chem. Toxicol. 2019, 124, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Okuyama, R.; Fujiwara, T.; Ohsumi, J. High glucose potentiates palmitate-induced NO-mediated cytotoxicity through generation of superoxide in clonal beta-cell HIT-T15. FEBS Lett. 2003, 545, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, J.H.; Fielding, B.A.; Evershed, R.; Berstan, R.; Currie, J.M.; Clark, A. Adverse physicochemical properties of tripalmitin in beta cells lead to morphological changes and lipotoxicity in vitro. Diabetologia 2005, 48, 1819–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Xia, P. Cellular Inhibitor of Apoptosis Protein-1 (cIAP1) Plays a Critical Role in β-Cell Survival under Endoplasmic Reticulum Stress: Promoting ubiquitination and degradation of C/EBP Homologous Protein (CHOP). J. Biol. Chem. 2012, 287, 32236–32245. [Google Scholar] [CrossRef] [Green Version]
- Sommerweiss, D.; Gorski, T.; Richter, S.; Garten, A.; Kiess, W. Oleate rescues INS-1E beta-cells from palmitate-induced apoptosis by preventing activation of the unfolded protein response. Biochem. Biophys. Res. Commun. 2013, 441, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Vasu, S.; McClenaghan, N.H.; McCluskey, J.T.; Flatt, P.R. Effects of lipotoxicity on a novel insulin-secreting human pancreatic beta-cell line, 1.1B4. Biol. Chem. 2013, 394, 909–918. [Google Scholar] [CrossRef]
- Nemecz, M.; Constantin, A.; Dumitrescu, M.; Alexandru, N.; Filippi, A.; Tanko, G.; Georgescu, A. The Distinct Effects of Palmitic and Oleic Acid on Pancreatic Beta Cell Function: The Elucidation of Associated Mechanisms and Effector Molecules. Front. Pharm. 2018, 9, 1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurado-Ruiz, E.; Alvarez-Amor, L.; Varela, L.M.; Berna, G.; Parra-Camacho, M.S.; Oliveras-Lopez, M.J.; Martinez-Force, E.; Rojas, A.; Hmadcha, A.; Soria, B.; et al. Extra virgin olive oil diet intervention improves insulin resistance and islet performance in diet-induced diabetes in mice. Sci. Rep. 2019, 9, 13. [Google Scholar] [CrossRef]
- Cnop, M.; Hannaert, J.C.; Hoorens, A.; Eizirik, D.L.; Pipeleers, D.G. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 2001, 50, 1771–1777. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Hannaert, J.C.; Pipeleers, D.G. Troglitazone does not protect rat pancreatic beta cells against free fatty acid-induced cytotoxicity. Biochem. Pharm. 2002, 63, 1281–1285. [Google Scholar] [CrossRef]
- Wrede, C.E.; Dickson, L.M.; Lingohr, M.K.; Briaud, I.; Rhodes, C.J. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J. Biol. Chem. 2002, 277, 49676–49684. [Google Scholar] [CrossRef] [Green Version]
- Maestre, I.; Jordan, J.; Calvo, S.; Reig, J.A.; Cena, V.; Soria, B.; Prentki, M.; Roche, E. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology 2003, 144, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, X.; Ran, X.; Chen, J.; Li, X.; Wu, W.; Huang, H.; Huang, H.; Long, Y.; Liang, J.; et al. Sterol regulatory element-binding protein-1c knockdown protected INS-1E cells from lipotoxicity. Diabetes Obes. Metab. 2010, 12, 35–46. [Google Scholar] [CrossRef]
- Tuo, Y.; Wang, D.; Li, S.; Chen, C. Long-term exposure of INS-1 rat insulinoma cells to linoleic acid and glucose in vitro affects cell viability and function through mitochondrial-mediated pathways. Endocrine 2011, 39, 128–138. [Google Scholar] [CrossRef]
- Plotz, T.; Krummel, B.; Laporte, A.; Pingitore, A.; Persaud, S.J.; Jorns, A.; Elsner, M.; Mehmeti, I.; Lenzen, S. The monounsaturated fatty acid oleate is the major physiological toxic free fatty acid for human beta cells. Nutr. Diabetes 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dhayal, S.; Morgan, N.G. Structure-activity relationships influencing lipid-induced changes in eIF2alpha phosphorylation and cell viability in BRIN-BD11 cells. FEBS Lett. 2011, 585, 2243–2248. [Google Scholar] [CrossRef] [Green Version]
- Bellini, L.; Campana, M.; Rouch, C.; Chacinska, M.; Bugliani, M.; Meneyrol, K.; Hainault, I.; Lenoir, V.; Denom, J.; Veret, J.; et al. Protective role of the ELOVL2/docosahexaenoic acid axis in glucolipotoxicity-induced apoptosis in rodent beta cells and human islets. Diabetologia 2018, 61, 1780–1793. [Google Scholar] [CrossRef] [Green Version]
- Sargsyan, E.; Artemenko, K.; Manukyan, L.; Bergquist, J.; Bergsten, P. Oleate protects beta-cells from the toxic effect of palmitate by activating pro-survival pathways of the ER stress response. Biochim. Biophys. Acta 2016, 1861, 1151–1160. [Google Scholar] [CrossRef]
- Plotz, T.; Hartmann, M.; Lenzen, S.; Elsner, M. The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutr. Metab. 2016, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Beeharry, N.; Chambers, J.A.; Green, I.C. Fatty acid protection from palmitic acid-induced apoptosis is lost following PI3-kinase inhibition. Apoptosis 2004, 9, 599–607. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Zhang, X.Y.; Zhang, L.; Zhang, M.L.; Li, L.; Luo, D.; Zhong, Y. Perilipin5 protects against lipotoxicity and alleviates endoplasmic reticulum stress in pancreatic beta-cells. Nutr. Metab. 2019, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, L.B.; Li, G.; Luo, T.H.; Gu, Y.Y.; Qian, L.; Fu, X.L.; Li, F.Y.; Li, J.P.; Luo, M. Palmitate activates AMP-activated protein kinase and regulates insulin secretion from beta cells. Biochem. Biophys. Res. Commun. 2007, 352, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Eberhard, C.E.; Screaton, R.A. Role of AMPK in pancreatic beta cell function. Mol. Cell. Endocrinol. 2013, 366, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.L.; Huang, S.L.; Leng, Y. AICAR and Metformin Exert AMPK-dependent Effects on INS-1E Pancreatic beta-cell Apoptosis via Differential Downstream Mechanisms. Int. J. Biol. Sci. 2015, 11, 1272–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, M.; Pechberty, S.; Bellini, L.; Gopel, S.O.; Campana, M.; Rouch, C.; Dairou, J.; Cosentino, C.; Fantuzzi, F.; Toivonen, S.; et al. Stearoyl CoA desaturase is a gatekeeper that protects human beta cells against lipotoxicity and maintains their identity. Diabetologia 2020, 63, 395–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janikiewicz, J.; Hanzelka, K.; Dziewulska, A.; Kozinski, K.; Dobrzyn, P.; Bernas, T.; Dobrzyn, A. Inhibition of SCD1 impairs palmitate-derived autophagy at the step of autophagosome-lysosome fusion in pancreatic beta-cells. J. Lipid. Res. 2015, 56, 1901–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briaud, I.; Harmon, J.S.; Kelpe, C.L.; Segu, V.B.; Poitout, V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes 2001, 50, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Tsai, T.H.; Li, L.; Saha, P.; Chan, L.; Chang, B.H.J. PLIN2 is a Key Regulator of the Unfolded Protein Response and Endoplasmic Reticulum Stress Resolution in Pancreatic beta Cells. Sci. Rep. 2017, 7, 12. [Google Scholar]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar] [CrossRef] [Green Version]
- Manukyan, L.; Ubhayasekera, S.; Bergquist, J.; Sargsyan, E.; Bergsten, P. Palmitate-Induced Impairments of beta-Cell Function Are Linked With Generation of Specific Ceramide Species via Acylation of Sphingosine. Endocrinology 2015, 156, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimabukuro, M.; Higa, M.; Zhou, Y.T.; Wang, M.Y.; Newgard, C.B.; Unger, R.H. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J. Biol. Chem. 1998, 273, 32487–32490. [Google Scholar] [CrossRef] [Green Version]
- Veret, J.; Coant, N.; Berdyshev, E.V.; Skobeleva, A.; Therville, N.; Bailbe, D.; Gorshkova, I.; Natarajan, V.; Portha, B.; Le Stunff, H. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 beta-cells. Biochem. J. 2011, 438, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolova-Karakashian, M.N.; Rozenova, K.A. Ceramide in Stress Response. In Sphingolipids as Signaling and Regulatory Molecules; Chalfant, C., Del Poeta, M., Eds.; Springer: Berlin, Germany, 2010; Volume 688, pp. 86–108. [Google Scholar]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blouin, C.M.; Prado, C.; Takane, K.K.; Lasnier, F.; Garcia-Ocana, A.; Ferre, P.; Dugail, I.; Hajduch, E. Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes 2010, 59, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.; Lee, H.; Goni, F.M.; Kolesnick, R.; Fernandez-Checa, J.C. Sphingolipids and cell death. Apoptosis 2007, 12, 923–939. [Google Scholar] [CrossRef]
- Nemcova-Furstova, V.; Balusikova, K.; Sramek, J.; James, R.F.; Kovar, J. Caspase-2 and JNK activated by saturated fatty acids are not involved in apoptosis induction but modulate ER stress in human pancreatic beta-cells. Cell. Physiol. Biochem. 2013, 31, 277–289. [Google Scholar] [CrossRef]
- Sramek, J.; Nemcova-Furstova, V.; Pavlikova, N.; Kovar, J. Effect of Saturated Stearic Acid on MAP Kinase and ER Stress Signaling Pathways during Apoptosis Induction in Human Pancreatic beta-Cells Is Inhibited by Unsaturated Oleic Acid. Int. J. Mol. Sci. 2017, 18, 2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Ullrich, S.; Gulbins, E. Ceramide formation as a target in beta-cell survival and function. Expert Opin. Ther. Targets 2011, 15, 1061–1071. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Thevissen, K.; Francois, I.E.; Winderickx, J.; Pannecouque, C.; Cammue, B.P. Ceramide involvement in apoptosis and apoptotic diseases. Mini. Rev. Med. Chem. 2006, 6, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Karnovsky, M.J.; Kleinfeld, A.M.; Hoover, R.L.; Klausner, R.D. The concept of lipid domains in membranes. J. Cell Biol. 1982, 94, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, G.; Cohen, O.; Daniel, B.; Sansone, A.; Petropoulou, P.I.; Filou, S.; Spyridonidis, A.; Pani, G.; De Spirito, M.; Chatgilialoglu, C.; et al. Fatty acid-related modulations of membrane fluidity in cells: Detection and implications. Free Radic. Res. 2016, 50, S40–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Sheng, W.; Sun, G.Y.; Lee, J.C. Effects of fatty acid unsaturation numbers on membrane fluidity and alpha-secretase-dependent amyloid precursor protein processing. Neurochem. Int. 2011, 58, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilon, M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016, 15, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamedova, L.K.; Yuan, K.; Laudick, A.N.; Fleming, S.D.; Mashek, D.G.; Bradford, B.J. Toll-like receptor 4 signaling is required for induction of gluconeogenic gene expression by palmitate in human hepatic carcinoma cells. J. Nutr. Biochem. 2013, 24, 1499–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Q.L.; Zou, C.P.; Zhong, P.; Lin, F.; Li, W.X.; Wang, L.T.; Zhang, Y.L.; Zheng, C.; Wang, Y.; Li, X.K.; et al. EGFR mediates hyperlipidemia-induced renal injury via regulating inflammation and oxidative stress: The detrimental role and mechanism of EGFR activation. Oncotarget 2016, 7, 24361–24373. [Google Scholar] [CrossRef] [Green Version]
- Vacaresse, N.; Lajoie-Mazenc, I.; Auge, N.; Suc, I.; Frisach, M.F.; Salvayre, R.; Negre-Salvayre, A. Activation of epithelial growth factor receptor pathway by unsaturated fatty acids. Circ. Res. 1999, 85, 892–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.E.; Sun, Y.; Niu, J.X.; Jarugumilli, G.K.; Wu, X. Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities. Cell Chem. Biol. 2018, 25, 817–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Li, X.J.; Guo, Q.H.; Wang, X.; Xue, B.; Sun, L.Q.; Meng, Q.T.; Lu, J.M.; Mu, Y.M. LRP16 gene protects mouse insulinoma MIN6 cells against fatty acid-induced apoptosis through Akt/FoxO1 signaling. Chin. Med. J. 2012, 125, 1695–1702. [Google Scholar] [PubMed]
- Quan, X.; Zhang, L.; Li, Y.; Liang, C. TCF2 attenuates FFA-induced damage in islet beta-cells by regulating production of insulin and ROS. Int. J. Mol. Sci. 2014, 15, 13317–13332. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Nie, M.; Chen, C.; Chen, X.; Zhang, M.; Yuan, G.; Yu, X.; Yang, Y. Protective action of liraglutide in beta cells under lipotoxic stress via PI3K/Akt/FoxO1 pathway. J. Cell. Biochem. 2014, 115, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Kang, J.; Cao, Y.; Fan, S.; Yang, H.; An, Y.; Pan, Y.; Tie, L.; Li, X. Curcumin attenuates palmitate-induced apoptosis in MIN6 pancreatic beta-cells through PI3K/Akt/FoxO1 and mitochondrial survival pathways. Apoptosis 2015, 20, 1420–1432. [Google Scholar] [CrossRef]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Spagnuolo, R.; Labarbuta, R.; Porreca, I.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; Perrini, S.; et al. The Myokine Irisin Is Released in Response to Saturated Fatty Acids and Promotes Pancreatic beta-Cell Survival and Insulin Secretion. Diabetes 2017, 66, 2849–2856. [Google Scholar] [CrossRef] [Green Version]
- Litwak, S.A.; Wali, J.A.; Pappas, E.G.; Saadi, H.; Stanley, W.J.; Varanasi, L.C.; Kay, T.W.H.; Thomas, H.E.; Gurzov, E.N. Lipotoxic Stress Induces Pancreatic beta-Cell Apoptosis through Modulation of Bcl-2 Proteins by the Ubiquitin-Proteasome System. J. Diabetes Res. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Cunha, D.A.; Gurzov, E.N.; Naamane, N.; Ortis, F.; Cardozo, A.K.; Bugliani, M.; Marchetti, P.; Eizirik, D.L.; Cnop, M. JunB protects beta-cells from lipotoxicity via the XBP1-AKT pathway. Cell Death Differ. 2014, 21, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- Buteau, J.; El-Assaad, W.; Rhodes, C.J.; Rosenberg, L.; Joly, E.; Prentki, M. Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia 2004, 47, 806–815. [Google Scholar]
- Tuttle, R.L.; Gill, N.S.; Pugh, W.; Lee, J.P.; Koeberlein, B.; Furth, E.E.; Polonsky, K.S.; Naji, A.; Birnbaum, M.J. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKB alpha. Nat. Med. 2001, 7, 1133–1137. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, E.; Fatrai, S.; Johnson, J.D.; Ohsugi, M.; Otani, K.; Han, Z.Q.; Polonsky, K.S.; Permutt, M.A. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J. Clin. Investig. 2004, 114, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Widenmaier, S.B.; Ao, Z.L.; Kim, S.J.; Warnock, G.; McIntosh, C.H.S. Suppression of p38 MAPK and JNK via Akt-mediated Inhibition of Apoptosis Signal-regulating Kinase 1 Constitutes a Core Component of the beta-Cell Pro-survival Effects of Glucose-dependent Insulinotropic Polypeptide. J. Biol. Chem. 2009, 284, 30372–30382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikin, R.; Maysinger, D.; Rosenberg, L. Cross-talk between phosphatidylinositol 3-kinase/AKT and c-Jun NH2-terminal kinase mediates survival of isolated human islets. Endocrinology 2004, 145, 4522–4531. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.B.; Tang, N.M.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Kitamura, Y.I. Role of FoxO proteins in pancreatic beta cells. Endocr. J. 2007, 54, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H.; Wright, C.V.E.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Tindall, J.T. FOXO factors: A matter of life and death. Future Oncol. 2006, 2, 83–89. [Google Scholar] [CrossRef]
- Wang, W.; Liu, Y.; Chen, Y.; Cao, C.; Xiang, Y.; Zhang, D.; Han, L.; Zhao, H.; Liu, G. Inhibition of Foxo1 mediates protective effects of ghrelin against lipotoxicity in MIN6 pancreatic beta-cells. Peptides 2010, 31, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of beta-cells via JNK, p38 MAPK and p53 pathways. PLoS ONE 2010, 5, e15726. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Yong, W.; Yu, J.; Zhang, X.; Lin, H.; Zhu, Y.; Han, X. Pdcd2l Promotes Palmitate-Induced Pancreatic Beta-Cell Apoptosis as a FoxO1 Target Gene. PLoS ONE 2016, 11, e0166692. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Winter, K.; Nian, C.; Tsuneoka, M.; Koda, Y.; McIntosh, C.H. Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J. Biol. Chem. 2005, 280, 22297–22307. [Google Scholar] [PubMed] [Green Version]
- Martinez, S.C.; Tanabe, K.; Cras-Meneur, C.; Abumrad, N.A.; Bernal-Mizrachi, E.; Permutt, M.A. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 2008, 57, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Cunha, D.A.; Igoillo-Esteve, M.; Gurzov, E.N.; Germano, C.M.; Naamane, N.; Marhfour, I.; Fukaya, M.; Vanderwinden, J.M.; Gysemans, C.; Mathieu, C.; et al. Death Protein 5 and p53-Upregulated Modulator of Apoptosis Mediate the Endoplasmic Reticulum Stress-Mitochondrial Dialog Triggering Lipotoxic Rodent and Human beta-Cell Apoptosis. Diabetes 2012, 61, 2763–2775. [Google Scholar] [CrossRef] [Green Version]
- Wrede, C.E.; Dickson, L.M.; Lingohr, M.K.; Briaud, I.; Rhodes, C.J. Fatty acid and phorbol ester-mediated interference of mitogenic signaling via novel protein kinase C isoforms in pancreatic beta-cells (INS-1). J. Mol. Endocrinol. 2003, 30, 271–286. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, M.; Zhang, S.; Yan, L.; Yang, C.; Lu, W.; Li, Y.; Cheng, H. The role of G protein-coupled receptor 40 in lipoapoptosis in mouse beta-cell line NIT-1. J. Mol. Endocrinol. 2007, 38, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.N.; Azevedo-Martins, A.K.; Amanso, A.M.; Carvalho, C.R.O.; Curi, R. Persistent activation of Akt or ERK prevents the toxicity induced by saturated and polyunsaturated fatty acids in RINm5F beta-cells. Toxicol. Vitr. 2008, 22, 1018–1024. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharm. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Panse, M.; Gerst, F.; Kaiser, G.; Teutsch, C.A.; Dolker, R.; Wagner, R.; Haring, H.U.; Ullrich, S. Activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) by free fatty acid receptor 1 (FFAR1/GPR40) protects from palmitate-induced beta cell death, but plays no role in insulin secretion. Cell. Physiol. Biochem. 2015, 35, 1537–1545. [Google Scholar] [CrossRef]
- Abaraviciene, S.M.; Lundquist, I.; Salehi, A. Rosiglitazone counteracts palmitate-induced beta-cell dysfunction by suppression of MAP kinase, inducible nitric oxide synthase and caspase 3 activities. Cell. Mol. Life Sci. 2008, 65, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Fontes, G.; Semache, M.; Hagman, D.K.; Tremblay, C.; Shah, R.; Rhodes, C.J.; Rutter, J.; Poitout, V. Involvement of Per-Arnt-Sim Kinase and Extracellular-Regulated Kinases-1/2 in Palmitate Inhibition of Insulin Gene Expression in Pancreatic beta-Cells. Diabetes 2009, 58, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Plaisance, V.; Perret, V.; Favre, D.; Abderrahmani, A.; Yang, J.Y.; Widmann, C.; Regazzi, R. Role of the transcriptional factor C/EBPbeta in free fatty acid-elicited beta-cell failure. Mol. Cell. Endocrinol. 2009, 305, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Qian, Y.; Xi, X.; Hu, X.; Zhu, J.; Han, X. Blockage of ceramide metabolism exacerbates palmitate inhibition of pro-insulin gene expression in pancreatic beta-cells. Mol. Cell. Biochem. 2010, 338, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.L.; Macrae, K.; Marley, A.E.; Hundal, H.S. Chronic effects of palmitate overload on nutrient-induced insulin secretion and autocrine signalling in pancreatic MIN6 beta cells. PLoS ONE 2011, 6, e25975. [Google Scholar] [CrossRef]
- Liu, L.; Liang, C.; Mei, P.C.; Zhu, H.; Hou, M.L.; Yu, C.L.; Song, Z.B.; Bao, Y.L.; Huang, Y.X.; Yi, J.W.; et al. Dracorhodin perchlorate protects pancreatic beta-cells against glucotoxicity- or lipotoxicity-induced dysfunction and apoptosis in vitro and in vivo. FEBS J. 2019, 286, 3718–3736. [Google Scholar] [CrossRef]
- Sramek, J.; Nemcova-Furstova, V.; Kovar, J. Kinase Signaling in Apoptosis Induced by Saturated Fatty Acids in Pancreatic beta-Cells. Int. J. Mol. Sci. 2016, 17, 1400. [Google Scholar] [CrossRef] [Green Version]
- Morgan, N.G.; Dhayal, S.; Diakogiannaki, E.; Welters, H.J. The cytoprotective actions of long-chain mono-unsaturated fatty acids in pancreatic beta-cells. Biochem. Soc. Trans. 2008, 36, 905–908. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Lavallard, V.J.; Meijer, A.J.; Codogno, P.; Gual, P. Autophagy, signaling and obesity. Pharm. Res. 2012, 66, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J.H. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvjeticanin, T.; Stojanovic, I.; Timotijevic, G.; Stosic-Grujicic, S.; Miljkovic, D. T cells cooperate with palmitic acid in induction of beta cell apoptosis. BMC Immunol. 2009, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Natalicchio, A.; Labarbuta, R.; Tortosa, F.; Biondi, G.; Marrano, N.; Peschechera, A.; Carchia, E.; Orlando, M.R.; Leonardini, A.; Cignarelli, A.; et al. Exendin-4 protects pancreatic beta cells from palmitate-induced apoptosis by interfering with GPR40 and the MKK4/7 stress kinase signalling pathway. Diabetologia 2013, 56, 2456–2466. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, T.; Zhang, D.; Leung, P.S. GPR120 protects lipotoxicity- induced pancreatic beta-cell dysfunction through regulation of PDX1 expression and inhibition of islet inflammation. Clin. Sci. 2019, 133, 101–116. [Google Scholar] [CrossRef]
- Zhou, L.; Cai, X.; Han, X.; Ji, L. P38 plays an important role in glucolipotoxicity-induced apoptosis in INS-1 cells. J. Diabetes Res. 2014, 2014, 834528. [Google Scholar] [CrossRef]
- Wei, X.; Gu, N.; Feng, N.; Guo, X.; Ma, X. Inhibition of p38 mitogen-activated protein kinase exerts a hypoglycemic effect by improving beta cell function via inhibition of beta cell apoptosis in db/db mice. J. Enzym. Inhib. Med. Chem. 2018, 33, 1494–1500. [Google Scholar] [CrossRef] [Green Version]
- Flores-Lopez, L.A.; Diaz-Flores, M.; Garcia-Macedo, R.; Avalos-Rodriguez, A.; Vergara-Onofre, M.; Cruz, M.; Contreras-Ramos, A.; Konigsberg, M.; Ortega-Camarillo, C. High glucose induces mitochondrial p53 phosphorylation by p38 MAPK in pancreatic RINm5F cells. Mol. Biol. Rep. 2013, 40, 4947–4958. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, U.; Matsuzaki, H.; Yamamoto, T. Protein kinase C delta (PKC delta): Activation mechanisms and functions. J. Biochem. 2002, 132, 831–839. [Google Scholar] [CrossRef]
- Qin, J.; Fang, N.; Lou, J.N.; Zhang, W.J.; Xu, S.Q.; Liu, H.L.; Fang, Q.; Wang, Z.; Liu, J.; Men, X.L.; et al. TRB3 Is Involved in Free Fatty Acid-Induced INS-1-Derived Cell Apoptosis via the Protein Kinase C delta Pathway. PLoS ONE 2014, 9, e96089. [Google Scholar]
- Welters, H.J.; Smith, S.A.; Tadayyon, M.; Scarpello, J.H.B.; Morgan, N.G. Evidence that protein kinase C delta is not required for palmitate-induced cytotoxicity in BRIN-BD11 beta-cells. J. Mol. Endocrinol. 2004, 32, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyland, M.E. Protein kinase C delta and apoptosis. Biochem. Soc. Trans. 2007, 35, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- De la Cadena, S.G.; Massieu, L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis 2016, 21, 763–777. [Google Scholar] [CrossRef]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress-mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.B.; Snyder, J.T.; Alonso, L.C. Atf6 alpha impacts cell number by influencing survival, death and proliferation. Mol. Metab. 2019, 27, S69–S80. [Google Scholar] [CrossRef] [PubMed]
- Martino, L.; Masini, M.; Novelli, M.; Beffy, P.; Bugliani, M.; Marselli, L.; Masiello, P.; Marchetti, P.; De Tata, V. Palmitate activates autophagy in INS-1E beta-cells and in isolated rat and human pancreatic islets. PLoS ONE 2012, 7, e36188. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Montano, P.; Rodriguez-Velazquez, E.; Ibarra-Lopez, E.; Frayde-Gomez, H.; Mas-Oliva, J.; Delgado-Coello, B.; Rivero, I.A.; Alatorre-Meda, M.; Aguilera, J.; Guevara-Olaya, L.; et al. Fatty Acid and Lipopolysaccharide Effect on Beta Cells Proteostasis and its Impact on Insulin Secretion. Cells 2019, 8, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, D.A.; Ladriere, L.; Ortis, F.; Igoillo-Esteve, M.; Gurzov, E.N.; Lupi, R.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Glucagon-like peptide-1 agonists protect pancreatic beta-cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 2009, 58, 2851–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachar, E.; Ariav, Y.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS ONE 2009, 4, e4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Q.; Fontes, G.; Saxena, G.; Poitout, V.; Shalev, A. Lack of TXNIP Protects Against Mitochondria-Mediated Apoptosis but Not Against Fatty Acid-Induced ER Stress-Mediated beta-Cell Death. Diabetes 2010, 59, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Gwiazda, K.S.; Yang, T.L.; Lin, Y.; Johnson, J.D. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E690–E701. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, E.; Sol, E.R.M.; Bergsten, P. UPR in palmitate-treated pancreatic beta-cells is not affected by altering oxidation of the fatty acid. Nutr. Metab. 2011, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Abdulkarim, B.; Hernangomez, M.; Igoillo-Esteve, M.; Cunha, D.A.; Marselli, L.; Marchetti, P.; Ladriere, L.; Cnop, M. Guanabenz Sensitizes Pancreatic beta Cells to Lipotoxic Endoplasmic Reticulum Stress and Apoptosis. Endocrinology 2017, 158, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Wan, F. Exendin-4 protects INS-1 cells against palmitate-induced apoptosis through the IRE1alpha-Xbp1 signaling pathway. Exp. Med. 2018, 16, 1029–1035. [Google Scholar]
- Allagnat, F.; Cunha, D.; Moore, F.; Vanderwinden, J.M.; Eizirik, D.L.; Cardozo, A.K. Mcl-1 downregulation by pro-inflammatory cytokines and palmitate is an early event contributing to beta-cell apoptosis. Cell Death. Differ. 2011, 18, 328–337. [Google Scholar] [CrossRef]
- Lu, H.M.; Hao, L.Y.; Li, S.T.; Lin, S.; Lv, L.; Chen, Y.; Cui, H.L.; Zi, T.Q.; Chu, X.; Na, L.X.; et al. Elevated circulating stearic acid leads to a major lipotoxic effect on mouse pancreatic beta cells in hyperlipidaemia via a miR-34a-5p-mediated PERK/p53-dependent pathway. Diabetologia 2016, 59, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.J.; Wu, J.H.; Sun, S.Y.; Zhou, J.Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic beta-cell injury. Sci. Rep. 2017, 7, 44746. [Google Scholar] [CrossRef] [Green Version]
- Bugliani, M.; Mossuto, S.; Grano, F.; Suleiman, M.; Marselli, L.; Boggi, U.; De Simone, P.; Eizirik, D.L.; Cnop, M.; Marchetti, P.; et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front. Endocrinol. 2019, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Ishigaki, S.; Fonseca, S.G.; Oslowski, C.M.; Jurczyk, A.; Shearstone, J.R.; Zhu, L.J.; Permutt, M.A.; Greiner, D.L.; Bortell, R.; Urano, F. AATF mediates an antiapoptotic effect of the unfolded protein response through transcriptional regulation of AKT1. Cell Death Differ. 2010, 17, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Allagnat, F.; Christulia, F.; Ortis, F.; Pirot, P.; Lortz, S.; Lenzen, S.; Eizirik, D.L.; Cardozo, A.K. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 2010, 53, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Miani, M.; Colli, M.L.; Ladriere, L.; Cnop, M.; Eizirik, D.L. Mild endoplasmic reticulum st XBP1) induces pancreatic beta cell dysfunction ress augments the proinflammatory effect of IL-1beta in pancreatic rat beta-cells via the IRE1alpha/XBP1s pathway. Endocrinology 2012, 153, 3017–3028. [Google Scholar] [CrossRef]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, beta cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef]
- Chan, J.Y.; Luzuriaga, J.; Maxwell, E.L.; West, P.K.; Bensellam, M.; Laybutt, D.R. The balance between adaptive and apoptotic unfolded protein responses regulates beta-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol. Cell. Endocrinol. 2015, 413, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.R.; Murakami, K.; Chen, N.J.; Saibil, S.D.; Matysiak-Zablocki, E.; Elford, A.R.; Bonnard, M.; Benchimol, S.; Jurisicova, A.; Yeh, W.C.; et al. DNA damage- and stress-induced apoptosis occurs independently of PIDD. Apoptosis 2009, 14, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Lee, Y.J.; Kang, Y.; Han, J.; Lim, O.K.; Jun, H.S. Exendin-4 inhibits glucolipotoxic ER stress in pancreatic beta cells via regulation of SREBP1c and C/EBP beta transcription factors. J. Endocrinol. 2013, 216, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Jung, I.R.; Choi, S.E.; Lee, S.M.; Lee, S.J.; Han, S.J.; Kim, H.J.; Kim, D.J.; Lee, K.W.; Karig, Y. Toxicity generated through inhibition of pyruvate carboxylase and carnitine palmitoyl transferase-1 is similar to high glucose/palmitate-induced glucolipotoxicity in INS-1 beta cells. Mol. Cell. Endocrinol. 2014, 383, 48–59. [Google Scholar] [CrossRef]
- Kitahara, A.; Takahashi, K.; Morita, N.; Murashima, T.; Onuma, H.; Sumitani, Y.; Tanaka, T.; Kondo, T.; Hosaka, T.; Ishida, H. The Novel Mechanisms Concerning the Inhibitions of Palmitate-Induced Proinflammatory Factor Releases and Endogenous Cellular Stress with Astaxanthin on MIN6-Cells. Mar. Drugs 2017, 15, 16. [Google Scholar]
- Prause, M.; Christensen, D.P.; Billestrup, N.; Mandrup-Poulsen, T. JNK1 Protects against Glucolipotoxicity-Mediated Beta-Cell Apoptosis. PLoS ONE 2014, 9, e87067. [Google Scholar] [CrossRef] [Green Version]
- Komiya, K.; Uchida, T.; Ueno, T.; Koike, M.; Abe, H.; Hirose, T.; Kawamori, R.; Uchiyama, Y.; Kominami, E.; Fujitani, Y.; et al. Free fatty acids stimulate autophagy in pancreatic beta-cells via JNK pathway. Biochem. Biophys. Res. Commun. 2010, 401, 561–567. [Google Scholar] [CrossRef]
- Lanuza-Masdeu, J.; Arevalo, M.I.; Vila, C.; Barbera, A.; Gomis, R.; Caelles, C. In vivo JNK activation in pancreatic beta-cells leads to glucose intolerance caused by insulin resistance in pancreas. Diabetes 2013, 62, 2308–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.H.; Lee, J.W.; Gao, B.; Jung, M.H. Synergistic activation of JNK/SAPK induced by TNF-alpha and IFN-gamma: Apoptosis of pancreatic beta-cells via the p53 and ROS pathway. Cell. Signal. 2005, 17, 1516–1532. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Choi, S.E.; Lee, J.H.; Lee, J.J.; Jung, I.R.; Lee, S.J.; Lee, K.W.; Kang, Y. Involvement of the TLR4 (Toll-like receptor4) signaling pathway in palmitate-induced INS-1 beta cell death. Mol. Cell. Biochem. 2011, 354, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Sramek, J.; Nemcova-Furstova, V.; Polak, J.; Kovar, J. Hypoxia Modulates Effects of Fatty Acids on NES2Y Human Pancreatic beta-cells. Int. J. Mol. Sci. 2019, 20, 3441. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Ma, J.; Wang, X.; Yang, W.; Zhang, J.; Ji, Q. Free fatty acid induces endoplasmic reticulum stress and apoptosis of beta-cells by Ca2+/calpain-2 pathways. PLoS ONE 2013, 8, e59921. [Google Scholar]
- Teodoro, T.; Odisho, T.; Sidorova, E.; Volchuk, A. Pancreatic beta-cells depend on basal expression of active ATF6alpha-p50 for cell survival even under nonstress conditions. Am. J. Physiol. Cell Physiol. 2012, 302, C992–C1003. [Google Scholar] [CrossRef]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef] [Green Version]
- Ahowesso, C.; Black, P.N.; Saini, N.; Montefusco, D.; Chekal, J.; Malosh, C.; Lindsley, C.W.; Stauffer, S.R.; DiRusso, C.C. Chemical inhibition of fatty acid absorption and cellular uptake limits lipotoxic cell death. Biochem. Pharm. 2015, 98, 167–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, B.; Bai, M.J.; Shen, W. Reduced Glutathione Protects Human Hepatocytes from Palmitate-Mediated Injury by Suppressing Endoplasmic Reticulum Stress Response. Hepato Gastroenterol. 2011, 58, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Chung, M.H.; Park, S.; Cha, J.; Baek, J.H.; Lee, S.Y.; Choi, S.Y. ER stress attenuation by Aloe-derived polysaccharides in the protection of pancreatic beta-cells from free fatty acid-induced lipotoxicity. Biochem. Biophys. Res. Commun. 2018, 500, 797–803. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Mihailidou, C.; Papavassiliou, A.G.; Kiaris, H. A crosstalk between p21 and UPR-induced transcription factor C/EBP homologous protein (CHOP) linked to type 2 diabetes. Biochimie 2014, 99, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Wali, J.A.; Rondas, D.; McKenzie, M.D.; Zhao, Y.; Elkerbout, L.; Fynch, S.; Gurzov, E.N.; Akira, S.; Mathieu, C.; Kay, T.W.H.; et al. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy-Thandavan, S.; Jiang, M.; Schoenlein, P.; Dong, Z. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Ren. Physiol. 2009, 297, F244–F256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.J.; Li, Y.B.; Wang, Y.; Liu, G.D.; Wang, J.; Zhu, X.O.; Pan, S.H. The role of autophagy in endoplasmic reticulum stress-induced pancreatic beta cell death. Autophagy 2012, 8, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.H.; Chen, L.X. Endoplasmic Reticulum Stress and Autophagy. In Autophagy: Biology and Diseases: Basic Science; Qin, Z.H., Ed.; Springer: Berlin, Germany, 2019; Volume 1206, pp. 167–177. [Google Scholar]
- Yuan, T.; Lupse, B.; Maedler, K.; Ardestani, A. mTORC2 Signaling: A Path for Pancreatic beta Cell’s Growth and Function. J. Mol. Biol. 2018, 430, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Song, S.L.; Tan, J.; Miao, Y.Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Hatanaka, M.; Maier, B.; Sims, E.K.; Templin, A.T.; Kulkarni, R.N.; Evans-Molina, C.; Mirmira, R.G. Palmitate Induces mRNA Translation and Increases ER Protein Load in Islet beta-Cells via Activation of the Mammalian Target of Rapamycin Pathway. Diabetes 2014, 63, 3404–3415. [Google Scholar] [CrossRef] [Green Version]
- Mir, S.U.; George, N.M.; Zahoor, L.; Harms, R.; Guinn, Z.; Sarvetnick, N.E. Inhibition of autophagic turnover in beta-cells by fatty acids and glucose leads to apoptotic cell death. J. Biol. Chem. 2015, 290, 6071–6085. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.E.; Lee, S.M.; Lee, Y.J.; Li, L.J.; Lee, S.J.; Lee, J.H.; Kim, Y.; Jun, H.S.; Lee, K.W.; Kang, Y. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology 2009, 150, 126–134. [Google Scholar] [CrossRef]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.S.; Chung, K.W.; Kim, J.W.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of Autophagy Diminishes Pancreatic beta Cell Mass and Function with Resultant Hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaniuk, N.A.; Kiraly, M.; Bates, H.; Vranic, M.; Volchuk, A.; Brumell, J.H. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 2007, 56, 930–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.C.; Kim, K.H.; Kim, G.H.; Kim, S.W.; Kim, H.L.; Lee, M.K.; et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujitani, Y.; Ebato, C.; Uchida, T.; Kawamori, R.; Watada, H. beta-cell autophagy: A novel mechanism regulating beta-cell function and mass: Lessons from beta-cell-specific Atg7-deficient mice. Islets 2009, 1, 151–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zummo, F.P.; Cullen, K.S.; Honkanen-Scott, M.; Shaw, J.A.M.; Lovat, P.E.; Arden, C. Glucagon-Like Peptide 1 Protects Pancreatic-Cells From Death by Increasing Autophagic Flux and Restoring Lysosomal Function. Diabetes 2017, 66, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, L.; Meshinchi, S.; Dias-Leme, C.; Raffin, D.; Johnson, J.D.; Treutelaar, M.K.; Burant, C.F. Islet microvasculature in islet hyperplasia and failure in a model of type 2 diabetes. Diabetes 2006, 55, 2965–2973. [Google Scholar] [CrossRef] [Green Version]
- Las, G.; Serada, S.B.; Wikstrom, J.D.; Twig, G.; Shirihai, O.S. Fatty acids suppress autophagic turnover in beta-cells. J. Biol. Chem. 2011, 286, 42534–42544. [Google Scholar] [CrossRef] [Green Version]
- Dhayal, S.; Zummo, F.P.; Anderson, M.W.; Thomas, P.; Welters, H.J.; Arden, C.; Morgan, N.G. Differential effects of saturated and unsaturated fatty acids on autophagy in pancreatic beta-cells. J. Mol. Endocrinol. 2019, 63, 285–296. [Google Scholar] [CrossRef]
- Chu, K.Y.; O’Reilly, L.; Mellet, N.; Meikle, P.J.; Bartley, C.; Biden, T.J. Oleate disrupts cAMP signaling, contributing to potent stimulation of pancreatic beta-cell autophagy. J. Biol. Chem. 2019, 294, 1218–1229. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wu, J.; Wu, H.; Liu, X.Z.; Chen, Y.J.; Wu, J.Y.; Hu, C.J.; Zou, D.J. Liraglutide protects pancreatic beta-cells against free fatty acids in vitro and affects glucolipid metabolism in apolipoprotein E-/- mice by activating autophagy. Mol. Med. Rep. 2015, 12, 4210–4218. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Yang, S.; Yang, L.; Cheng, Y.; Zhang, H. Interleukin-22 Alleviated Palmitate-Induced Endoplasmic Reticulum Stress in INS-1 Cells through Activation of Autophagy. PLoS ONE 2016, 11, e0146818. [Google Scholar] [CrossRef]
- Lin, N.; Niu, Y.X.; Zhang, W.W.; Li, X.Y.; Yang, Z.; Su, Q. microRNA-802 is involved in palmitate-induced damage to pancreatic beta cells through repression of sirtuin 6. Int. J. Clin. Exp. Pathol. 2017, 10, 11300–11307. [Google Scholar]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Wehinger, S.; Ortiz, R.; Diaz, M.I.; Aguirre, A.; Valenzuela, M.; Llanos, P.; Mc Master, C.; Leyton, L.; Quest, A.F. Phosphorylation of caveolin-1 on tyrosine-14 induced by ROS enhances palmitate-induced death of beta-pancreatic cells. Biochim. Biophys. Acta 2015, 1852, 693–708. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Chen, S.Y.; Deng, L.D.; Feng, L.P.; Huang, L.Z.; Yu, R.R. Antioxidant effect of mogrosides against oxidative stress induced by palmitic acid in mouse insulinoma NIT-1 cells. Braz. J. Med. Biol. Res. 2013, 46, 949–955. [Google Scholar] [CrossRef]
- Shen, X.; Yang, L.; Yan, S.; Wei, W.; Liang, L.; Zheng, H.; Cai, X. The effect of FFAR1 on pioglitazone-mediated attenuation of palmitic acid-induced oxidative stress and apoptosis in betaTC6 cells. Metabolism 2014, 63, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Kim, C.H.; Kim, K.Y.; Cheon, H.G. Protective effects of arachidonic acid against palmitic acid-mediated lipotoxicity in HIT-T15 cells. Mol. Cell Biochem. 2012, 364, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Chen, H.; Zhang, H.; Wan, X.; Su, Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine 2012, 42, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Lameloise, N.; Muzzin, P.; Prentki, M.; Assimacopoulos-Jeannet, F. Uncoupling protein 2: A possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes 2001, 50, 803–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, L.R.; Rebelato, E.; Graciano, M.F.; Abdulkader, F.; Curi, R.; Carpinelli, A.R. Oleic acid modulates metabolic substrate channeling during glucose-stimulated insulin secretion via NAD(P)H oxidase. Endocrinology 2011, 152, 3614–3621. [Google Scholar] [CrossRef] [Green Version]
- Koshkin, V.; Dai, F.F.; Robson-Doucette, C.A.; Chan, C.B.; Wheeler, M.B. Limited mitochondrial permeabilization is an early manifestation of palmitate-induced lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2008, 283, 7936–7948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Steneberg, P.; Rubins, N.; Bartoov-Shifman, R.; Walker, M.D.; Edlund, H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005, 1, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.K.; Sadasivuni, M.K.; Yateesh, A.N.; Neelima, K.; Mrudula, S.; Reddy, M.; Smitha, R.; Biswas, S.; Chandravanshi, B.; Pallavi, P.M.; et al. Activation of GPR40 attenuates chronic inflammation induced impact on pancreatic beta-cells health and function. BMC Cell Biol. 2014, 15, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61–R75. [Google Scholar] [CrossRef] [Green Version]
- Lovis, P.; Roggli, E.; Laybutt, R.; Gattesco, S.; Yang, J.Y.; Widmann, C.; Abderrahmani, A.; Regazzi, R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008, 57, 2728–2736. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Sun, Y.; Zhou, Y.C.; Zhang, Y.; Zhang, T.; Li, Y.T.; You, W.Y.; Chang, X.A.; Yuan, L.; Han, X. MicroRNA-24 promotes pancreatic beta cells toward dedifferentiation to avoid endoplasmic reticulum stress-induced apoptosis. J. Mol. Cell Biol. 2019, 11, 747–760. [Google Scholar] [CrossRef]
- Lin, X.; Guan, H.; Huang, Z.; Liu, J.; Li, H.; Wei, G.; Cao, X.; Li, Y. Downregulation of Bcl-2 expression by miR-34a mediates palmitate-induced Min6 cells apoptosis. J. Diabetes Res. 2014, 2014, 258695. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.B.; Wang, M.N.; Li, Q.; Guo, L.; Yang, Y.M.; Li, P.J.; Wang, W.; Zhang, J.C. MicroRNA-34a contributes to the protective effects of glucagon-like peptide-1 against lipotoxicity in INS-1 cells. Chin. Med. J. 2012, 125, 4202–4208. [Google Scholar]
- Li, Y.; Xu, X.J.; Liang, Y.; Liu, S.Y.; Xiao, H.S.; Li, F.; Cheng, H.; Fu, Z.Z. miR-375 enhances palmitate-induced lipoapoptosis in insulin-secreting NIT-1 cells by repressing myotrophin (V1) protein expression. Int. J. Clin. Exp. Pathol. 2010, 3, 254–264. [Google Scholar] [PubMed]
- Liu, Y.; Dong, J.Y.; Ren, B. MicroRNA-182-5p contributes to the protective effects of thrombospondin 1 against lipotoxicity in INS-1 cells. Exp. Med. 2018, 16, 5272–5279. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Yu, Y.; Zhang, Y.J.; Li, Y.L.; Chu, X.; Lu, H.M.; Sun, C.H. Overexpression of miR-297b-5p protects against stearic acid-induced pancreatic beta-cell apoptosis by targeting LATS2. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E430–E439. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Guo, R.; Zhang, Y.J.; Shi, H.B.; Sun, H.R.; Chu, X.; Wu, X.Y.; Lu, H.M.; Sun, C.H. miRNA-mRNA profile and regulatory network in stearic acid-treated beta-cell dysfunction. J. Endocrinol. 2020, 246, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.; Cheng, Y.; Fu, Y.; Zhao, H.; Tang, M.; Zhao, H.; Lin, N.; Shi, X.; Lei, Y.; et al. Mesenchymal stem cell-derived exosomes protect beta cells against hypoxia-induced apoptosis via miR-21 by alleviating ER stress and inhibiting p38 MAPK phosphorylation. Stem Cell Res. Ther. 2020, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, A.; Takada, M.; Nakayama, K.; Ishikawa, T. cGMP-independent anti-apoptotic effect of nitric oxide on thapsigargin-induced apoptosis in the pancreatic beta-cell line INS-1. Life Sci. 2008, 83, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Bachar, E.; Ariav, Y.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Neuronal nitric oxide synthase protects the pancreatic beta cell from glucolipotoxicity-induced endoplasmic reticulum stress and apoptosis. Diabetologia 2010, 53, 2177–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piro, S.; Anello, M.; Di, P.C.; Lizzio, M.N.; Patane, G.; Rabuazzo, A.M.; Vigneri, R.; Purrello, M.; Purrello, F. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: Possible role of oxidative stress. Metabolism 2002, 51, 1340–1347. [Google Scholar] [CrossRef]
- Sargsyan, E.; Ortsater, H.; Thorn, K.; Bergsten, P. Diazoxide-induced beta-cell rest reduces endoplasmic reticulum stress in lipotoxic beta-cells. J. Endocrinol. 2008, 199, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium efflux from the endoplasmic reticulum leads to beta-cell death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Sun, P.; Wang, T.; Chen, K.; Zhu, W.; Wang, H. Inhibition of Calcium Influx Reduces Dysfunction and Apoptosis in Lipotoxic Pancreatic beta-Cells via Regulation of Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0132411. [Google Scholar]
- Vogel, J.; Yin, J.; Su, L.; Wang, S.X.; Zessis, R.; Fowler, S.; Chiu, C.H.; Wilson, A.C.; Chen, A.; Zecri, F.; et al. A Phenotypic Screen Identifies Calcium Overload as a Key Mechanism of beta-Cell Glucolipotoxicity. Diabetes 2020, 69, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Rakatzi, I.; Mueller, H.; Ritzeler, O.; Tennagels, N.; Eckel, J. Adiponectin counteracts cytokine- and fatty acid-induced apoptosis in the pancreatic beta-cell line INS-1. Diabetologia 2004, 47, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melloul, D. Role of NF-kappaB in beta-cell death. Biochem. Soc. Trans. 2008, 36, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kowluru, A. CD36 mediates lipid accumulation in pancreatic beta cells under the duress of glucolipotoxic conditions: Novel roles of lysine deacetylases. Biochem. Biophys. Res. Commun. 2018, 495, 2221–2226. [Google Scholar] [CrossRef]
- Johnson, J.D.; Han, Z.; Otani, K.; Ye, H.; Zhang, Y.; Wu, H.; Horikawa, Y.; Misler, S.; Bell, G.I.; Polonsky, K.S. RyR2 and calpain-10 delineate a novel apoptosis pathway in pancreatic islets. J. Biol. Chem. 2004, 279, 24794–24802. [Google Scholar] [CrossRef] [Green Version]
- Nemcova-Furstova, V.; Balusikova, K.; Halada, P.; Pavlikova, N.; Sramek, J.; Kovar, J. Stearate-Induced Apoptosis in Human Pancreatic beta-Cells is Associated with Changes in Membrane Protein Expression and These Changes are Inhibited by Oleate. Proteom. Clin. Appl. 2019, 13, e1800104. [Google Scholar] [CrossRef]
- Yamani, L.; Li, B.; Larose, L. Nck1 deficiency improves pancreatic beta cell survival to diabetes-relevant stresses by modulating PERK activation and signaling. Cell. Signal. 2015, 27, 2555–2567. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.; Tharakan, B.; Bhat, G.K. Caspases—An update. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 151, 10–27. [Google Scholar] [CrossRef]
- Edlich, F. BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef]
- Gu, J.Q.; Wei, Q.; Zheng, H.Z.; Meng, X.; Zhang, J.; Wang, D.F. Exendin-4 Promotes Survival of Mouse Pancreatic beta-Cell Line in Lipotoxic Conditions, through the Extracellular Signal-Related Kinase 1/2 Pathway. J. Diabetes Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Kim, M.H.; Kwon, H.J.; Choi, S.Y.; Kwon, H.Y. Protective Effects of Oleic Acid Against Palmitic Acid-Induced Apoptosis in Pancreatic AR42J Cells and Its Mechanisms. Korean J. Physiol. Pharmacol. 2013, 17, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.T.; Wen, B.; Ning, Z.W.; Zhai, L.X.; Liao, C.H.; Lin, C.Y.; Mu, H.X.; Bian, Z.X. Cyclocarya paliurus tea leaves enhances pancreatic beta cell preservation through inhibition of apoptosis. Sci. Rep. 2017, 7, 13. [Google Scholar]
- Cunha, D.A.; Cito, M.; Carlsson, P.O.; Vanderwinden, J.M.; Molkentin, J.D.; Bugliani, M.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Thrombospondin 1 protects pancreatic beta-cells from lipotoxicity via the PERK-NRF2 pathway. Cell Death Differ. 2016, 23, 1995–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, T.C.; Paula, F.M.; Villate, O.; Colli, M.L.; Moura, R.F.; Cunha, D.A.; Marselli, L.; Marchetti, P.; Cnop, M.; Julier, C.; et al. GLIS3, a Susceptibility Gene for Type 1 and Type 2 Diabetes, Modulates Pancreatic Beta Cell Apoptosis via Regulation of a Splice Variant of the BH3-Only Protein Bim. PLoS Genet. 2013, 9, e1003532. [Google Scholar] [CrossRef]
- Tan, C.; Voss, U.; Svensson, S.; Erlinge, D.; Olde, B. High glucose and free fatty acids induce beta cell apoptosis via autocrine effects of ADP acting on the P2Y(13) receptor. Purinergic Signal. 2013, 9, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Grishko, V.; Rachek, L.; Musiyenko, S.; Ledoux, S.P.; Wilson, G.L. Involvement of mtDNA damage in free fatty acid-induced apoptosis. Free Radic. Biol. Med. 2005, 38, 755–762. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis—Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Su, Y.X.; Deng, H.C. Lipoapoptosis Pathways in Pancreatic beta-Cells and the Anti-Apoptosis Mechanisms of Adiponectin. Horm. Metab. Res. 2014, 46, 722–727. [Google Scholar]
- Lingohr, M. Decreasing IRS-2 expression in pancreatic β-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol. Cell. Endocrinol. 2003, 209, 17–31. [Google Scholar] [CrossRef]
- Marchetti, P.; Del, G.S.; Marselli, L.; Lupi, R.; Masini, M.; Pollera, M.; Bugliani, M.; Boggi, U.; Vistoli, F.; Mosca, F.; et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J. Clin. Endocrinol. Metab. 2004, 89, 5535–5541. [Google Scholar] [CrossRef] [Green Version]
- Liadis, N.; Salmena, L.; Kwan, E.; Tajmir, P.; Schroer, S.A.; Radziszewska, A.; Li, X.; Sheu, L.; Eweida, M.; Xu, S.; et al. Distinct in vivo roles of caspase-8 in beta-cells in physiological and diabetes models. Diabetes 2007, 56, 2302–2311. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.F.; Zhang, Z.Y.; Li, Q. DR5 but not miRNA-181 or miRNA-211 is involved in ER stress-mediated apoptosis induced by palmitate in islet beta cells. Int. J. Clin. Exp. Pathol. 2017, 10, 7692–7698. [Google Scholar]
- Bagnati, M.; Ogunkolade, B.W.; Marshall, C.; Tucci, C.; Hanna, K.; Jones, T.A.; Bugliani, M.; Nedjai, B.; Caton, P.W.; Kieswich, J.; et al. Glucolipotoxicity initiates pancreatic beta-cell death through TNFR5/CD40-mediated STAT1 and NF-kappa B activation. Cell Death Dis. 2016, 7, 8. [Google Scholar] [CrossRef]
- Neubauer, H.; Setiadi, P.; Gunesdogan, B.; Pinto, A.; Borgel, J.; Mugge, A. Influence of glycaemic control on platelet bound CD40-CD40L system, P-selectin and soluble CD40 ligand in Type 2 diabetes. Diabet. Med. 2010, 27, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Fingas, C.D.; Meng, X.W.; Finnberg, N.; El-Deiry, W.S.; Kaufmann, S.H.; Gores, G.J. Death Receptor 5 Signaling Promotes Hepatocyte Lipoapoptosis. J. Biol. Chem. 2011, 286, 39336–39348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.; Radziszewska, A.; Schroer, S.A.; Liadis, N.; Liu, Y.; Zhang, Y.; Lam, P.P.; Sheu, L.; Hao, Z.; Gaisano, H.Y.; et al. Deletion of Fas in the pancreatic beta-cells leads to enhanced insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1304–E1312. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.S.; Lindblom, K.R.; Robeson, A.; Stevens, R.D.; Ilkayeva, O.R.; Newgard, C.B.; Kornbluth, S.; Andersen, J.L. Metabolomic Profiling Reveals a Role for Caspase-2 in Lipoapoptosis. J. Biol. Chem. 2013, 288, 14463–14475. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Q.F.; Xiao, X.W.; Prasadan, K.; Chen, C.D.; Ming, Y.C.; Fusco, J.; Gangopadhyay, N.N.; Ricks, D.; Gittes, G.K. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S.; Hatae, N.; Yano, T.; Ruike, Y.; Kimura, M.; Hirasawa, A.; Tsujimoto, G. Free fatty acids inhibit serum deprivation-induced apoptosis through GPR120 in a murine enteroendocrine cell line STC-1. J. Biol. Chem. 2005, 280, 19507–19515. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šrámek, J.; Němcová-Fürstová, V.; Kovář, J. Molecular Mechanisms of Apoptosis Induction and Its Regulation by Fatty Acids in Pancreatic β-Cells. Int. J. Mol. Sci. 2021, 22, 4285. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084285
Šrámek J, Němcová-Fürstová V, Kovář J. Molecular Mechanisms of Apoptosis Induction and Its Regulation by Fatty Acids in Pancreatic β-Cells. International Journal of Molecular Sciences. 2021; 22(8):4285. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084285
Chicago/Turabian StyleŠrámek, Jan, Vlasta Němcová-Fürstová, and Jan Kovář. 2021. "Molecular Mechanisms of Apoptosis Induction and Its Regulation by Fatty Acids in Pancreatic β-Cells" International Journal of Molecular Sciences 22, no. 8: 4285. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084285