
Nusinersen Modulates Proteomics Profiles of Cerebrospinal Fluid in Spinal Muscular Atrophy Type 1 Patients

 , , , and
, , , and
Abstract
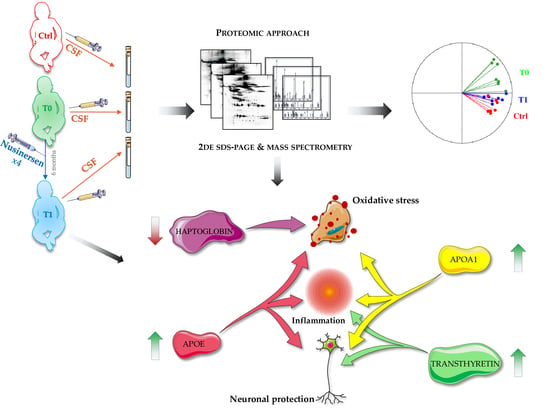
:
1. Introduction
2. Results
2.1. Clinical Data
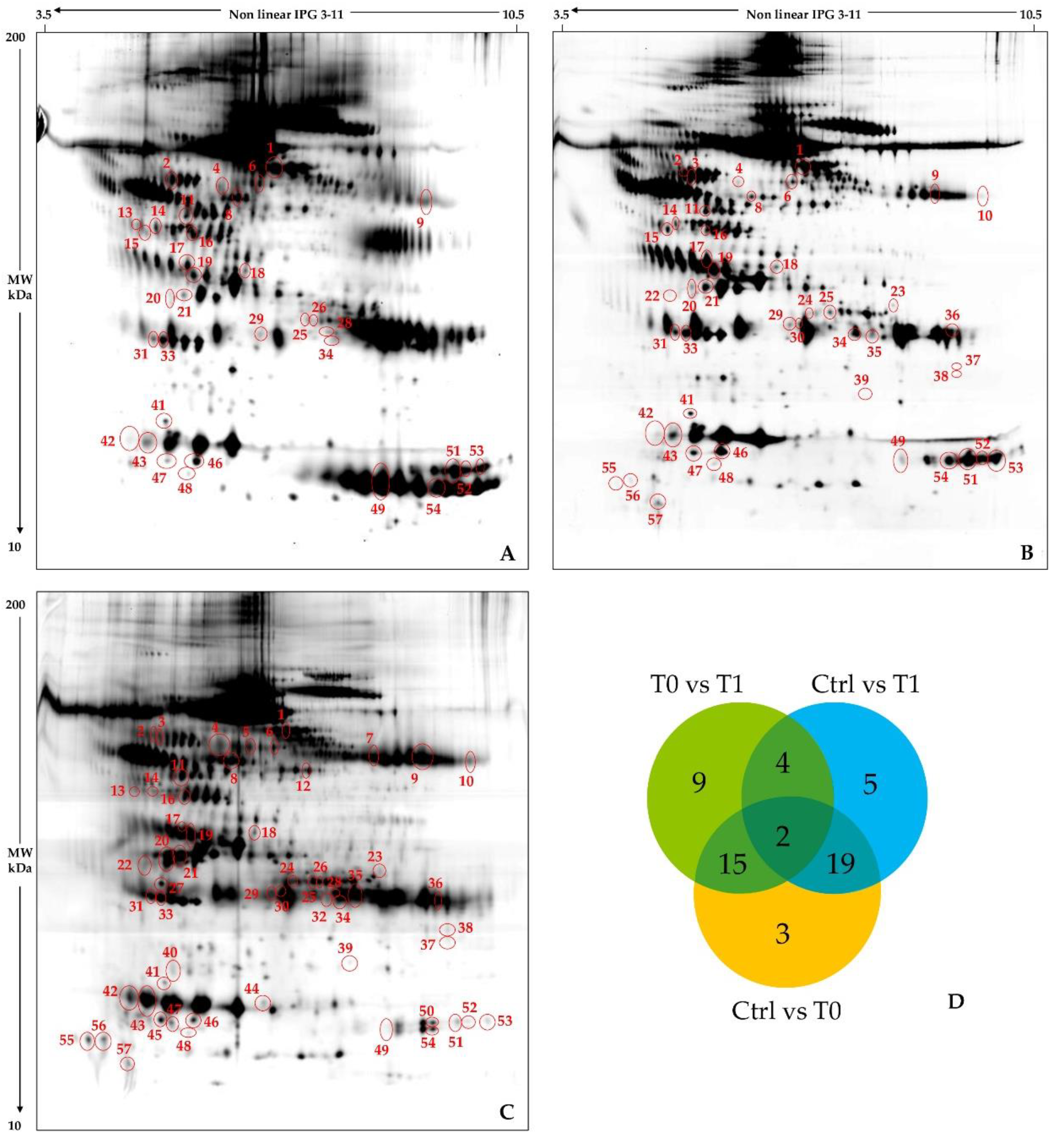
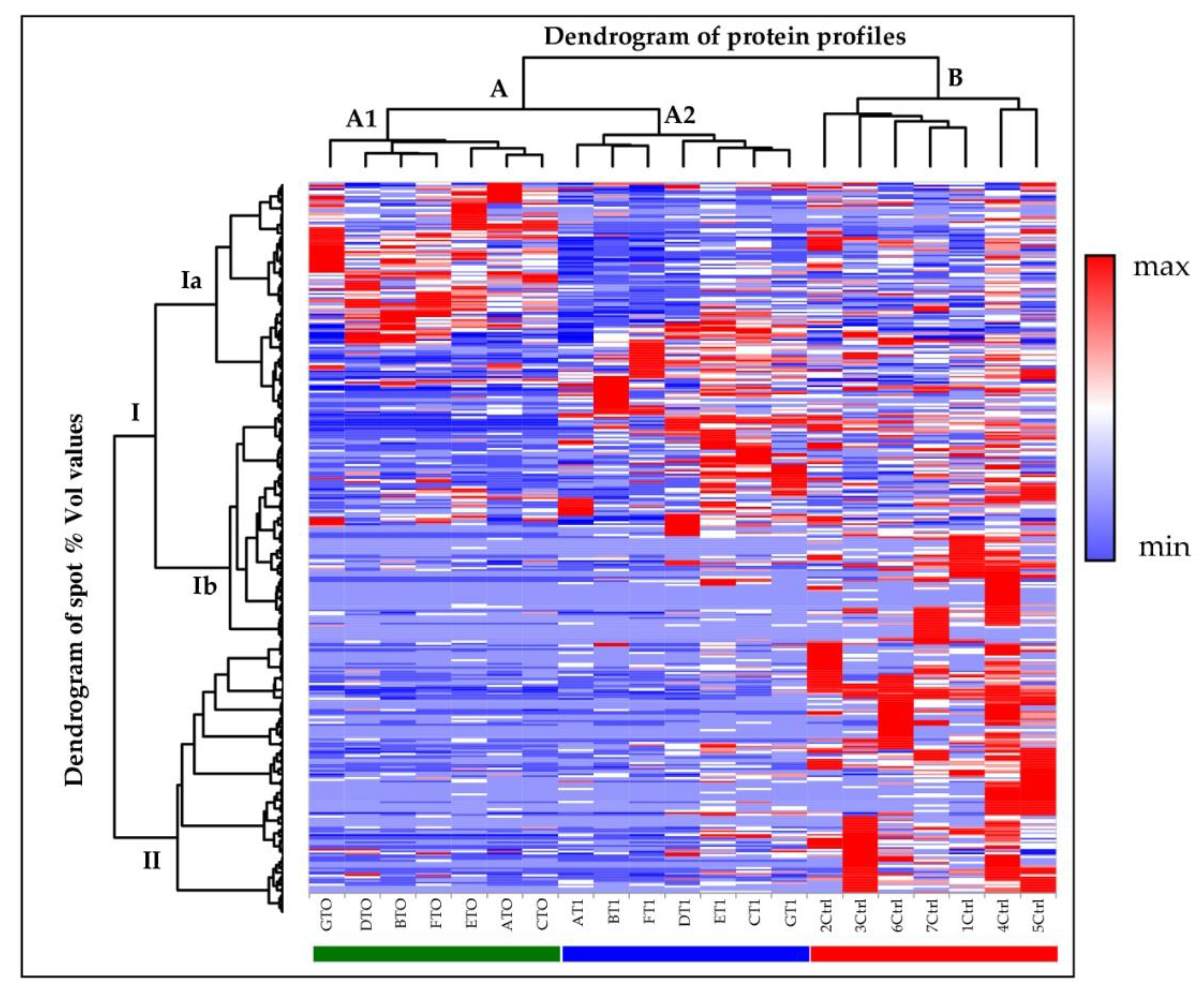
2.2. Cerebrospinal Fluid Proteomics Profiles
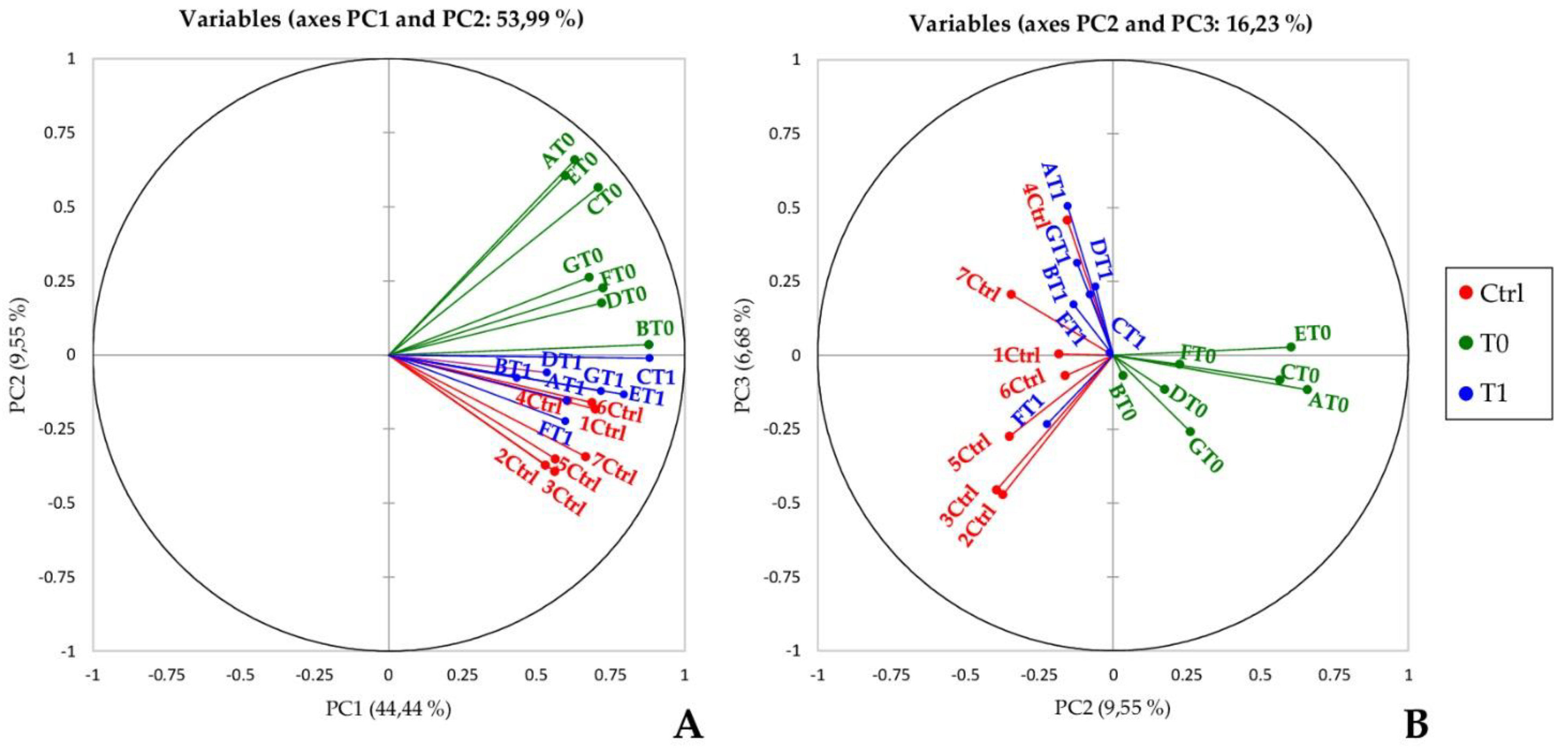
2.3. Principal Component Analysis and Hierarchical Clustering
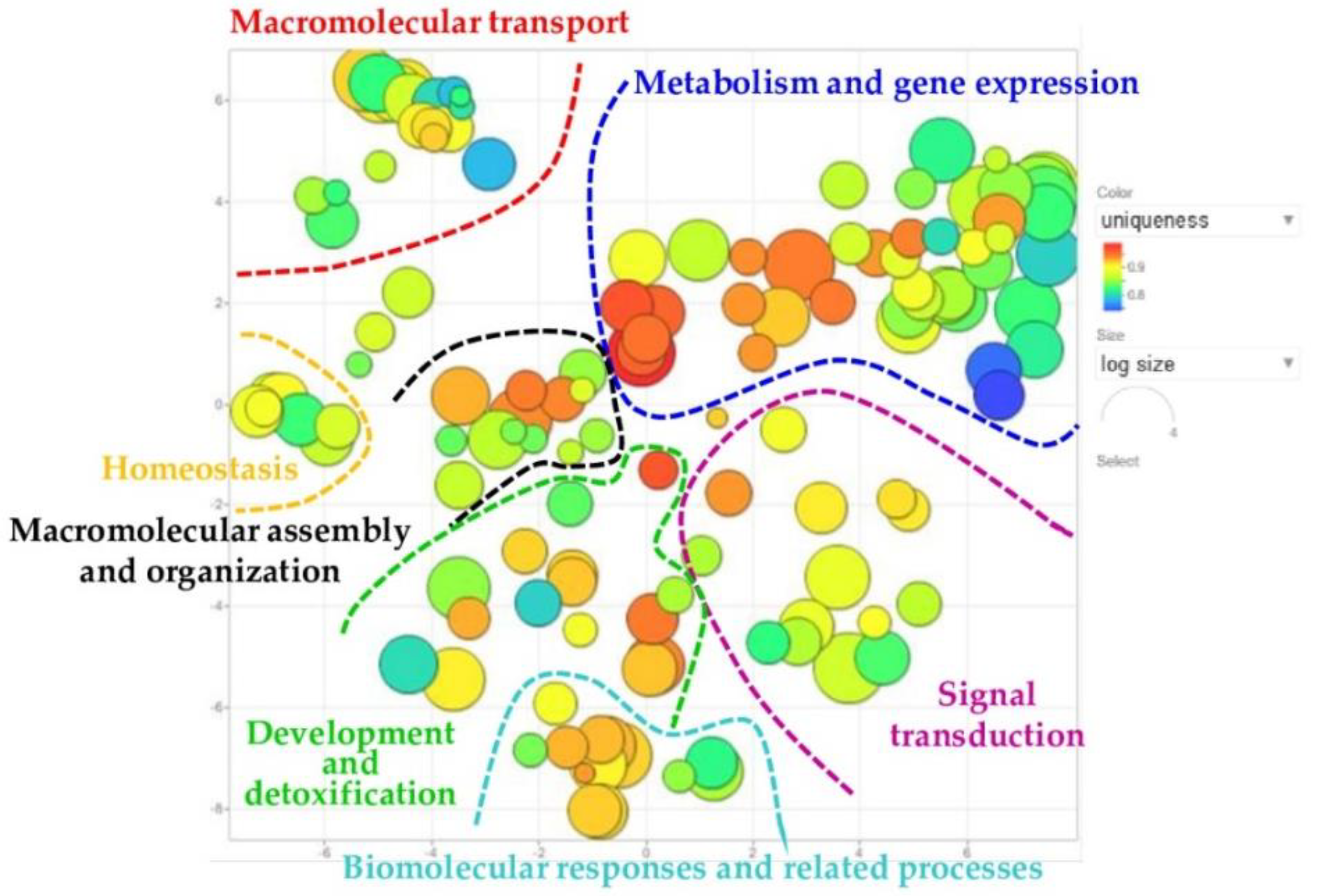
2.4. REVIGO Analysis on Biological Process GO-Terms Annotating Identified Proteins of Interest
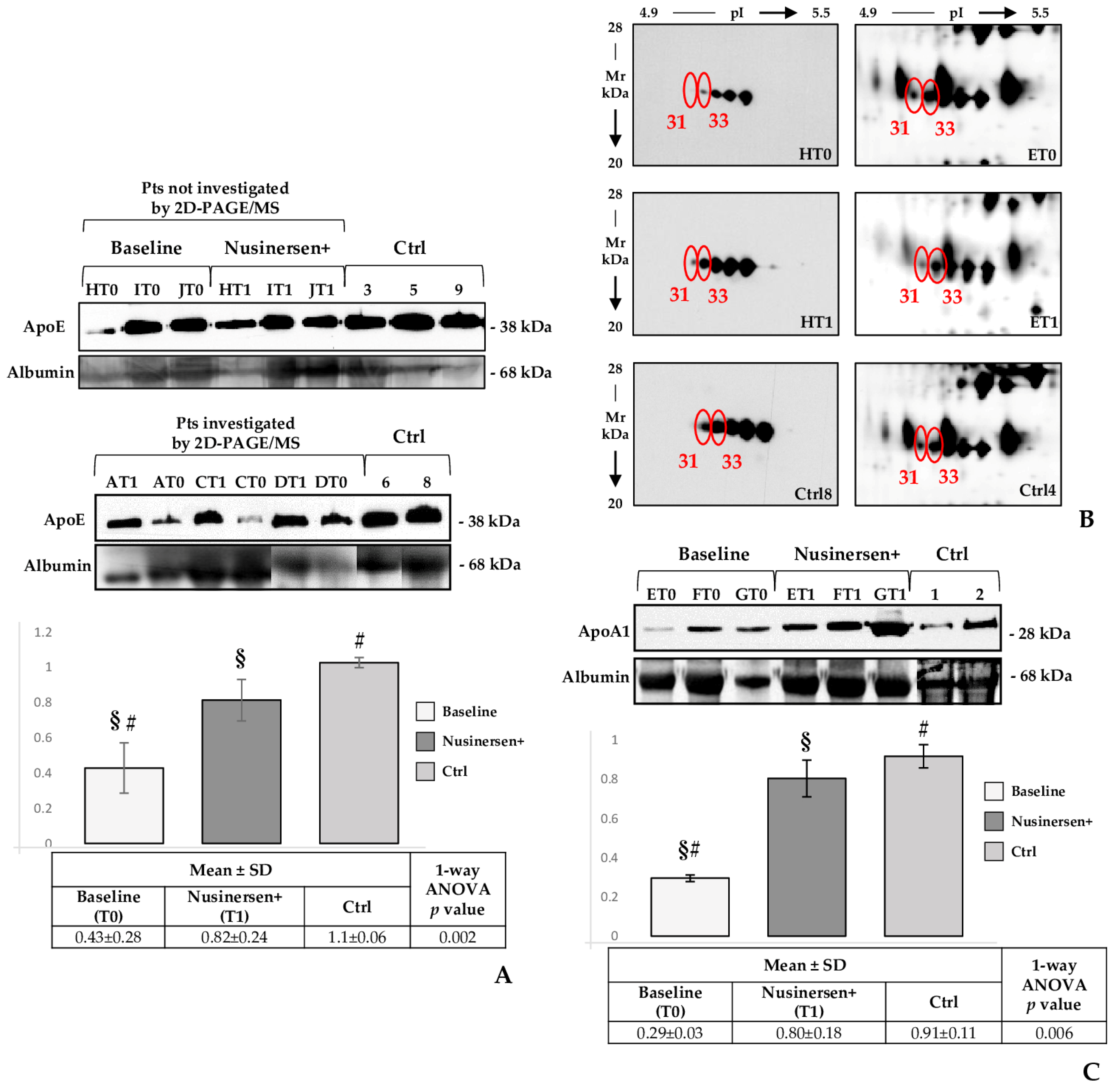
2.5. Western Blot Profiles of APOE, APOA1 and Transthyretin in CSF Samples from SMA Type 1 Patients (T0 and T1) and Control Subjects
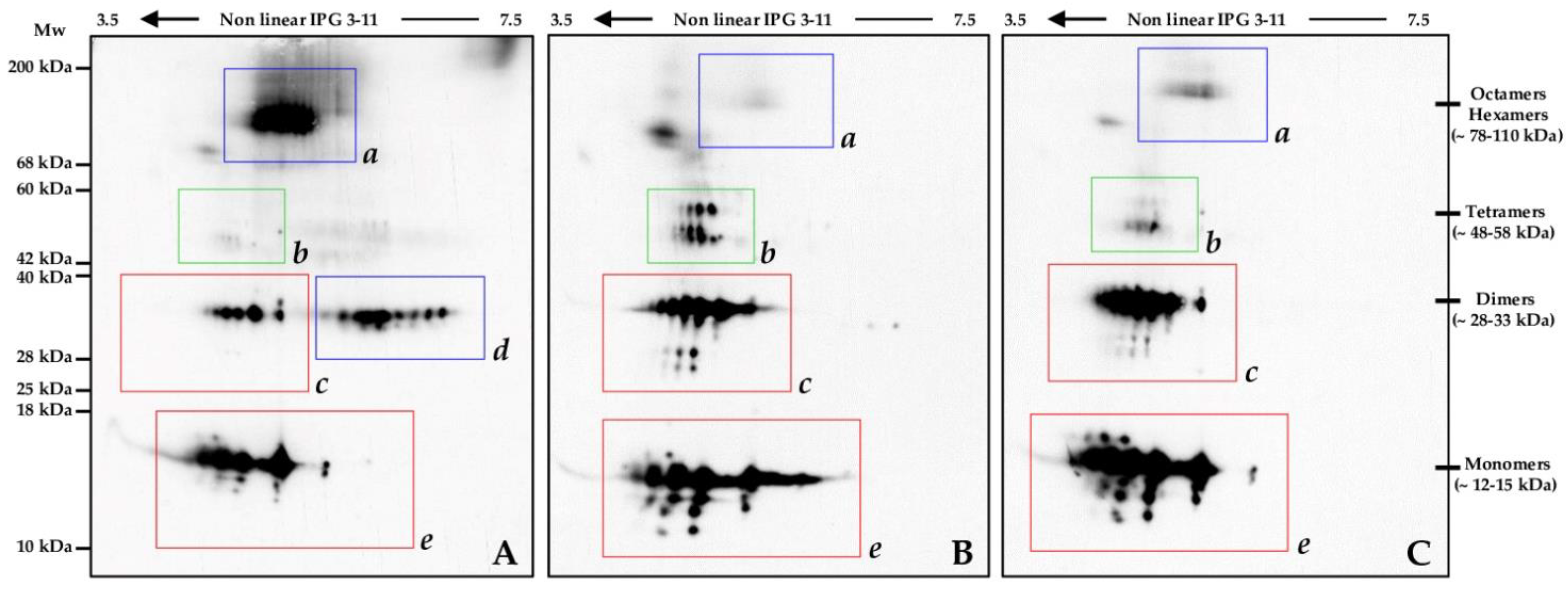
2.6. Oxidized Protein Pattern in Two SMA Type 1 Patients before and after Nusinersen Therapy
3. Discussion
3.1. Are APOE and APOA1 Crucial Allies of Nusinersen in Restoring Nervous System Plasticity in SMA Type 1 Patients?
3.2. APOE, APOA1 and Haptoglobin May Support Nusinersen in Inflammation and Oxidative Stress Balancing
3.3. Nusinersen Treatment Highly Influences Transthyretin Pattern in the Investigated Cohort of SMA Type 1 Patients
3.4. Concluding Remarks
4. Materials and Methods
4.1. Participants and Cerebrospinal Fluid Sampling
4.2. Cerebrospinal Fluid Preparation for Proteomics Analysis
4.3. Two-Dimensional Gel Electrophoresis
4.4. Image Analysis and Statistics
4.5. Mass Spectrometry by MALDI-TOF
4.6. Gene Ontology (GO) Clustering
4.7. Western Blot
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ogino, S.; Wilson, R.B.; Gold, B. New Insights on the Evolution of the SMN1 and SMN2 Region: Simulation and Meta-Analysis for Allele and Haplotype Frequency Calculations. Eur. J. Hum. Genet. 2004, 12, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, Incidence and Carrier Frequency of 5q-Linked Spinal Muscular Atrophy—A Literature Review. Orphanet J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy: A Timely Review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN Deficiency Causes Tissue-Specific Perturbations in the Repertoire of SnRNAs and Widespread Defects in Splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, L.; Burns, J.; Warman Chardon, J.; Kothary, R.; Parks, R. Spinal Muscular Atrophy: More than a Disease of Motor Neurons? Curr. Mol. Med. 2016, 16, 779–792. [Google Scholar] [CrossRef]
- Ando, S.; Osanai, D.; Takahashi, K.; Nakamura, S.; Shimazawa, M.; Hara, H. Survival Motor Neuron Protein Regulates Oxidative Stress and Inflammatory Response in Microglia of the Spinal Cord in Spinal Muscular Atrophy. J. Pharmacol. Sci. 2020, 144, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Rindt, H.; Feng, Z.; Mazzasette, C.; Glascock, J.J.; Valdivia, D.; Pyles, N.; Crawford, T.O.; Swoboda, K.J.; Patitucci, T.N.; Ebert, A.D.; et al. Astrocytes Influence the Severity of Spinal Muscular Atrophy. Hum. Mol. Genet. 2015, 24, 4094–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Rossol, W.; et al. The Human Centromeric Survival Motor Neuron Gene (SMN2) Rescues Embryonic Lethality in Smn(-/-) Mice and Results in a Mouse with Spinal Muscular Atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shababi, M.; Lorson, C.L.; Rudnik-Schöneborn, S.S. Spinal Muscular Atrophy: A Motor Neuron Disorder or a Multi-Organ Disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Gama-Carvalho, M.; Garcia-Vaquero, M.; Pinto, F.; Besse, F.; Weis, J.; Voigt, A.; Schulz, J.B.; De Las Rivas, J. Linking Amyotrophic Lateral Sclerosis and Spinal Muscular Atrophy through RNA-Transcriptome Homeostasis: A Genomics Perspective. J. Neurochem. 2017, 141, 12–30. [Google Scholar] [CrossRef] [Green Version]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the SMA Gene SMN1 from the Copy Gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative Analyses of SMN1 and SMN2 Based on Real-Time LightCycler PCR: Fast and Highly Reliable Carrier Testing and Prediction of Severity of Spinal Muscular Atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a Critical Exon of Human Survival Motor Neuron Is Regulated by a Unique Silencer Element Located in the Last Intron. Mol. Cell Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [Green Version]
- Berciano, M.T.; Puente-Bedia, A.; Medina-Samamé, A.; Rodríguez-Rey, J.C.; Calderó, J.; Lafarga, M.; Tapia, O. Nusinersen Ameliorates Motor Function and Prevents Motoneuron Cajal Body Disassembly and Abnormal Poly(A) RNA Distribution in a SMA Mouse Model. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of Infantile-Onset Spinal Muscular Atrophy with Nusinersen: A Phase 2, Open-Label, Dose-Escalation Study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Aragon-Gawinska, K.; Daron, A.; Ulinici, A.; Vanden Brande, L.; Seferian, A.; Gidaro, T.; Scoto, M.; Deconinck, N.; Servais, L. SMA-Registry Study Group Sitting in Patients with Spinal Muscular Atrophy Type 1 Treated with Nusinersen. Dev. Med. Child. Neurol. 2020, 62, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Pane, M.; Coratti, G.; Sansone, V.A.; Messina, S.; Bruno, C.; Catteruccia, M.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. Nusinersen in Type 1 Spinal Muscular Atrophy: Twelve-Month Real-World Data. Ann. Neurol. 2019, 86, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, A.; Stolte, B.; Kizina, K.; Bolz, S.; Schlag, M.; Thimm, A.; Kleinschnitz, C.; Hagenacker, T. Neurofilament Heavy Chain and Tau Protein Are Not Elevated in Cerebrospinal Fluid of Adult Patients with Spinal Muscular Atrophy during Loading with Nusinersen. Int. J. Mol. Sci. 2019, 20, 5397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a Potential Biomarker for Spinal Muscular Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Latzer, P.; Schmid, D.; Warnken, U.; Saffari, A.; Ziegler, A.; Kollmer, J.; Möhlenbruch, M.; Ulfert, C.; Herweh, C.; et al. Cerebrospinal Fluid Proteomic Profiling in Nusinersen-Treated Patients with Spinal Muscular Atrophy. J. Neurochem. 2020, 153, 650–661. [Google Scholar] [CrossRef]
- Fuller, H.R.; Gillingwater, T.H.; Wishart, T.M. Commonality amid Diversity: Multi-Study Proteomic Identification of Conserved Disease Mechanisms in Spinal Muscular Atrophy. Neuromuscul. Disord. 2016, 26, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, C.A.; Lamont, D.J.; Hunter, G.; Wishart, T.M.; Gillingwater, T.H. Label-Free Proteomics Identifies Calreticulin and GRP75/Mortalin as Peripherally Accessible Protein Biomarkers for Spinal Muscular Atrophy. Genome Med. 2013, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Šoltić, D.; Bowerman, M.; Stock, J.; Shorrock, H.K.; Gillingwater, T.H.; Fuller, H.R. Multi-Study Proteomic and Bioinformatic Identification of Molecular Overlap between Amyotrophic Lateral Sclerosis (ALS) and Spinal Muscular Atrophy (SMA). Brain Sci. 2018, 8, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-Y.; Whye, D.; Glazewski, L.; Choe, L.; Kerr, D.; Lee, K.H.; Mason, R.W.; Wang, W. Proteomic Assessment of a Cell Model of Spinal Muscular Atrophy. BMC Neurosci. 2011, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Dubowitz, V. Chaos in Classification of the Spinal Muscular Atrophies of Childhood. Neuromuscul. Disord. 1991, 1, 77–80. [Google Scholar] [CrossRef]
- Pane, M.; Palermo, C.; Messina, S.; Sansone, V.A.; Bruno, C.; Catteruccia, M.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. An Observational Study of Functional Abilities in Infants, Children, and Adults with Type 1 SMA. Neurology 2018, 91, e696–e703. [Google Scholar] [CrossRef]
- Finkel, R.; Bertini, E.; Muntoni, F.; Mercuri, E. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul. Disord. 2015, 25, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue Damage in the Amyloidoses: Transthyretin Monomers and Nonnative Oligomers Are the Major Cytotoxic Species in Tissue Culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, A.K.R.; Hughes, R.M.; Wi, S.; Hung, I.; Gan, Z.; Kelly, J.W.; Lim, K.H. Transthyretin Aggregation Pathway toward the Formation of Distinct Cytotoxic Oligomers. Sci. Rep. 2019, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Araki, S.; Arai, N.; Kumada, S.; Itoh, M.; Tamagawa, K.; Oda, M.; Morimatsu, Y. Oxidative Stress and Disturbed Glutamate Transport in Spinal Muscular Atrophy. Brain Dev. 2002, 24, 770–775. [Google Scholar] [CrossRef]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.-C. Motor Neuron Mitochondrial Dysfunction in Spinal Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grune, T. Oxidized Protein Aggregates: Formation and Biological Effects. Free Radic. Biol. Med. 2020, 150, 120–124. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein Carbonyl Groups as Biomarkers of Oxidative Stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Olsson, B.; Alberg, L.; Cullen, N.C.; Michael, E.; Wahlgren, L.; Kroksmark, A.-K.; Rostasy, K.; Blennow, K.; Zetterberg, H.; Tulinius, M. NFL Is a Marker of Treatment Response in Children with SMA Treated with Nusinersen. J. Neurol. 2019, 266, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, N.; et al. Nusinersen Treatment and Cerebrospinal Fluid Neurofilaments: An Explorative Study on Spinal Muscular Atrophy Type 3 Patients. J. Cell Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef] [Green Version]
- Fonteh, A.N.; Harrington, R.J.; Huhmer, A.F.; Biringer, R.G.; Riggins, J.N.; Harrington, M.G. Identification of Disease Markers in Human Cerebrospinal Fluid Using Lipidomic and Proteomic Methods. Dis. Markers 2006, 22, 39–64. [Google Scholar] [CrossRef] [Green Version]
- Spagnuolo, M.S.; Donizetti, A.; Iannotta, L.; Aliperti, V.; Cupidi, C.; Bruni, A.C.; Cigliano, L. Brain-Derived Neurotrophic Factor Modulates Cholesterol Homeostasis and Apolipoprotein E Synthesis in Human Cell Models of Astrocytes and Neurons. J. Cell Physiol. 2018, 233, 6925–6943. [Google Scholar] [CrossRef] [PubMed]
- Tsui-Pierchala, B.A.; Encinas, M.; Milbrandt, J.; Johnson, E.M. Lipid Rafts in Neuronal Signaling and Function. Trends Neurosci. 2002, 25, 412–417. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Deguise, M.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal Fatty Acid Metabolism Is a Core Component of Spinal Muscular Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamim, A.; Mahmood, T.; Ahsan, F.; Kumar, A.; Bagga, P. Lipids: An Insight into the Neurodegenerative Disorders. Clin. Nutr. Exp. 2018, 20, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Alecu, I.; Bennett, S.A.L. Dysregulated Lipid Metabolism and Its Role in α-Synucleinopathy in Parkinson’s Disease. Front. Neurosci. 2019, 13, 328. [Google Scholar] [CrossRef]
- Kao, Y.-C.; Ho, P.-C.; Tu, Y.-K.; Jou, I.-M.; Tsai, K.-J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Mahley, R.W. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Mahoney-Sanchez, L.; Belaidi, A.A.; Bush, A.I.; Ayton, S. The Complex Role of Apolipoprotein E in Alzheimer’s Disease: An Overview and Update. J. Mol. Neurosci. 2016, 60, 325–335. [Google Scholar] [CrossRef]
- Lynch, J.R.; Morgan, D.; Mance, J.; Matthew, W.D.; Laskowitz, D.T. Apolipoprotein E Modulates Glial Activation and the Endogenous Central Nervous System Inflammatory Response. J. Neuroimmunol. 2001, 114, 107–113. [Google Scholar] [CrossRef]
- Geffin, R.; McCarthy, M. Aging and Apolipoprotein E in HIV Infection. J. Neurovirol. 2018, 24, 529–548. [Google Scholar] [CrossRef] [Green Version]
- Flowers, S.A.; Rebeck, G.W. APOE in the Normal Brain. Neurobiol. Dis. 2020, 136, 104724. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of Beta-Amyloid Is Facilitated by Apolipoprotein E and Circulating High-Density Lipoproteins in Bioengineered Human Vessels. Elife 2017, 6, e29595. [Google Scholar] [CrossRef]
- Zhou, A.L.; Swaminathan, S.K.; Curran, G.L.; Poduslo, J.F.; Lowe, V.J.; Li, L.; Kandimalla, K.K. Apolipoprotein A-I Crosses the Blood-Brain Barrier through Clathrin-Independent and Cholesterol-Mediated Endocytosis. J. Pharmacol. Exp. Ther. 2019, 369, 481–488. [Google Scholar] [CrossRef]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the Brain: Implications for Neurological and Psychiatric Disorders. Clin. Lipidol. 2010, 51, 555–573. [Google Scholar] [CrossRef] [Green Version]
- Dal Magro, R.; Simonelli, S.; Cox, A.; Formicola, B.; Corti, R.; Cassina, V.; Nardo, L.; Mantegazza, F.; Salerno, D.; Grasso, G.; et al. The Extent of Human Apolipoprotein A-I Lipidation Strongly Affects the β-Amyloid Efflux Across the Blood-Brain Barrier in Vitro. Front. Neurosci. 2019, 13, 419. [Google Scholar] [CrossRef]
- Gardner, L.A.; Levin, M.C. Importance of Apolipoprotein A-I in Multiple Sclerosis. Front. Pharmacol. 2015, 6, 278. [Google Scholar] [CrossRef] [Green Version]
- Navab, M.; Anantharamaiah, G.M.; Fogelman, A.M. The Role of High-Density Lipoprotein in Inflammation. Trends Cardiovasc. Med. 2005, 15, 158–161. [Google Scholar] [CrossRef]
- Martínez-López, D.; Camafeita, E.; Cedó, L.; Roldan-Montero, R.; Jorge, I.; García-Marqués, F.; Gómez-Serrano, M.; Bonzon-Kulichenko, E.; Blanco-Vaca, F.; Blanco-Colio, L.M.; et al. APOA1 Oxidation Is Associated to Dysfunctional High-Density Lipoproteins in Human Abdominal Aortic Aneurysm. EBioMedicine 2019, 43, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Lane-Donovan, C.; Philips, G.T.; Herz, J. More than Cholesterol Transporters: Lipoprotein Receptors in CNS Function and Neurodegeneration. Neuron 2014, 83, 771–787. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, M.B.; Saha, S.; Mohanty, P.K.; Mukhopadhyay, K.K.; Mukhopadhyay, D. Increased Expression of ApoA1 after Neuronal Injury May Be Beneficial for Healing. Mol. Cell Biochem. 2017, 424, 45–55. [Google Scholar] [CrossRef]
- Hoe, H.-S.; Harris, D.C.; Rebeck, G.W. Multiple Pathways of Apolipoprotein E Signaling in Primary Neurons. J. Neurochem. 2005, 93, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.-R.; Remaley, A.T.; Feuerborn, R.; Wolinnska, I.; Engel, T.; von Eckardstein, A.; Assmann, G. Apolipoprotein A-I Activates Cdc42 Signaling through the ABCA1 Transporter. J. Lipid Res. 2006, 47, 794–803. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.G. Distinct Roles for ERK1 and ERK2 in Pathophysiology of CNS. Front. Biol. 2012, 7, 267–276. [Google Scholar] [CrossRef]
- Yin, C.; Guo, Z.-D.; He, Z.-Z.; Wang, Z.-Y.; Sun, X.-C. Apolipoprotein E Affects In Vitro Axonal Growth and Regeneration via the MAPK Signaling Pathway. Cell Transplant. 2019, 28, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, M.B.; Basu, M.; Iswarari, S.; Mukhopadhyay, K.K.; Sardar, K.P.; Acharyya, B.; Mohanty, P.K.; Mukhopadhyay, D. CSF Proteomics of Secondary Phase Spinal Cord Injury in Human Subjects: Perturbed Molecular Pathways Post Injury. PLoS ONE 2014, 9, e110885. [Google Scholar] [CrossRef]
- Ghaiad, H.R.; Elmazny, A.N.; Nooh, M.M.; El-Sawalhi, M.M.; Shaheen, A.A. Long Noncoding RNAs APOA1-AS, IFNG-AS1, RMRP and Their Related Biomolecules in Egyptian Patients with Relapsing-Remitting Multiple Sclerosis: Relation to Disease Activity and Patient Disability. J. Adv. Res. 2020, 21, 141–150. [Google Scholar] [CrossRef]
- Lewis, T.L.; Cao, D.; Lu, H.; Mans, R.A.; Su, Y.R.; Jungbauer, L.; Linton, M.F.; Fazio, S.; LaDu, M.J.; Li, L. Overexpression of Human Apolipoprotein A-I Preserves Cognitive Function and Attenuates Neuroinflammation and Cerebral Amyloid Angiopathy in a Mouse Model of Alzheimer Disease. J. Biol. Chem. 2010, 285, 36958–36968. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yin, X.; Yu, H.; Liu, X.; Yang, F.; Yao, J.; Jin, H.; Yang, P. Quantitative Proteomic Analysis of Serum Proteins in Patients with Parkinson’s Disease Using an Isobaric Tag for Relative and Absolute Quantification Labeling, Two-Dimensional Liquid Chromatography, and Tandem Mass Spectrometry. Analyst 2012, 137, 490–495. [Google Scholar] [CrossRef]
- Riddell, D.R.; Graham, A.; Owen, J.S. Apolipoprotein E Inhibits Platelet Aggregation through the L-Arginine: Nitric Oxide Pathway. Implications for Vascular Disease. J. Biol. Chem. 1997, 272, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Bian, J.-T.; Grizelj, I.; Cavka, A.; Phillips, S.A.; Makino, A.; Mazzone, T. Apolipoprotein E Enhances Endothelial-NO Production by Modulating Caveolin 1 Interaction with Endothelial NO Synthase. Hypertension 2012, 60, 1040–1046. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F.; DiNicolantonio, J.J. Neuroprotective Potential of High-Dose Biotin. Med. Hypotheses 2017, 109, 145–149. [Google Scholar] [CrossRef]
- Wherlock, M.; Mellor, H. The Rho GTPase Family: A Racs to Wrchs Story. J. Cell Sci. 2002, 115, 239–240. [Google Scholar]
- Leemhuis, J.; Bouché, E.; Frotscher, M.; Henle, F.; Hein, L.; Herz, J.; Meyer, D.K.; Pichler, M.; Roth, G.; Schwan, C.; et al. Reelin Signals through Apolipoprotein E Receptor 2 and Cdc42 to Increase Growth Cone Motility and Filopodia Formation. J. Neurosci. 2010, 30, 14759–14772. [Google Scholar] [CrossRef]
- Casey, C.S.; Atagi, Y.; Yamazaki, Y.; Shinohara, M.; Tachibana, M.; Fu, Y.; Bu, G.; Kanekiyo, T. Apolipoprotein E Inhibits Cerebrovascular Pericyte Mobility through a RhoA Protein-Mediated Pathway. J. Biol. Chem. 2015, 290, 14208–14217. [Google Scholar] [CrossRef] [Green Version]
- Kyriakou, K.; Lederer, C.W.; Kleanthous, M.; Drousiotou, A.; Malekkou, A. Acid Ceramidase Depletion Impairs Neuronal Survival and Induces Morphological Defects in Neurites Associated with Altered Gene Transcription and Sphingolipid Content. Int. J. Mol. Sci. 2020, 21, 1607. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, M.; Beauvais, A.; Anderson, C.L.; Kothary, R. Rho-Kinase Inactivation Prolongs Survival of an Intermediate SMA Mouse Model. Hum. Mol. Genet. 2010, 19, 1468–1478. [Google Scholar] [CrossRef]
- Kariya, S.; Park, G.-H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced SMN Protein Impairs Maturation of the Neuromuscular Junctions in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef] [Green Version]
- Nölle, A.; Zeug, A.; van Bergeijk, J.; Tönges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The Spinal Muscular Atrophy Disease Protein SMN Is Linked to the Rho-Kinase Pathway via Profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef] [Green Version]
- Coque, E.; Raoul, C.; Bowerman, M. ROCK Inhibition as a Therapy for Spinal Muscular Atrophy: Understanding the Repercussions on Multiple Cellular Targets. Front. Neurosci. 2014, 8, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, P.J.; Gillingwater, T.H. Chapter 8—Axonal and Neuromuscular Junction Pathology in Spinal Muscular Atrophy. In Spinal Muscular Atrophy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 133–151. ISBN 978-0-12-803685-3. [Google Scholar]
- Wishart, T.M.; Mutsaers, C.A.; Riessland, M.; Reimer, M.M.; Hunter, G.; Hannam, M.L.; Eaton, S.L.; Fuller, H.R.; Roche, S.L.; Somers, E.; et al. Dysregulation of Ubiquitin Homeostasis and β-Catenin Signaling Promote Spinal Muscular Atrophy. J. Clin. Investig. 2014, 124, 1821–1834. [Google Scholar] [CrossRef] [Green Version]
- Ramser, J.; Ahearn, M.E.; Lenski, C.; Yariz, K.O.; Hellebrand, H.; von Rhein, M.; Clark, R.D.; Schmutzler, R.K.; Lichtner, P.; Hoffman, E.P.; et al. Rare Missense and Synonymous Variants in UBE1 Are Associated with X-Linked Infantile Spinal Muscular Atrophy. Am. J. Hum. Genet. 2008, 82, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dlamini, N.; Josifova, D.J.; Paine, S.M.L.; Wraige, E.; Pitt, M.; Murphy, A.J.; King, A.; Buk, S.; Smith, F.; Abbs, S.; et al. Clinical and Neuropathological Features of X-Linked Spinal Muscular Atrophy (SMAX2) Associated with a Novel Mutation in the UBA1 Gene. Neuromuscul. Disord. 2013, 23, 391–398. [Google Scholar] [CrossRef]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, Beta-Catenin, and Cadherin Pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, A.; Motolese, M.; Iacovelli, L.; Caraci, F.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; Gaviraghi, G.; Caricasole, A. Inhibition of the Canonical Wnt Signaling Pathway by Apolipoprotein E4 in PC12 Cells. J. Neurochem. 2006, 98, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, A.; Yaniv, A.; Gazit, A. The Low Density Lipoprotein Receptor-1, LRP1, Interacts with the Human Frizzled-1 (HFz1) and down-Regulates the Canonical Wnt Signaling Pathway. J. Biol. Chem. 2004, 279, 17535–17542. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, D.; Le Verche, V.; Jacquier, A.; Ikiz, B.; Przedborski, S.; Re, D.B. Inflammation in ALS and SMA: Sorting out the Good from the Evil. Neurobiol. Dis. 2010, 37, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Wan, B.; Feng, P.; Guan, Z.; Sheng, L.; Liu, Z.; Hua, Y. A Severe Mouse Model of Spinal Muscular Atrophy Develops Early Systemic Inflammation. Hum. Mol. Genet. 2018, 27, 4061–4076. [Google Scholar] [CrossRef]
- Abati, E.; Citterio, G.; Bresolin, N.; Comi, G.P.; Corti, S. Glial Cells Involvement in Spinal Muscular Atrophy: Could SMA Be a Neuroinflammatory Disease? Neurobiol. Dis. 2020, 140, 104870. [Google Scholar] [CrossRef]
- Maezawa, I.; Nivison, M.; Montine, K.S.; Maeda, N.; Montine, T.J. Neurotoxicity from Innate Immune Response Is Greatest with Targeted Replacement of E4 Allele of Apolipoprotein E Gene and Is Mediated by Microglial P38MAPK. FASEB J. 2006, 20, 797–799. [Google Scholar] [CrossRef]
- Urushitani, M.; Inoue, R.; Nakamizo, T.; Sawada, H.; Shibasaki, H.; Shimohama, S. Neuroprotective Effect of Cyclic GMP against Radical-Induced Toxicity in Cultured Spinal Motor Neurons. J. Neurosci. Res. 2000, 61, 443–448. [Google Scholar] [CrossRef]
- Somers, E.; Lees, R.D.; Hoban, K.; Sleigh, J.N.; Zhou, H.; Muntoni, F.; Talbot, K.; Gillingwater, T.H.; Parson, S.H. Vascular Defects and Spinal Cord Hypoxia in Spinal Muscular Atrophy. Ann. Neurol. 2016, 79, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Hyka, N.; Dayer, J.M.; Modoux, C.; Kohno, T.; Edwards, C.K.; Roux-Lombard, P.; Burger, D. Apolipoprotein A-I Inhibits the Production of Interleukin-1beta and Tumor Necrosis Factor-Alpha by Blocking Contact-Mediated Activation of Monocytes by T Lymphocytes. Blood 2001, 97, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, M.B.; Mukhopadhyay, D. Possible Role of Apolipoprotein A1 in Healing and Cell Death after Neuronal Injury. Front. Biosci. 2016, 8, 460–477. [Google Scholar] [CrossRef]
- Furlaneto, C.J.; Ribeiro, F.P.; Hatanaka, E.; Souza, G.M.; Cassatella, M.A.; Campa, A. Apolipoproteins A-I and A-II Downregulate Neutrophil Functions. Lipids 2002, 37, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Righy, C.; Turon, R.; de Freitas, G.; Japiassú, A.M.; de Castro Faria Neto, H.C.; Bozza, M.; Oliveira, M.F.; Bozza, F.A. Hemoglobin Metabolism By-Products Are Associated with an Inflammatory Response in Patients with Hemorrhagic Stroke. Rev. Bras. Ter. Intensiv. 2018, 30, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.M.; Kumar, A.P.; Yadav, A.N.; Rajkumar, K.; Mvs, S.; Burgula, S. Haptoglobin Improves Acute Phase Response and Endotoxin Tolerance in Response to Bacterial LPS. Immunol. Lett. 2019, 207, 17–27. [Google Scholar] [CrossRef]
- Tseng, C.F.; Lin, C.C.; Huang, H.Y.; Liu, H.C.; Mao, S.J.T. Antioxidant Role of Human Haptoglobin. Proteomics 2004, 4, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Hametner, S.; Facchiano, F.; Marastoni, D.; Rossi, S.; Castellaro, M.; Poli, A.; Lattanzi, F.; Visconti, A.; Nicholas, R.; et al. Iron Homeostasis, Complement, and Coagulation Cascade as CSF Signature of Cortical Lesions in Early Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2150–2163. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Cao, S.; Hua, Y.; Keep, R.F.; Huang, Y.; Xi, G. CD163 Expression in Neurons After Experimental Intracerebral Hemorrhage. Stroke 2017, 48, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-H.; Tseng, M.-Y.; Ro, L.-S.; Lyu, R.-K.; Tai, Y.-H.; Chang, H.-S.; Wu, Y.-R.; Huang, C.-C.; Hsu, W.-C.; Kuo, H.-C.; et al. Analyses of Haptoglobin Level in the Cerebrospinal Fluid and Serum of Patients with Neuromyelitis Optica and Multiple Sclerosis. Clin. Chim. Acta 2013, 417, 26–30. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.H.; Bozorgmehr, J. Haptoglobin Phenotypes in Health and Disorders. Am. J. Clin. Pathol. 2004, 121, S97–S104. [Google Scholar] [CrossRef]
- Salvatore, A.; Cigliano, L.; Carlucci, A.; Bucci, E.M.; Abrescia, P. Haptoglobin Binds Apolipoprotein E and Influences Cholesterol Esterification in the Cerebrospinal Fluid. J. Neurochem. 2009, 110, 255–263. [Google Scholar] [CrossRef]
- Conti, A.; Sanchez-Ruiz, Y.; Bachi, A.; Beretta, L.; Grandi, E.; Beltramo, M.; Alessio, M. Proteome Study of Human Cerebrospinal Fluid Following Traumatic Brain Injury Indicates Fibrin(Ogen) Degradation Products as Trauma-Associated Markers. J. Neurotrauma 2004, 21, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L.; Inzitari, D. Abnormal Penetration of Haptoglobin through the Blood-Brain-Barrier (BBB) into the Cerebro-Spinal Fluid (CSF) in Alzheimer’s Disease Patients. Acta Neurol. Scand. 1995, 91, 225. [Google Scholar] [PubMed]
- Lewin, A.; Hamilton, S.; Witkover, A.; Langford, P.; Nicholas, R.; Chataway, J.; Bangham, C.R.M. Free Serum Haemoglobin Is Associated with Brain Atrophy in Secondary Progressive Multiple Sclerosis. Wellcome Open Res. 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-R.; Liu, S.-L.; Qin, Z.-Y.; Liu, F.-J.; Qin, Y.-J.; Bai, S.-M.; Chen, Z.-Y. Comparative Proteomics Analysis of Cerebrospinal Fluid of Patients with Guillain-Barré Syndrome. Cell Mol. Neurobiol. 2008, 28, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-C.; Wu, Y.-R.; Tseng, M.-Y.; Chen, Y.-C.; Hsieh, S.-Y.; Chen, C.-M. Increased Prothrombin, Apolipoprotein A-IV, and Haptoglobin in the Cerebrospinal Fluid of Patients with Huntington’s Disease. PLoS ONE 2011, 6, e15809. [Google Scholar] [CrossRef] [PubMed]
- Vejda, S.; Posovszky, C.; Zelzer, S.; Peter, B.; Bayer, E.; Gelbmann, D.; Schulte-Hermann, R.; Gerner, C. Plasma from Cancer Patients Featuring a Characteristic Protein Composition Mediates Protection against Apoptosis. Mol. Cell Proteom. 2002, 1, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric Oxide and Macrophage Function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Matschke, V.; Theiss, C.; Matschke, J. Oxidative Stress: The Lowest Common Denominator of Multiple Diseases. Neural. Regen. Res. 2019, 14, 238–241. [Google Scholar] [CrossRef]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial Membrane Potential Regulates Matrix Configuration and Cytochrome c Release during Apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Liz, M.A.; Leite, S.C.; Juliano, L.; Saraiva, M.J.; Damas, A.M.; Bur, D.; Sousa, M.M. Transthyretin Is a Metallopeptidase with an Inducible Active Site. Biochem. J. 2012, 443, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, J.; Lehmensiek, V.; Mogel, H.; Pfeifle, M.; Dorst, J.; Hendrich, C.; Ludolph, A.C.; Tumani, H. Proteome Analysis Reveals Candidate Markers of Disease Progression in Amyotrophic Lateral Sclerosis (ALS). Neurosci. Lett. 2010, 468, 23–27. [Google Scholar] [CrossRef]
- Vieira, M.; Saraiva, M.J. Transthyretin: A Multifaceted Protein. Biomol. Concepts 2014, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Liz, M.A.; Coelho, T.; Bellotti, V.; Fernandez-Arias, M.I.; Mallaina, P.; Obici, L. A Narrative Review of the Role of Transthyretin in Health and Disease. Neurol. Ther. 2020, 9, 395–402. [Google Scholar] [CrossRef]
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausín, K.; Lachen-Montes, M.; Santamaría, E.; Fernandez-Irigoyen, J.; Jericó, I. Neuroanatomical Quantitative Proteomics Reveals Common Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 2018, 20, 4. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-H.; Jung, E.S.; Sohn, J.-H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human Serum Transthyretin Levels Correlate Inversely with Alzheimer’s Disease. J. Alzheimers Dis. 2011, 25, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Argüelles, S.; Venero, J.L.; García-Rodriguez, S.; Tomas-Camardiel, M.; Ayala, A.; Cano, J.; Machado, A. Use of Haptoglobin and Transthyretin as Potential Biomarkers for the Preclinical Diagnosis of Parkinson’s Disease. Neurochem. Int. 2010, 57, 227–234. [Google Scholar] [CrossRef]
- Landi, C.; Santinelli, L.; Bianchi, L.; Shaba, E.; Ceccarelli, G.; Cavallari, E.N.; Borrazzo, C.; Pinacchio, C.; Scagnolari, C.; Vullo, V.; et al. Cognitive Impairment and CSF Proteome Modification after Oral Bacteriotherapy in HIV Patients. J. Neurovirol. 2020, 26, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal Fluid Transthyretin: Aging and Late Onset Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K. Transthyretin Sequesters Amyloid Beta Protein and Prevents Amyloid Formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.R.; Cabrito, I.; Soares, H.R.; Costelha, S.; Teixeira, A.; Wittelsberger, A.; Stortelers, C.; Vanlandschoot, P.; Saraiva, M.J. Delivery of an Anti-Transthyretin Nanobody to the Brain through Intranasal Administration Reveals Transthyretin Expression and Secretion by Motor Neurons. J. Neurochem. 2018, 145, 393–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, C.E.; Saraiva, M.J.; Sousa, M.M. Transthyretin Enhances Nerve Regeneration. J. Neurochem. 2007, 103, 831–839. [Google Scholar] [CrossRef]
- Fleming, C.E.; Mar, F.M.; Franquinho, F.; Saraiva, M.J.; Sousa, M.M. Transthyretin Internalization by Sensory Neurons Is Megalin Mediated and Necessary for Its Neuritogenic Activity. J. Neurosci. 2009, 29, 3220–3232. [Google Scholar] [CrossRef]
- Glanzman, A.M.; Mazzone, E.; Main, M.; Pelliccioni, M.; Wood, J.; Swoboda, K.J.; Scott, C.; Pane, M.; Messina, S.; Bertini, E.; et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test Development and Reliability. Neuromuscul. Disord. 2010, 20, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, P.; Poland, J.; Schnölzer, M.; Rabilloud, T. A New Silver Staining Apparatus and Procedure for Matrix-Assisted Laser Desorption/Ionization-Time of Flight Analysis of Proteins after Two-Dimensional Electrophoresis. Proteomics 2001, 1, 835–840. [Google Scholar] [CrossRef]
- Gharahdaghi, F.; Weinberg, C.R.; Meagher, D.A.; Imai, B.S.; Mische, S.M. Mass Spectrometric Identification of Proteins from Silver-Stained Polyacrylamide Gel: A Method for the Removal of Silver Ions to Enhance Sensitivity. Electrophoresis 1999, 20, 601–605. [Google Scholar] [CrossRef]
- Bianchi, L.; Lorenzoni, P.; Bini, L.; Weber, E.; Tani, C.; Rossi, A.; Agliano, M.; Pallini, V.; Sacchi, G. Protein Expression Profiles of Bos Taurus Blood and Lymphatic Vessel Endothelial Cells. Proteomics 2007, 7, 1600–1614. [Google Scholar] [CrossRef]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A Web-Based Tool for Gene Ontology Searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntley, R.P.; Sawford, T.; Mutowo-Meullenet, P.; Shypitsyna, A.; Bonilla, C.; Martin, M.J.; O’Donovan, C. The GOA Database: Gene Ontology Annotation Updates for 2015. Nucleic Acids Res. 2015, 43, D1057–D1063. [Google Scholar] [CrossRef] [PubMed]
- Bini, L.; Schvartz, D.; Carnemolla, C.; Besio, R.; Garibaldi, N.; Sanchez, J.-C.; Forlino, A.; Bianchi, L. Intracellular and Extracellular Markers of Lethality in Osteogenesis Imperfecta: A Quantitative Proteomic Approach. Int. J. Mol. Sci. 2021, 22, 429. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic Transfer of Proteins from Polyacrylamide Gels to Nitrocellulose Sheets: Procedure and Some Applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [Green Version]
- Magi, B.; Bianchi, L. Immunoblotting of 2-DE Separated Proteins. In The Protein Protocols Handbook; Walker, J.M., Ed.; Springer Protocols Handbooks; Humana Press: Totowa, NJ, USA, 2009; pp. 641–662. ISBN 978-1-59745-198-7. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pts a | Sex M/F | Age at Symptoms Onset (Months) | Age at Treatment Onset (Months) | CHOP INTEND T0 | CHOP INTEND T1 | ΔCHOP INTEND (T1-T0) | SMN2 Copy Number | SMA Type 1 Decimal System b | SMA Type 1 ABC System c | Respiratory Support d T0 (Hours) | Nutritional SUPPORT e T0 | Respiratory Support d T1 (Hours) | Nutritional Support e T1 | Sample T0 | Sample T1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt. 1 | M | 0 | 2 | 21 | 35 | 14 | 2 | 1.5 | A | NIV (12) | None | NIV (18) | NGT | AT0 | AT1 |
| Pt. 2 | M | 3 | 17 | 22 | 35 | 13 | 3 | 1.5 | B | Nocturnal NIV (8) | PEG | Nocturnal NIV (8) | PEG | BT0 | BT1 |
| Pt. 3 | M | 5 | 25 | 43 | 46 | 3 | 3 | 1.9 | C | Nocturnal NIV (8) | None | Nocturnal NIV (8) | None | CT0 | CT1 |
| Pt. 4 | M | 5 | 28 | 42 | 47 | 5 | 3 | 1.9 | C | None | None | None | None | DT0 | DT1 |
| Pt. 5 | F | 4 | 7 | 39 | 48 | 9 | 2 | 1.9 | C | Nocturnal NIV (8) | None | Nocturnal NIV (8) | None | ET0 | ET1 |
| Pt. 6 | F | 3 | 5 | 24 | 35 | 11 | 2 | 1.5 | B | Nocturnal NIV (8) | PEG | Nocturnal NIV (8) | PEG | FT0 | FT1 |
| Pt. 7 | M | 2 | 3 | 30 | 48 | 18 | 2 | 1.5 | B | Nocturnal NIV (6) | None | Nocturnal NIV (4) | None | GT0 | GT1 |
| Pt. 8 | M | 2 | 3 | 23 | 36 | 13 | 2 | 1.5 | B | Nocturnal NIV (8) | PEG | Nocturnal NIV (8) | PEG | HT0 | HT1 |
| Pt. 9 | M | 0 | 2 | 16 | 36 | 10 | 2 | 1.1 | A | NIV (24) | PEG | NIV (24) | PEG | IT0 | IT1 |
| Pt. 10 | M | 1 | 3 | 9 | 35 | 16 | 2 | 1.5 | A | NIV (10) | OGT | NIV (18) | PEG | JT0 | JT1 |
| Spot N. a | Protein Description b | Ctrl Spot Mean % Vol ± SD c | T0 Spot Mean % Vol ± SD c | T1 Spot Mean % Vol ± SD c | AC d | Experimental pI; Mw e | Theoretical pI; Mw f | Mascot Search Results g | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Score | No. of Matched Peptides | Sequence Coverage (%) | ||||||||
| 31 | Apolipoprotein A1 | 174 ± 46 # | 73 ± 19 # | 102 ± 38 | P02647 | 5.22; 24559 | 5.56; 30777 | 181 | 24 | 61 |
| 33 | Apolipoprotein A1 | 238 ± 63 # | 114 ± 48 # § | 234 ± 43 § | P02647 | 5.34; 24211 | 5.56; 30777 | 170 | 21 | 56 |
| 17 | Apolipoprotein E | 120 ± 49 # * | 257 ± 106 # | 254 ± 148 * | P02649 | 5.49; 36422 | 5.65; 36154 | 111 | 18 | 45 |
| 19 | Apolipoprotein E | 506 ± 191 # * | 214 ± 149 # | 242 ± 128 * | P02649 | 5.52; 34722 | 5.65; 36154 | 82 | 13 | 29 |
| 34 | Hemoglobin sub. β | 376 ± 163 | 566 ± 42 § | 213 ± 107 § # | P68871 | 6.31; 24629 | 6.74; 15998 | 99 | 12 | 82 |
| 35 | Hemoglobin sub. β | 462 ± 294 * | 0 | 210 ± 179 * | P68871 | 6.46; 24629 | 6.74; 15998 | 56 | 6 | 39 |
| 36 | Hemoglobin sub. β | 72 ± 50 | 0 | 139 ± 88 | P68871 | 7.65; 24073 | 6.74; 15998 | 83 | 8 | 62 |
| 49 | Hemoglobin sub. β | 123 ± 141 # | 1284 ± 1151 # | 928 ± 983 | P68871 | 6.75; 12024 | 6.74; 15998 | 100 | 10 | 80 |
| 50 | Mix: Hemoglobin sub. β Hemoglobin sub. α | 57 ± 30 | 0 | 0 | P68871 P69905 | 7.55; 12589 | 6.74; 15998 8.72; 15257 | 119 58 | 12 6 | 82 42 |
| 51 | Hemoglobin sub. α | 91 ± 79 # * | 522 ± 128 # | 485 ± 260 * | P69905 | 7.95; 12589 | 8.72; 15257 | 100 | 9 | 50 |
| 53 | Hemoglobin sub. α | 46 ± 52 # * | 294 ± 103 # | 565 ± 303 * | P69905 | 8.42; 12686 | 8.72; 15257 | 104 | 9 | 50 |
| 54 | Hemoglobin sub. α | 93 ± 31 * | 172 ± 111 | 254 ± 214 * | P69906 | 7.55; 12116 | 8.72; 15257 | 70 | 7 | 50 |
| 16 | Haptoglobin | 152 ± 96 | 185 ± 102 § | 44 ± 37 § | P00738 | 5.49; 41071 | 6.13; 45205 | 54 | 11 | 25 |
| 20 | Transthyretin | 278 ± 331 # | 15 ± 14 # § | 116 ± 82 § | P02766 | 5.39; 30717 | 5.49; 15887 | 67 | 8 | 65 |
| 43 | Transthyretin | 533 ± 203 # * | 129 ± 60 # § | 263 ± 137 * § | P02766 | 5.17; 14121 | 5.49; 15887 | 70 | 8 | 50 |
| 46 | Transthyretin | 145 ± 49 * | 227 ± 87 | 315 ± 143 * | P02766 | 5.54; 12816 | 5.49; 15887 | 116 | 10 | 65 |
| 48 | Transthyretin | 19 ± 12 * | 27 ± 16 | 54 ± 37 * | P02766 | 5.52; 11902 | 5.49; 15887 | 95 | 10 | 65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchi, L.; Sframeli, M.; Vantaggiato, L.; Vita, G.L.; Ciranni, A.; Polito, F.; Oteri, R.; Gitto, E.; Di Giuseppe, F.; Angelucci, S.; et al. Nusinersen Modulates Proteomics Profiles of Cerebrospinal Fluid in Spinal Muscular Atrophy Type 1 Patients. Int. J. Mol. Sci. 2021, 22, 4329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094329
Bianchi L, Sframeli M, Vantaggiato L, Vita GL, Ciranni A, Polito F, Oteri R, Gitto E, Di Giuseppe F, Angelucci S, et al. Nusinersen Modulates Proteomics Profiles of Cerebrospinal Fluid in Spinal Muscular Atrophy Type 1 Patients. International Journal of Molecular Sciences. 2021; 22(9):4329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094329
Chicago/Turabian StyleBianchi, Laura, Maria Sframeli, Lorenza Vantaggiato, Gian Luca Vita, Annamaria Ciranni, Francesca Polito, Rosaria Oteri, Eloisa Gitto, Fabrizio Di Giuseppe, Stefania Angelucci, and et al. 2021. "Nusinersen Modulates Proteomics Profiles of Cerebrospinal Fluid in Spinal Muscular Atrophy Type 1 Patients" International Journal of Molecular Sciences 22, no. 9: 4329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094329