
Role of Thrombopoietin Receptor Agonists in Inherited Thrombocytopenia


Abstract
:1. Introduction
2. Pathogenesis, Diagnosis and General Management of IT
2.1. Pathogenesis
2.2. Diagnosis
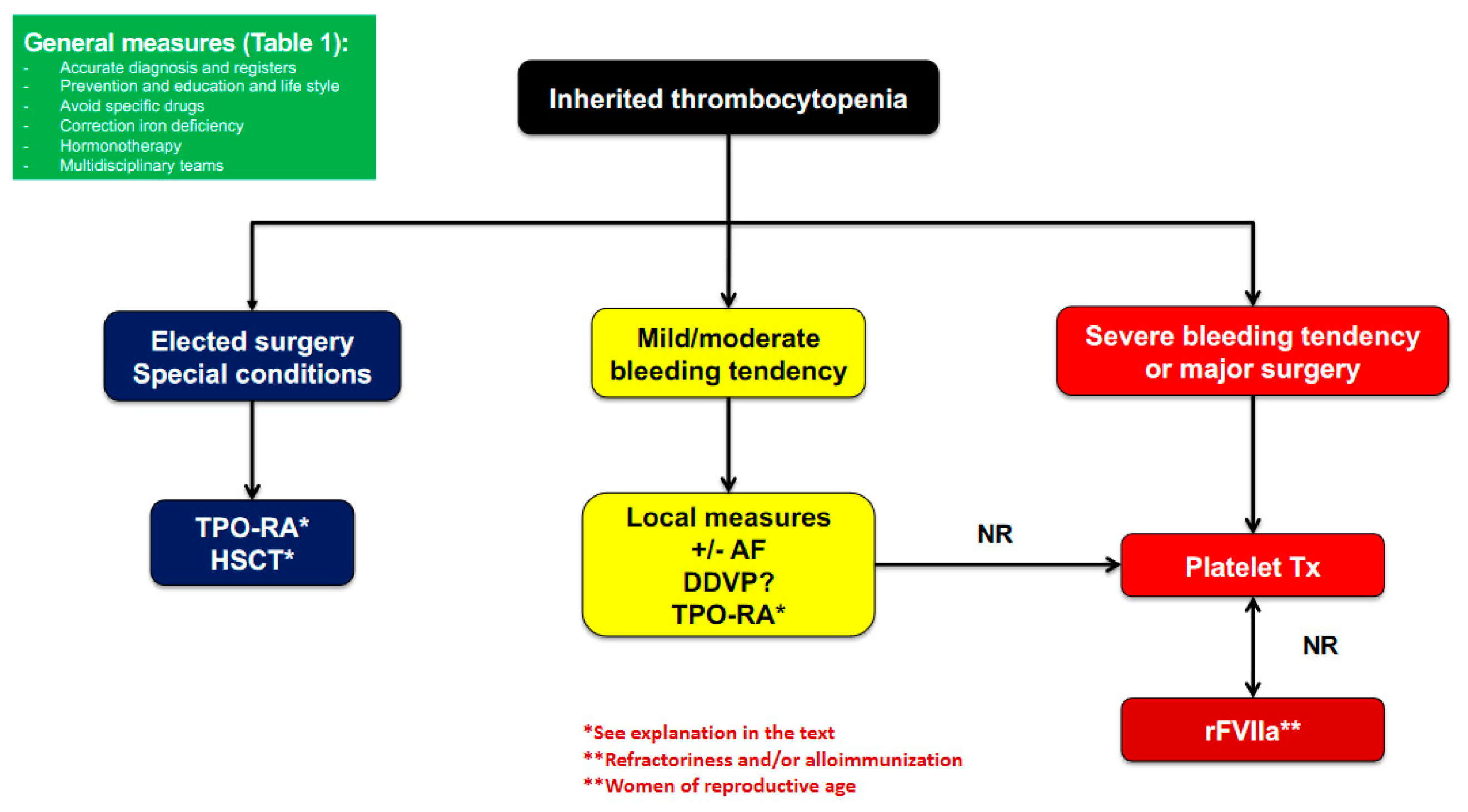
2.3. Management and Treatment
2.3.1. General Measures
2.3.2. Prohemostatic Drugs
2.3.3. Platelet Transfusion
2.3.4. Splenectomy
2.3.5. Curative Therapies
3. TPO-RA and Rationale for IT Treatment
4. Clinical Experience with TPO-RA for the Treatment of IT
4.1. Efficacy and Safety
4.2. Management of ITs in the Surgical Setting
4.3. TPO-RA Use in IT Patients with Special Particularities
5. Perspectives and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | Aplastic anemia |
| AE | Adverse event |
| AF | Antifibrinolytic |
| BAT | Bleeding assessment tool |
| BDPLT18 | Bleeding platelet type 18 |
| BMF | Bone marrow failure |
| BSS | Bernard–Soulier syndrome |
| CAMT | Congenital amegakaryocytic thrombocytopenia |
| CR | Complete response |
| DDAVP | Desmopressin |
| GPS | Gray platelet syndrome |
| GT | Glanzmann thrombasthenia |
| HSCT | Hematopoietic stem cell transplantation |
| HTS | High throughput sequencing |
| IPD | Inherited platelet disorders |
| IPFD | Inherited platelet function disorders |
| ISTH | International Society of Thrombosis and Hemostasis |
| IT | Inherited thrombocytopenia |
| ITP | Immune thrombocytopenia |
| IVIG | Intravenous immunoglobulin |
| LMWH | Low molecular weight heparin |
| MK | Megakaryocyte |
| PC | Platelet count |
| PR | Partial response |
| RD | Related disorders |
| rFVIIa | Recombinant activated factor VII |
| RT | Related thrombocytopenia |
| TPO-RA | Thrombopoietin receptor agonist |
| WAS | Wiskott–Aldrich syndrome |
| WHO | World Health Organization |
| XLT | X-linked thrombocytopenia |
References
- Rodeghiero, F.; Pecci, A.; Balduini, C.L. Thrombopoietin Receptor Agonists in Hereditary Thrombocytopenias. J. Thromb. Haemost. 2018, 16, 1700–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastida, J.M.; Benito, R.; Lozano, M.L.; Marín-Quilez, A.; Janusz, K.; Martín-Izquierdo, M.; Hernández-Sánchez, J.; Palma-Barqueros, V.; Hernández-Rivas, J.M.; Rivera, J.; et al. Molecular Diagnosis of Inherited Coagulation and Bleeding Disorders. Semin. Thromb. Hemost. 2019, 45, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Balduini, C.L.; Savoia, A.; Seri, M. Inherited Thrombocytopenias Frequently Diagnosed in Adults. J. Thromb. Haemost. 2013, 11, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Pluthero, F.G.; Kahr, W.H.A. Recent Advances in Inherited Platelet Disorders. Curr. Opin. Hematol. 2019, 26, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Almazni, I.; Stapley, R.; Morgan, N.V. Inherited Thrombocytopenia: Update on Genes and Genetic Variants Which may be Associated with Bleeding. Front. Cardiovasc. Med. 2019, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bolton-Maggs, P.H.B.; Chalmers, E.A.; Collins, P.W.; Harrison, P.; Kitchen, S.; Liesner, R.J.; Minford, A.; Mumford, A.D.; Parapia, L.A.; Perry, D.J.; et al. A Review of Inherited Platelet Disorders with Guidelines for their Management on Behalf of the UKHCDO. Br. J. Haematol. 2006, 135, 603–633. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W.; Cooper, N.; Rodeghiero, F.; Godeau, B.; Bussel, J.B. Thrombopoietin Receptor Agonists: Ten Years Later. Haematologica 2019, 104, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, D.; Bastida, J.M.; Lopez-Corral, L.; Sanchez-Guijo, F.; Cabrero, M.; Martin, A.; Perez, E.; Lopez-Parra, M.; Avendaño, A.; Veiga, A.; et al. Usefulness of Eltrombopag for Treating Thrombocytopenia after Allogeneic Stem Cell Transplantation. Bone Marrow Transpl. 2019, 54, 757–761. [Google Scholar] [CrossRef]
- Gonzalez-Porras, J.R.; Bastida, J.M. Eltrombopag in Immune Thrombocytopenia: Efficacy Review and Update on Drug Safety. Ther. Adv. Vaccines 2018, 9, 263–285. [Google Scholar] [CrossRef]
- Bento, L.; Bastida, J.M.; García-Cadenas, I.; García-Torres, E.; Rivera, D.; Bosch-Vilaseca, A.; De Miguel, C.; Martínez-Muñoz, M.E.; Fernández-Avilés, F.; Roldán, E.; et al. Thrombopoietin Receptor Agonists for Severe Thrombocytopenia after Allogeneic Stem Cell Transplantation: Experience of the Spanish Group of Hematopoietic Stem Cell Transplant. Biol. Blood Marrow Transpl. 2019, 25, 1825–1831. [Google Scholar] [CrossRef]
- Nurden, A.T.; Nurden, P. Inherited Thrombocytopenias: History, Advances and Perspectives. Haematologica 2020, 105, 2004–2019. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Validation of the ISTH/SSC Bleeding Assessment Tool for Inherited Platelet Disorders: A Communication from the Platelet Physiology SSC. J. Thromb. Haemost. 2020, 18, 732–739. [Google Scholar] [CrossRef]
- Pecci, A. Pathogenesis and Management of Inherited Thrombocytopenias: Rationale for the Use of Thrombopoietin-Receptor Agonists. Int. J. Hematol. 2013, 98, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastida, J.M.; Benito, R.; González-Porras, J.R.; Rivera, J. ABCG5 and ABCG8 Gene Variations Associated with Sitosterolemia and Platelet Dysfunction. Platelets 2020. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Benito, R.; Janusz, K.; Díez-Campelo, M.; Hernández-Sánchez, J.M.; Marcellini, S.; Girós, M.; Rivera, J.; Lozano, M.L.; Hortal, A.; et al. Two Novel Variants of the ABCG5 gene Cause Xanthelasmas and Macrothrombocytopenia: A Brief Review of Hematologic Abnormalities of Sitosterolemia. J. Thromb. Haemost. 2017, 15, 1859–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurden, A.T.; Nurden, P. High-Throughput Sequencing for Rapid Diagnosis of Inherited Platelet Disorders: A Case for a European Consensus. Haematologica 2018, 103, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greinacher, A.; Pecci, A.; Kunishima, S.; Althaus, K.; Nurden, P.; Balduini, C.L.; Bakchoul, T. Diagnosis of Inherited Platelet Disorders on a Blood Smear: A Tool to Facilitate Worldwide Diagnosis of Platelet Disorders. J. Thromb. Haemost. 2017, 15, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing High-Throughput Sequencing into Mainstream Genetic Diagnosis Practice in Inherited Platelet Disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef]
- Simeoni, I.; Stephens, J.C.; Hu, F.; Deevi, S.V.V.; Megy, K.; Bariana, T.K.; Lentaigne, C.; Schulman, S.; Sivapalaratnam, S.; Vries, M.J.A.; et al. A High-Throughput Sequencing Test for Diagnosing Inherited Bleeding, Thrombotic, and Platelet Disorders. Blood 2016, 127, 2791–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ver Donck, F.; Downes, K.; Freson, K. Strengths and Limitations of High-Throughput Sequencing for the Diagnosis of Inherited Bleeding and Platelet Disorders. J. Thromb. Haemost. 2020, 18, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Romasko, E.J.; Devkota, B.; Biswas, S.; Jayaraman, V.; Rajagopalan, R.; Dulik, M.C.; Thom, C.S.; Choi, J.; Jairam, S.; Scarano, M.I.; et al. Utility and Limitations of Exome Sequencing in the Molecular Diagnosis of Pediatric Inherited Platelet Disorders. Am. J. Hematol. 2018, 93, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A.; Eekels, J.J.M. Diagnosis of Hereditary Platelet Disorders in the Era of Next-Generation Sequencing: “Primum Non Nocere”. J. Thromb. Haemost. 2019, 17, 551–554. [Google Scholar] [CrossRef]
- Downes, K.; Borry, P.; Ericson, K.; Gomez, K.; Greinacher, A.; Lambert, M.; Leinoe, E.; Noris, P.; Van Geet, C.; Freson, K. Clinical Management, Ethics and Informed Consent Related to Multi-Gene Panel-Based High Throughput Sequencing Testing for Platelet Disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2020, 18, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Bastida Bermejo, J.M.; Hernández-Rivas, J.M.; González-Porras, J.R. Novel Approaches for Diagnosing Inherited Platelet Disorders. Med. Clínica Engl. Ed. 2017, 148, 71–77. [Google Scholar] [CrossRef]
- Fasulo, M.R.; Biguzzi, E.; Abbattista, M.; Stufano, F.; Pagliari, M.T.; Mancini, I.; Gorski, M.M.; Cannavò, A.; Corgiolu, M.; Peyvandi, F.; et al. The ISTH Bleeding Assessment Tool and the Risk of Future Bleeding. J. Thromb. Haemost. 2018, 16, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodeghiero, F.; Pabinger, I.; Ragni, M.; Abdul-Kadir, R.; Berntorp, E.; Blanchette, V.; Bodó, I.; Casini, A.; Gresele, P.; Lassila, R.; et al. Fundamentals for a Systematic Approach to Mild and Moderate Inherited Bleeding Disorders: An EHA Consensus Report. HemaSphere 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Weyand, A.C.; James, P.D. Sexism in the Management of Bleeding Disorders. Res. Pract. Thromb. Haemost. 2021, 5, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.E.; Toledano, R.D.; Houle, T.; Beilin, Y.; MacEachern, M.; McCabe, M.; Rector, D.; Cooper, J.P.; Gernsheimer, T.; Landau, R.; et al. Lumbar Neuraxial Procedures in Thrombocytopenic Patients Across Populations: A Systematic Review and Meta-Analysis. J. Clin. Anesth. 2020, 61. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.; Kasthuri, R.S.; Bergmeier, W. Platelet Transfusion for Patients with Platelet Dysfunction: Effectiveness, Mechanisms, and Unanswered Questions. Curr. Opin. Hematol. 2020, 27, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Poon, M.C. Inherited Platelet Functional Disorders: General Principles and Practical Aspects of Management. Transfus. Apher. Sci. 2018, 57, 494–501. [Google Scholar] [CrossRef]
- Dupuis, A.; Gachet, C. Inherited Platelet Disorders: Management of the Bleeding Risk. Transfus. Clin. Biol. 2018, 25, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Orsini, S.; Noris, P.; Bury, L.; Heller, P.G.; Santoro, C.; Kadir, R.A.; Butta, N.C.; Falcinelli, E.; Cid, A.R.; Fabris, F.; et al. Bleeding Risk of Surgery and its Prevention in Patients with Inherited Platelet Disorders. Haematologica 2017, 102, 1192–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bury, L.; Falcinelli, E.; Gresele, P. Learning the Ropes of Platelet Count Regulation: Inherited Thrombocytopenias. J. Clin. Med. 2021, 10, 533. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T. Acquired Antibodies to αIIbβ3 in Glanzmann Thrombasthenia: From Transfusion and Pregnancy to Bone Marrow Transplants and Beyond. Transfus. Med. Rev. 2018, 32, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lozano, M.L.; Cook, A.; Bastida, J.M.; Paul, D.S.; Iruin, G.; Cid, A.R.; Adan-Pedroso, R.; González-Porras, J.R.; Hernández-Rivas, J.M.; Fletcher, S.J.; et al. Novel Mutations in RASGRP2, which Encodes CalDAG-GEFI, Abrogate Rap1 Activation, Causing Platelet Dysfunction. Blood 2016, 128, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Grainger, J.D.; Thachil, J.; Will, A.M. How we Treat the Platelet Glycoprotein Defects; Glanzmann Thrombasthenia and Bernard Soulier Syndrome in Children and Adults. Br. J. Haematol. 2018, 182, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cid, J.; Guijarro, F.; Carbassé, G.; Lozano, M. 24-H Continuous Infusion of Platelets for Patients with Platelet Transfusion Refractoriness. Br. J. Haematol. 2018, 181, 386–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.H.; Piatt, R.; Dhenge, A.; Lozano, M.L.; Palma-Barqueros, V.; Rivera, J.; Bergmeier, W. Impaired Hemostatic Activity of Healthy Transfused Platelets in Inherited and Acquired Platelet Disorders: Mechanisms and Implications. Sci. Transl. Med. 2019, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Khoreva, A.; Abramova, I.; Deripapa, E.; Rodina, Y.; Roppelt, A.; Pershin, D.; Larin, S.; Voronin, K.; Maschan, A.; Novichkova, G.; et al. Efficacy of Romiplostim in Treatment of Thrombocytopenia in Children with Wiskott–Aldrich Syndrome. Br. J. Haematol. 2021, 192, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Favier, R.; Roussel, X.; Audia, S.; Bordet, J.C.; De Maistre, E.; Hirsch, P.; Neuhart, A.; Bedgedjian, I.; Gkalea, V.; Favier, M.; et al. Correction of Severe Myelofibrosis, Impaired Platelet Functions and Abnormalities in a Patient with Gray Platelet Syndrome Successfully Treated by Stem Cell Transplantation. Platelets 2020, 31, 536–540. [Google Scholar] [CrossRef]
- Cid, A.R.; Montesinos, P.; Sánchez-Guiu, I.; Haya, S.; Lorenzo, J.I.; Sanz, J.; Moscardo, F.; Puig, N.; Planelles, D.; Bonanad, S.; et al. Allogeneic Hematopoietic Cell Transplantation in an Adult Patient with Glanzmann Thrombasthenia. Clin. Case Rep. 2017, 5, 1887–1890. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, L.M.; Petrovic, A.; Brazauskas, R.; Liu, X.; Griffith, L.M.; Ochs, H.D.; Bleesing, J.J.; Edwards, S.; Dvorak, C.C.; Chaudhury, S.; et al. Excellent Outcomes Following Hematopoietic cell Transplantation for Wiskott-Aldrich Syndrome: A PIDTC Report. Blood 2020, 135, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Tristán-Manzano, M.; Sánchez-Gilabert, A.; Santilli, G.; Galy, A.; Thrasher, A.J.; Martin, F. WAS Promoter-Driven Lentiviral Vectors Mimic Closely the Lopsided WASP Expression during Megakaryocytic Differentiation. Mol. Ther. Methods Clin. Dev. 2020, 19, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Romito, M.; Rivers, E.; Turchiano, G.; Blattner, G.; Vetharoy, W.; Ladon, D.; Andrieux, G.; Zhang, F.; Zinicola, M.; et al. Targeted Gene Correction of Human Hematopoietic Stem Cells for the Treatment of Wiskott-Aldrich Syndrome. Nat. Commun. 2020, 11, 4034. [Google Scholar] [CrossRef] [PubMed]
- Makris, M. Thrombopoietin Receptor Agonists for the Treatment of Inherited Thrombocytopenia. Haematologica 2020, 105, 536–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaninetti, C.; Gresele, P.; Bertomoro, A.; Klersy, C.; de Candia, E.; Veneri, D.; Barozzi, S.; Fierro, T.; Alberelli, M.A.; Musella, V.; et al. Eltrombopag for the Treatment of Inherited Thrombocytopenias: A Phase II Clinical Trial. Haematologica 2020, 105, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Sánchez, J.M.; Bastida, J.M.; Alonso-López, D.; Benito, R.; González-Porras, J.R.; De Las Rivas, J.; Hernández Rivas, J.M.; Rodríguez-Vicente, A.E. Transcriptomic Analysis of Patients with Immune Thrombocytopenia Treated with Eltrombopag. Platelets 2020, 31, 993–1000. [Google Scholar] [CrossRef]
- Currao, M.; Balduini, C.L.; Balduini, A. High Doses of Romiplostim Induce Proliferation and Reduce Proplatelet Formation by Human Megakaryocytes. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Di Buduo, C.A.; Currao, M.; Pecci, A.; Kaplan, D.L.; Balduini, C.L.; Balduini, A. Revealing Eltrombopag’s Promotion of Human Megakaryopoiesis Through AKT/ERK-Dependent Pathway Activation. Haematologica 2016, 101, 1479–1488. [Google Scholar] [CrossRef] [Green Version]
- Balduini, A.; Raslova, H.; Di Buduo, C.A.; Donada, A.; Ballmaier, M.; Germeshausen, M.; Balduini, C.L. Clinic, Pathogenic Mechanisms and Drug Testing of Two Inherited Thrombocytopenias, ANKRD26-Related Thrombocytopenia and MYH9-Related Diseases. Eur. J. Med. Genet. 2018, 61, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Spinler, K.R.; Shin, J.W.; Lambert, M.P.; Discher, D.E. Myosin-II Repression Favors pre/proplatelets but Shear Activation Generates Platelets and Fails in Macrothrombocytopenia. Blood 2015, 125, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanouchi, J.; Hato, T.; Kunishima, S.; Niiya, T.; Nakamura, H.; Yasukawa, M. A Novel MYH9 Mutation in a Patient with MYH9 Disorders and Platelet Size-Specific Effect of Romiplostim on Macrothrombocytopenia. Ann. Hematol. 2015, 94, 1599–1600. [Google Scholar] [CrossRef] [PubMed]
- Westbury, S.K.; Downes, K.; Burney, C.; Lozano, M.L.; Obaji, S.G.; Toh, C.H.; Sevivas, T.; Morgan, N.V.; Erber, W.N.; Kempster, C.; et al. Phenotype Description and Response to Thrombopoietin Receptor Agonist in DIAPH1-Related Disorder. Blood Adv. 2018, 2, 2341–2346. [Google Scholar] [CrossRef] [PubMed]
- Porrazzo, M.; Baldacci, E.; Ferretti, A.; Miulli, E.; Chistolini, A.; Pecci, A.; Mazzucconi, M.G.; Foà, R.; Santoro, C. The Role of an Accurate Diagnosis of Inherited Thrombocytopenia as the Basis for an Effective Treatment. A Case of MYH9 Syndrome Treated with a TPO-RA. Haemophilia 2019, 25, e288–e290. [Google Scholar] [CrossRef]
- Pecci, A.; Gresele, P.; Klersy, C.; Savoia, A.; Noris, P.; Fierro, T.; Bozzi, V.; Mezzasoma, A.M.; Melazzini, F.; Balduini, C.L. Eltrombopag for the Treatment of the Inherited Thrombocytopenia Deriving from MYH9 Mutations. Blood 2010, 116, 5832–5837. [Google Scholar] [CrossRef] [Green Version]
- Gerrits, A.J.; Leven, E.A.; Frelinger, A.L.; Brigstocke, S.L.; Berny-Lang, M.A.; Mitchell, W.B.; Revel-Vilk, S.; Tamary, H.; Carmichael, S.L.; Barnard, M.R.; et al. Effects of Eltrombopag on Platelet Count and Platelet Activation in Wiskott-Aldrich Syndrome/X-Linked Thrombocytopenia. Blood 2015, 126, 1367–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psaila, B.; Bussel, J.B.; Linden, M.D.; Babula, B.; Li, Y.; Barnard, M.R.; Tate, C.; Mathur, K.; Frelinger, A.L.; Michelson, A.D. In vivo Effects of Eltrombopag on Platelet Function in Immune Thrombocytopenia: No Evidence of Platelet Activation. Blood 2012, 119, 4066–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaninetti, C.; Barozzi, S.; Bozzi, V.; Gresele, P.; Balduini, C.L.; Pecci, A. Eltrombopag in Preparation for Surgery in Patients with Severe MYH9-Related Thrombocytopenia. Am. J. Hematol. 2019, 94, E199–E201. [Google Scholar] [CrossRef] [Green Version]
- Pecci, A.; Barozzi, S.; D’Amico, S.; Balduini, C.L. Short-Term Eltrombopag for Surgical Preparation of a Patient with Inherited Thrombocytopenia Deriving from MYH9 Mutation. Thromb. Haemost. 2012, 107, 1188–1189. [Google Scholar] [CrossRef] [PubMed]
- Favier, R.; Feriel, J.; Favier, M.; Denoyelle, F.; Martignetti, J.A. First Successful Use of Eltrombopag Before Surgery in a Child with MYH9-Related Thrombocytopenia. Pediatrics 2013, 132. [Google Scholar] [CrossRef] [Green Version]
- Fiore, M.; Saut, N.; Alessi, M.C.; Viallard, J.F. Successful Use of Eltrombopag for Surgical Preparation in a Patient with ANKRD26-Related Thrombocytopenia. Platelets 2016, 27, 828–829. [Google Scholar] [CrossRef]
- Paciullo, F.; Bury, L.; Gresele, P. Eltrombopag to Allow Chemotherapy in a Patient with MYH9-Related Inherited Thrombocytopenia and Pancreatic Cancer. Int. J. Hematol. 2020, 112, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Favier, R.; De Carne, C.; Elefant, E.; Lapusneanu, R.; Gkalea, V.; Rigouzzo, A. Eltrombopag to Treat Thrombocytopenia During Last Month of Pregnancy in a Woman With MYH9-Related Disease. A&A Pract. 2018, 10, 10–12. [Google Scholar] [CrossRef]
- Gröpper, S.; Althaus, K.; Najm, J.; Haase, S.; Aul, C.; Greinacher, A.; Giagounidis, A. A Patient with Fechtner Syndrome Successfully Treated with Romiplostim. Thromb. Haemost. 2012, 107, 590–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbolini, D.J.; Chun, Y.; Latimer, M.; Kunishima, S.; Fixter, K.; Valecha, B.; Tan, P.; Chew, L.P.; Kile, B.T.; Burt, R.; et al. Diagnosis and Treatment of MYH9-RD in an Australasian Cohort with Thrombocytopenia. Platelets 2018, 29, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Del Rey, M.; Revilla, N.; Benito, R.; Perez-Andrés, M.; González, B.; Riesco, S.; Janusz, K.; Padilla, J.; Hortal Benito-Sendin, A.; et al. Wiskott–Aldrich Syndrome in a Child Presenting with Macrothrombocytopenia. Platelets 2017, 28, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Fraczkiewicz, J.; Sȩga-Pondel, D.; Kazanowska, B.; Ussowicz, M. Eltrombopag Therapy in Children with Rare Disorders Associated with Thrombocytopenia. J. Pediatr. Hematol. Oncol. 2020, 42, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoumen, K.; Fabre, M.; Ducastelle-Lepretre, S.; Favier, R.; Ballerini, P.; Bordet, J.C.; Dargaud, Y. Eltrombopag for the Treatment of Severe Inherited Thrombocytopenia. Acta Haematol. 2020, 1–6. [Google Scholar] [CrossRef]
- Gabelli, M.; Marzollo, A.; Notarangelo, L.D.; Basso, G.; Putti, M.C. Eltrombopag Use in a Patient with Wiskott–Aldrich Syndrome. Pediatr. Blood Cancer 2017, 64, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.; Ben-Harosh, M.; Sirin, M.; Stein, J.; Dgany, O.; Kaplelushnik, J.; Hoenig, M.; Pannicke, U.; Lorenz, M.; Schwarz, K.; et al. Bone Marrow Failure Unresponsive to Bone Marrow Transplant is Caused by Mutations in Thrombopoietin. Blood 2017, 130, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Pecci, A.; Ragab, I.; Bozzi, V.; De Rocco, D.; Barozzi, S.; Giangregorio, T.; Ali, H.; Melazzini, F.; Sallam, M.; Alfano, C.; et al. Thrombopoietin Mutation in Congenital Amegakaryocytic Thrombocytopenia Treatable with Romiplostim. EMBO Mol. Med. 2018, 10, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Paciullo, F.; Bury, L.; Noris, P.; Falcinelli, E.; Melazzini, F.; Orsini, S.; Zaninetti, C.; Abdul-Kadir, R.; Obeng-Tuudah, D.; Heller, P.G.; et al. Antithrombotic Prophylaxis for Surgery-Associated Venous Thromboembolism Risk Disorders. In Patients the SPATA-DVT with Inherited Study Platelet. Haematologica 2020, 105, 1948–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussel, J.; Kulasekararaj, A.; Cooper, N.; Verma, A.; Steidl, U.; Semple, J.W.; Will, B. Mechanisms and Therapeutic Prospects of Thrombopoietin Receptor Agonists. Semin. Hematol. 2019, 56, 262–278. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Educational measures | Avoid trauma or bleeding risk situations (sports with strong contact or high risk of falls). Appropriate oral hygiene. Personal identification of the disorder (identification plates, etc.). Avoid drugs and foods with antiplatelet potential. | |
| Preventive measures | Inclusion in registries. Hepatitis B vaccination. Assessment of liver function at diagnosis. Routine medical follow-up, including asymptomatic patients, but with disorders at high risk of developing syndromic pathology or neoplasms. Specialized and multidisciplinary care for hemorrhagic risk interventions. Prenatal diagnosis and bleeding prevention plan in childbirth, surgeries, dental interventions, or antiplatelet treatments due to cardiovascular risk. | |
| Control of moderate active bleeding Prevention of bleeding in low hemorrhagic risk interventions Prophylactic treatment in patients with moderate disorders | Topical Measures | Mild wounds: compression and application of gelatin sponges or gauzes soaked in tranexamic acid Epistaxis/gum bleeding: nose pads, fibrin sealants, fibrin-coated/collagen sponges, mouthwash with tranexamic acid, anterior or posterior packing. Dental interventions: splint of soft acrylic assist, fibrin sealants. |
| Antifibrinolytic drugs | Control of mild—moderate bleeding (epistaxis or menorrhagia) and in prevention of bleeding in minor dental interventions. Treatment begins the day before and continues for 3–5 days. Dose: Tranexamic acid: oral 15–25 mg/kg/8 h; iv 10 mg/kg/8 h if more severe bleeding; for mouthwash (10 mL at 5%); e-aminocaproic acid: oral 4–8 g/8 h; iv 4 g/8 h | |
| Desmopressin (DDAVP) | Few data in inherited thrombocytopenia and not routinely recommended. Not recommended in PT-VWD, or in patients with atherosclerosis. Drug of choice in patients with mild/moderate IPFD undergoing dental interventions and minor surgeries. Causes fluid retention. Risk of desensitization with repeated doses. Dose: I.V. (0.2–0.3 ug/Kg of DDVAP in saline [4 ug/mL] in 30 min, start 1 h before); subcutaneous (0.3 ug/kg); Intranasal spray (150 ug/dose) | |
| Control of moderate to severe bleeding, prevention of bleeding in high bleeding risk interventions, preventive treatment in patients with severe disorders | Platelet transfusion | Essential for control of severe bleeding, in severe thrombocytopenia, for prevention of bleeding in major surgery and for the management of childbirth in a woman with severe platelet dysfunction. Preferable leucocyte-depleted platelet concentrates from single donor and/or HLA identical if possible, to reduce the risk of alloimmunization or if the patient already has anti-HLA antibodies; Recommended platelet counts: (If platelet dysfunction or severe bleeding history, individualization is mandatory). >30 × 109/L for dental extractions and minor dental interventions; >50−80 × 109/L for major surgery, deliveries, or caesarean sections; >100 × 109/L for eye and brain surgery. |
| Recombinant active Factor VII (rFVIIa) | Approval in GT patients with platelet transfusion refractoriness. Off-label use in other IPDs. May also be considered as an off-label drug to be used in BSS (risk of alloantibodies and ineffective platelet transfusion. Potentially useful, in combination with antifibrinolytics, in the control of bleeding in childbirth of women with severe platelet dysfunction. Dose: I.V. 90–120 ug/Kg before the procedure and then repeated doses every 90–120 min. The required number of doses is variable depending on the risk of the procedure and the patient characteristics. | |
| Increase of platelet count stably or transiently before surgery or invasive interventions | Splenectomy | To be considered only in WAS and XLT and a patient personalized basis. May reduce bleeding complications but worsens the immunodeficiency and the rate of severe infections. It should be avoided in WAS patients who have undergone or are candidates of HSCT. |
| TPO-RA (Eltrombopag, Romiplostim) | To be considered for short-term use (for instance to increase in numbers before elective surgery) in WAS, MYH9-RD, or ANKRD26-RT. Long-term treatment has been successfully used in some patients with MYH9-RD and WAS Romiplostim was successfully used in THPO-RT. | |
| Potential curative treatments | Allogeneic hematopoietic stem cell transplantation | To be considered in severe ITs at high risk of transformation to bone marrow failure or malignant disease and with high early mortality. Treatment of choice in CAMT and severe WAS. Successfully used in some severe cases of TAR, RUSAT, BSS and GT (about 60 cases). |
| Gene therapy | Clinical trial in WAS. Preclinical studies in other IPD (mainly GT). | |
| Disease | Type of Study | TPO-RA | Dose | N | Mean PC | Indication | Type of Response | Treatment Duration | Adverse Events (n) | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| MYH9-RD | Phase II | Eltrombopag | 50–75 mg/d | 12 | 31.2 × 109/L | Efficacy and safety of short-term course | R: 92% (CR: 67%) B: 80% | 3–6 w | Headache (2) Dry mouth (1) | [55] |
| WAS/XLT | Phase II | Eltrombopag | 0.8/kg/d–75 mg/d | 8 | 19 × 109/L | Efficacy and safety | R: 62.5% (CR: 50%) B: 75% | 20–187 w | Transaminitis (1) | [56] |
| MYH9-RD (9) ANKRD26-RT (9) WAS/XLT (3) mBSS (2) ITGB3-RT (1) | Phase II | Eltrombopag | 25–75 mg/d | 24 | 40 × 109/L | Efficacy and safety of short and long-term course | R: 91.3% (CR: 47%) B: 83% | 3–6 w | Headache (4) Bone pain (2) Creatinine, nr (1) | [46] |
| WAS/XLT | Observational | Romiplostim | 9 mcg/kg/w | 67 | 21 × 109/L | Efficacy and safety for bridging to HSCT | R: 60% (CR: 33%) B: 100% | 1–12 m | Thrombocytosis (2) Arterial thrombosis, nr (1) | [39] |
| Disease (n) | Type of Study | TPO-RA | Dose and Weeks before Surgery | Mean PC | Surgery | PC Day of Surgery (d) | Time to Response | Adverse Events | Ref |
|---|---|---|---|---|---|---|---|---|---|
| MYH9-RD (1) | Case report | Eltrombopag | 50 mg/d (3 w) | 19 × 109/L | Osteotomy | 195 × 109/L (19) CR | 10–12 d | No LMWH | [58] |
| MYH9-RD (1) | Case report | Eltrombopag | 25 m/d (1 w) 50 mg/d (3 w) | 10 × 109/L | Tympanoplasty | 77 × 109/L (33) PR | 33 d | No - | [59] |
| MYH9-RD (1) | Case report | Romiplostim | 1–5 mcg/kg/w (5 w) | 25 × 109/L | Craniotomy | 84 × 109/L (42) PR | 6 w | No - | [52] |
| ANKRD26-RT (1) | Case report | Eltrombopag | 50 mg/d (4 w) 75 mg/d (1 w) | 16 × 109/L | Lumbar recalibration | 93 × 109/L (35) PR | 2 w (lost) 5 w | No LMWH not reported | [60] |
| DIAPH1-RD (1) | Case report | Eltrombopag | 50 mg/d (3 w) 75 mg/d (1 w) | 29 × 109/L | Hip arthroplasty | 72 × 109/L (28) PR | 3 w | No LMWH not reported | [53] |
| MYH9-RD (1) | Case report | Eltrombopag | 50 mg/d (4 w) | 32 × 109/L | Ovariectomy | 153 × 109/L (28) | 2 w | No LMWH | [54] |
| MYH9-RD (1) | Case report | Eltrombopag | 75 mg/d (-) | 20 × 109/L | Urgent endoscopic treatment | 93 × 109/L (-) PR | - | No | [61] |
| MYH9-RD (1) | Case report | Romiplostim | 9 mcg/kg/w (5 w) + Prednisone | 6 × 109/L | General | 115 × 109/L (42) CR | 5 w | No | [64] |
| MYH9-RD (5) | Case series | Eltrombopag | 50 mg/d (3 w) | 19 × 109/L | Osteotomy | 180 × 109/L (20) CR | 3 w | No LMWH not reported | [57] |
| 20 × 109/L | Osteotomy | 172 × 109/L (21) CR | 3 w | No LMWH not reported | |||||
| 23 × 109/L | Percutaneous kidney biopsy | 161 × 109/L (21) CR | 3 w | No LMWH not reported | |||||
| 75 mg/d (3 w) | 15 × 109/L | Hysterectomy and bilateral annexectomy | 75 × 109/L (21) PR | 3 w | No LMWH not reported | ||||
| 17 × 109/L | Cochlear implantation | 78 × 109/L (22) PR | 3 w | No - | |||||
| 75 mg/d (3 w) | 7109/L | Dental extraction | 100 × 109/L (21) CR | 3 w | Headache - | ||||
| 9 × 109/L | Periodontal surgery | 120 × 109/L (21) CR | 3 w | Headache - | |||||
| 10 × 109/L | Dental extraction | 95 × 109/L (21) CR | 3 w | No - | |||||
| 10 × 109/L | Periodontal surgery | 132 × 109/L (22) CR | 3 w | No - | |||||
| 75 mg/d (3 w) | 25 × 109/L | Cochlear implantation | 104 × 109/L (23) CR | 3 w | No - | ||||
| 75 mg/d (3 w) | 5 × 109/L | Biopsy of tonsillar tumor | 11 × 109/L (21) NR | No | No - |
| Disease (n) | Type of Study | TPO-RA | Dose | Mean PC | Special Situation | Type of Response | Treatment Duration | Adverse Events (n) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| MYH9-RD (1) | Case report | Eltrombopag | 50–75 mg/d | 20 × 109/L | Chemotherapy Endoscopic treatment | CR | 2 m | No | [61] |
| MYH9-RD (1) | Case report | Eltrombopag | 50 mg/d | 30 × 109/L | Gestation | CR | 24 d | No LMWH | [62] |
| MYH9-RD (1) | Case report | Romiplostim | 10mcg/kg/w | 7 × 109/L | Misdiagnosis of ITP (2nd lines) | 60 × 109/L PR | 21 w | No | [63] |
| WAS (1) | Case report | Romiplostim and Eltrombopag | 10 mcg/kg/w 25–75 mg/d | 5 × 109/L | Misdiagnosis of ITP (2nd lines) | NR Bleeding response | 16 w | No | [65] |
| MYH9-RD (1) | Case report | Romiplostim | 6.3 mcg/kg/w | 6 × 109/L | Misdiagnosis of ITP (3th lines) | PR | 41 m | No | [64] |
| DiGeorge syndrome (1) | Case report | Eltrombopag | 25–50 mg/d | 2 × 109/L | Misdiagnosis of ITP (4th lines) | CR | 13 m | No | [66] |
| PT syndrome (1) | Case report | Eltrombopag | 50–150 mg/d | 9–20 × 109/L | Misdiagnosis of ITP (1st line) | PR and no bleeding | 23 m | No | [67] |
| WAS (1) | Case report | Eltrombopag | 0.8–5 mg/kg/d | 10 × 109/L | Bridging to HSCT | PR and reduced bleeding and platelet transfusion | 32 w | No | [68] |
| WAS (1) | Case report | Eltrombopag | 25 mg/d | 20 × 109/L | Engraftment failure post HSCT | NR | 7 m | No | [66] |
| THPO-RT (2) | Case series | Romiplostim | 5 mcg/kg/w | Both 21 × 109/L | BMF & HSCT failure | CR and no bleeding | 2 y 13 w | No | [69] |
| THPO-RT (3) | Case series | Romiplostim | 4 mcg/kg/m | 3–27 × 109/L | BMF & Transfusions | CR and no bleeding | >6 y | No | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastida, J.M.; Gonzalez-Porras, J.R.; Rivera, J.; Lozano, M.L. Role of Thrombopoietin Receptor Agonists in Inherited Thrombocytopenia. Int. J. Mol. Sci. 2021, 22, 4330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094330
Bastida JM, Gonzalez-Porras JR, Rivera J, Lozano ML. Role of Thrombopoietin Receptor Agonists in Inherited Thrombocytopenia. International Journal of Molecular Sciences. 2021; 22(9):4330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094330
Chicago/Turabian StyleBastida, José María, José Ramón Gonzalez-Porras, José Rivera, and María Luisa Lozano. 2021. "Role of Thrombopoietin Receptor Agonists in Inherited Thrombocytopenia" International Journal of Molecular Sciences 22, no. 9: 4330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094330