
Chrysin Inhibits TNFα-Induced TSLP Expression through Downregulation of EGR1 Expression in Keratinocytes

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
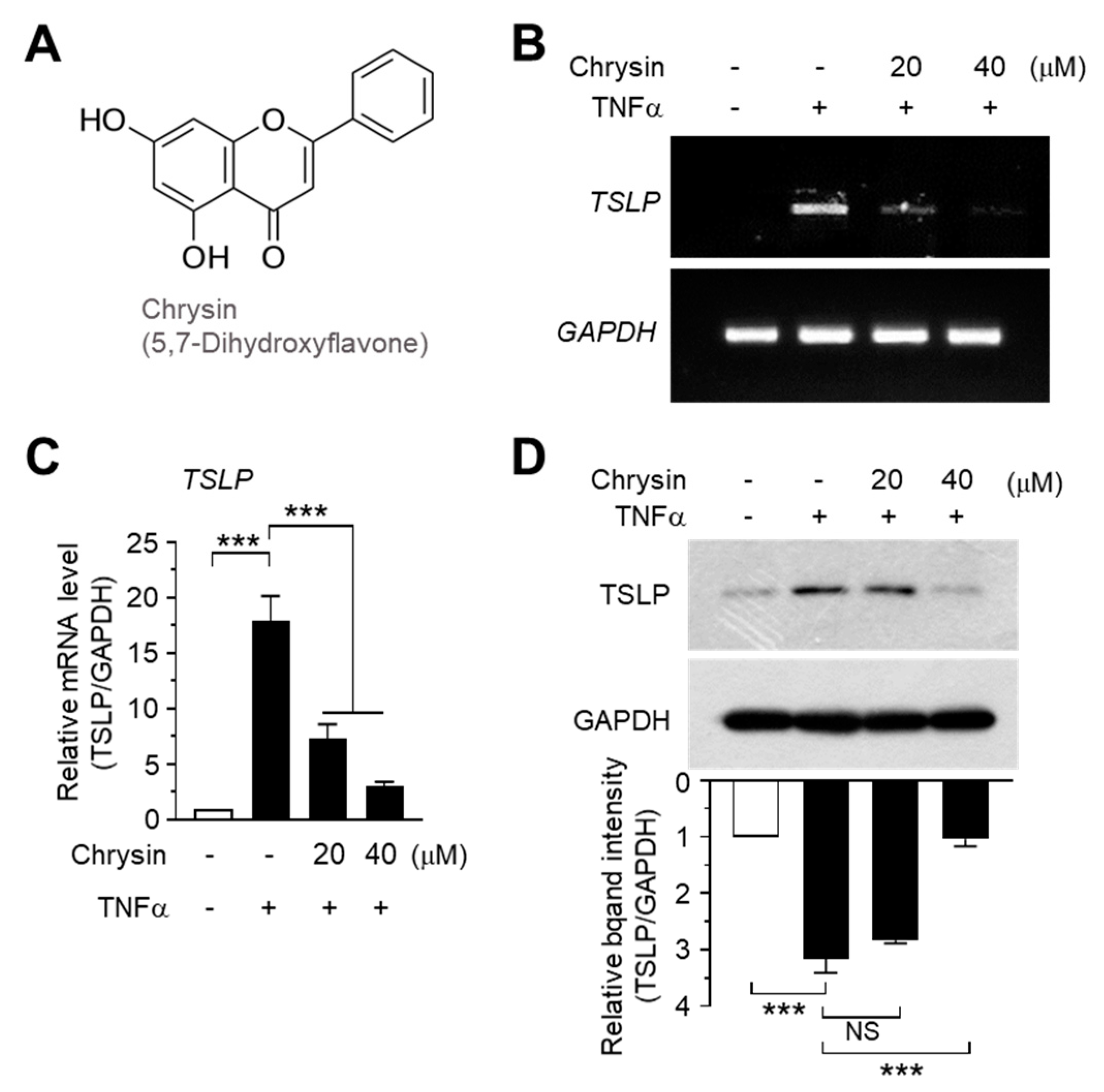
2.1. Chrysin Inhibits TNFα-Induced TSLP Expression in HaCaT Keratinocytes
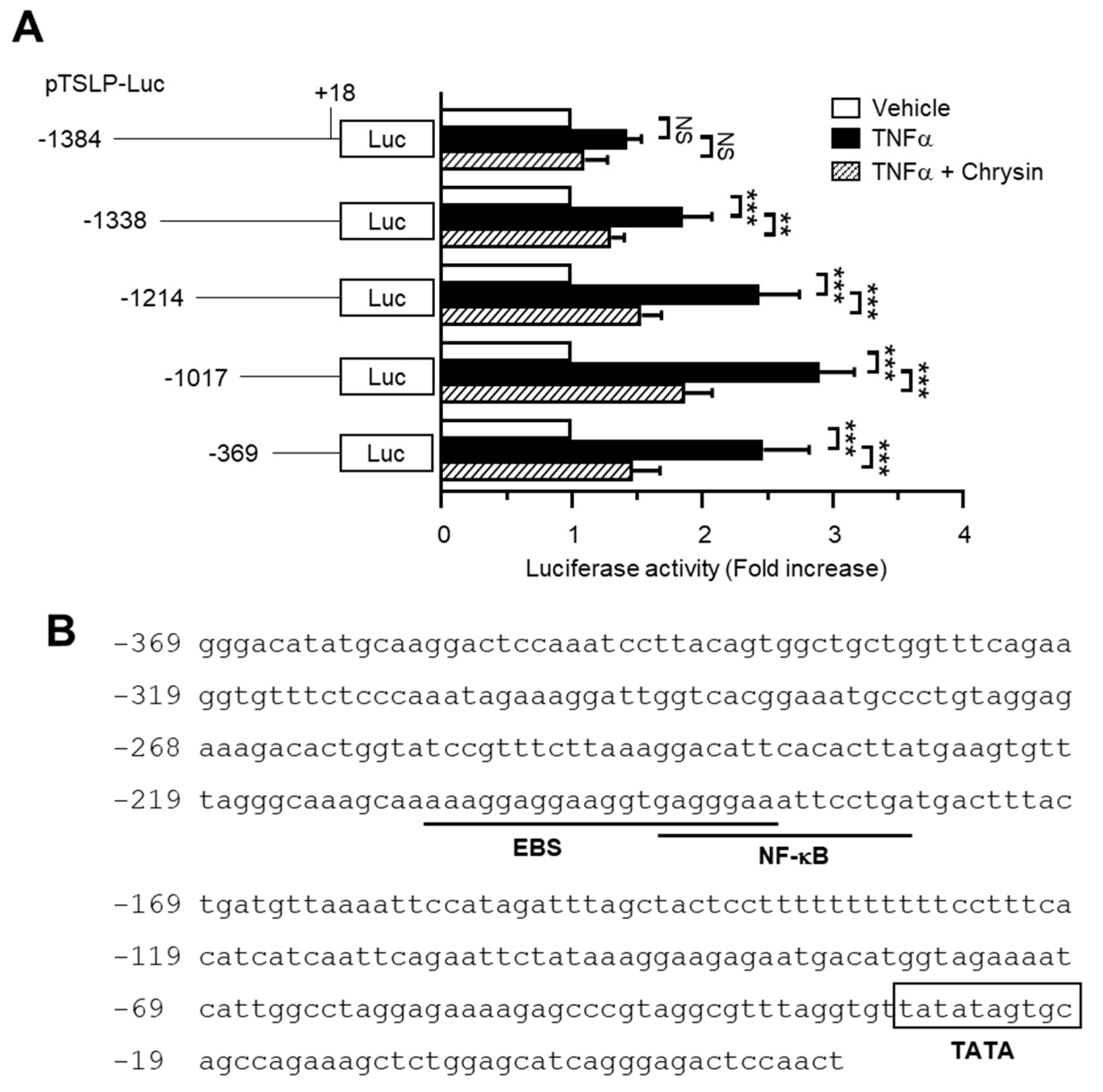
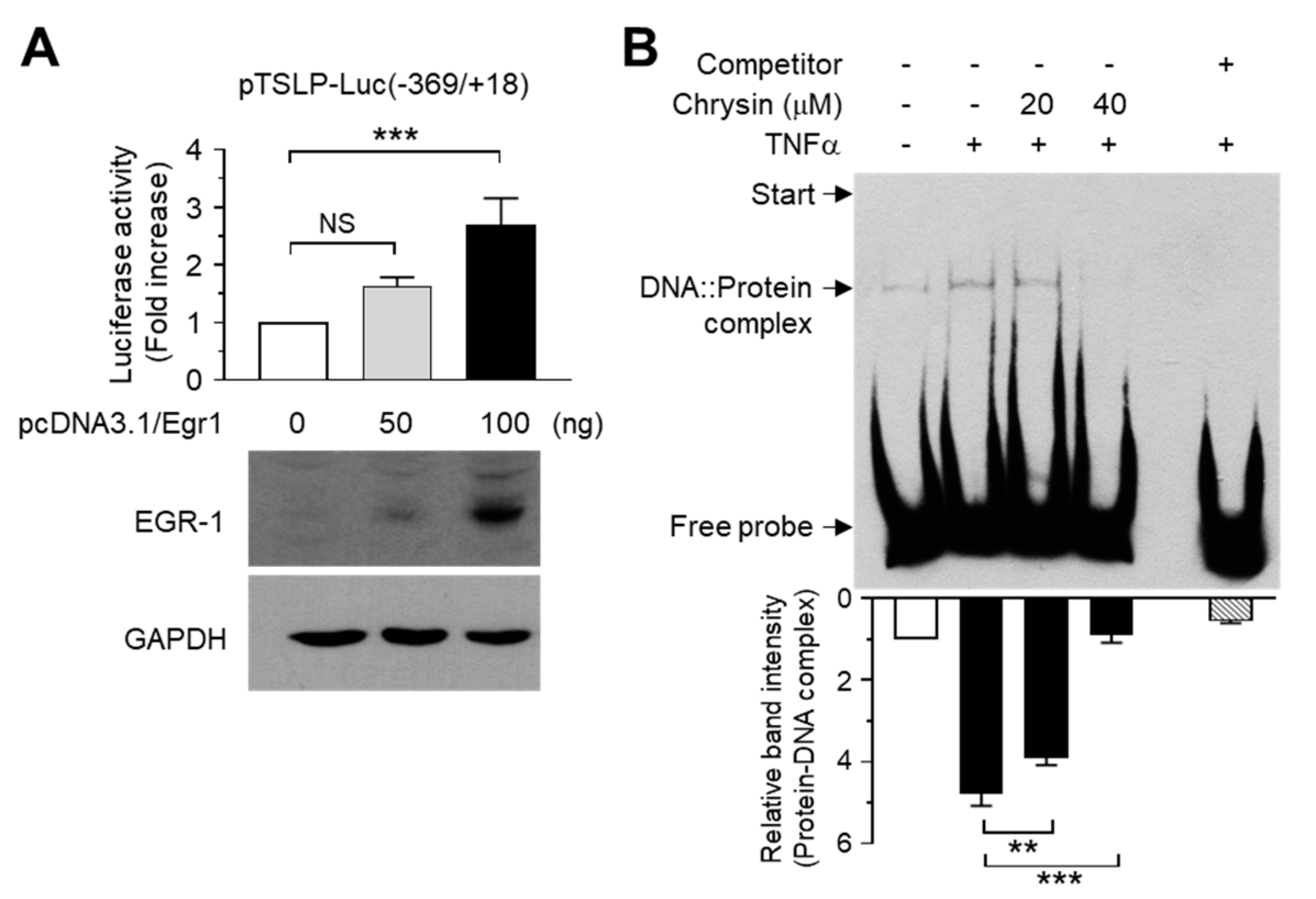
2.2. The Chrysin Response Element Is Located between the −369 and +18 Positions in the TSLP Promoter
2.3. Chrysin Inhibits the DNA-Binding Activity of EGR1
2.4. Chrysin Downregulates EGR1 Expression to Inhibit TNFα-Induced TSLP Expression
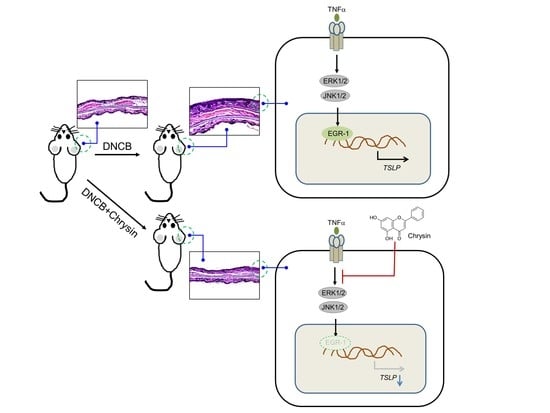
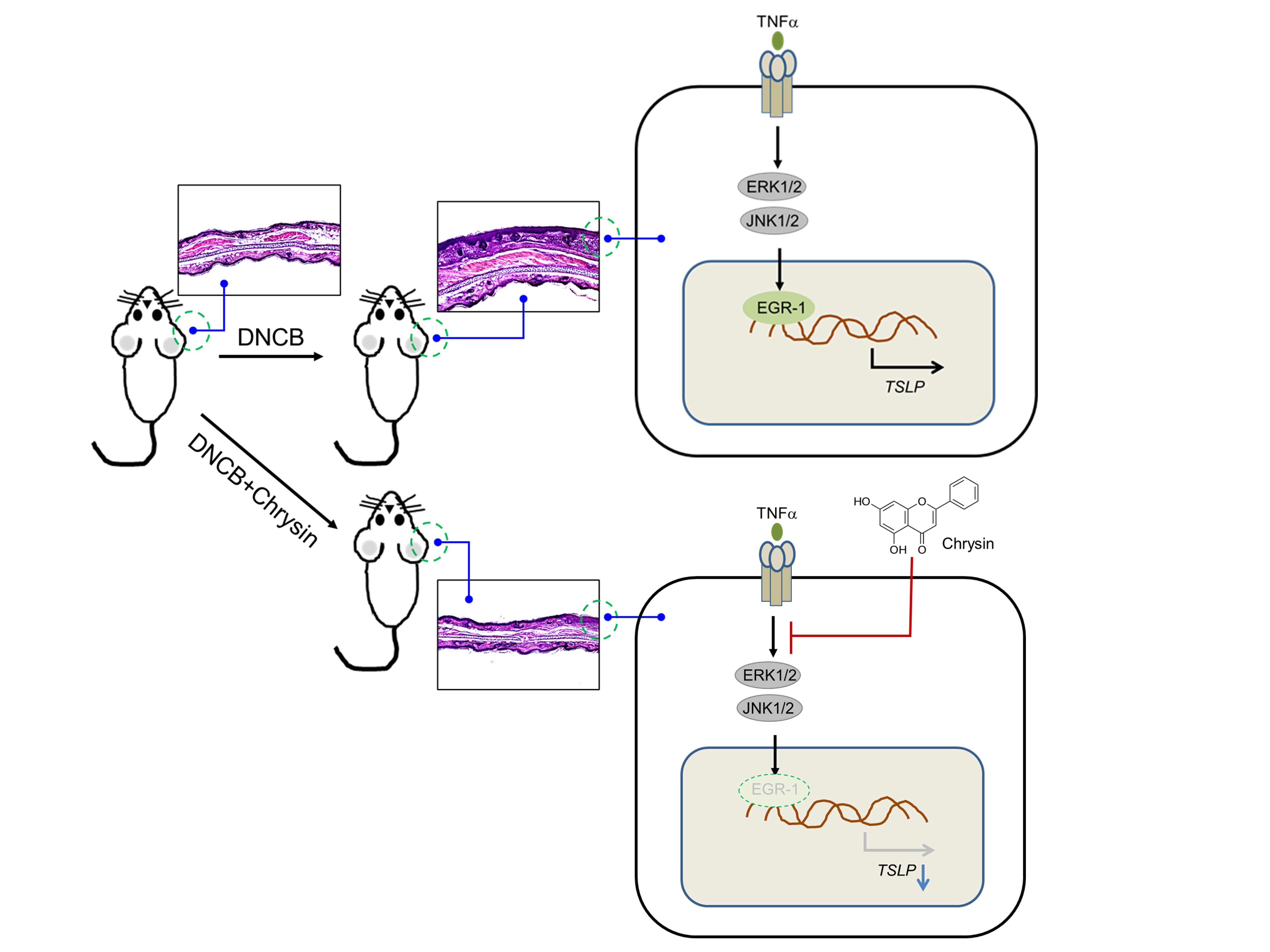
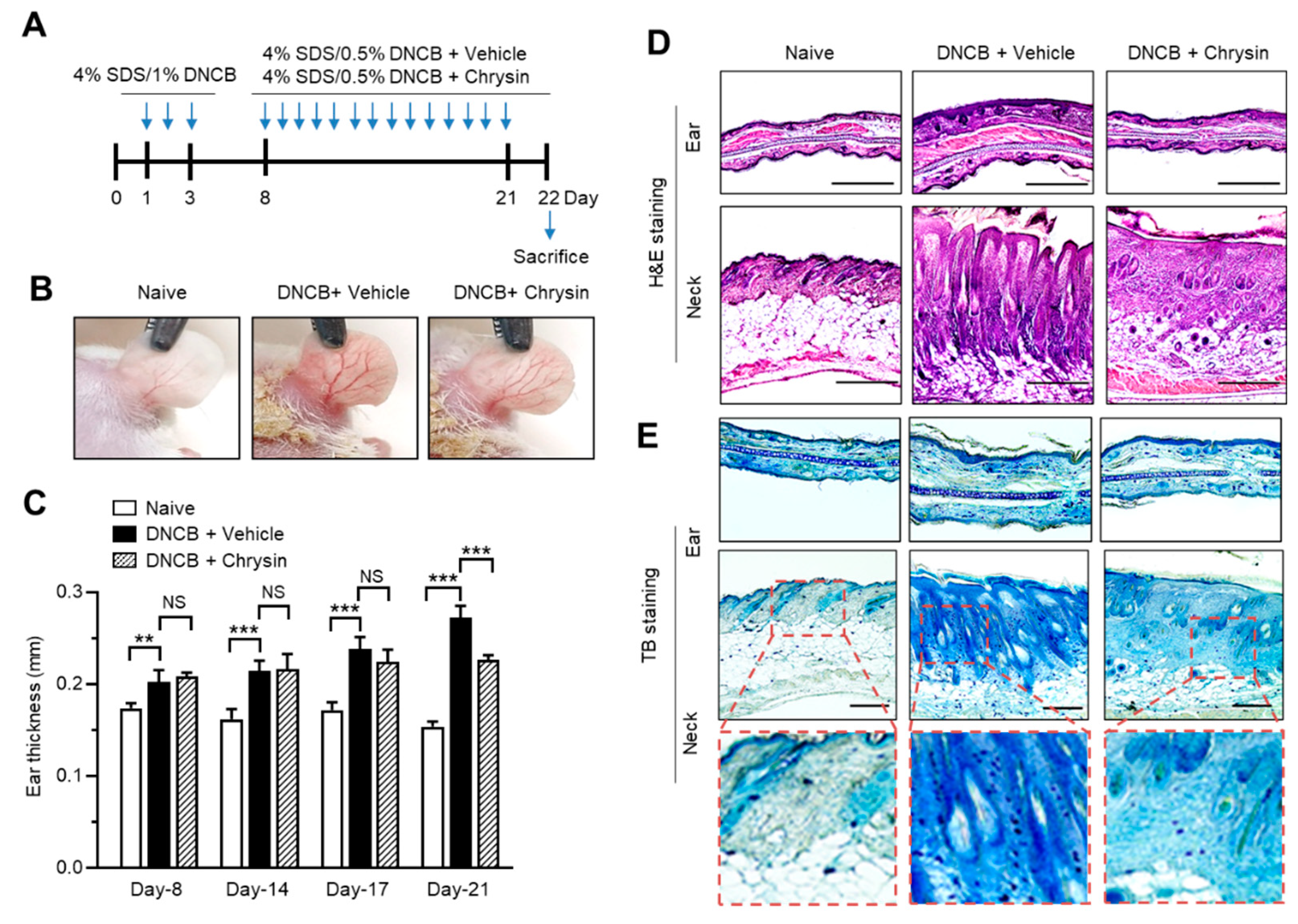
2.5. Oral Administration of Chrysin Attenuates 2,4-Dinitrochlorobenzene (DNCB)-Induced AD-Like Skin Lesions in BALB/c Mice
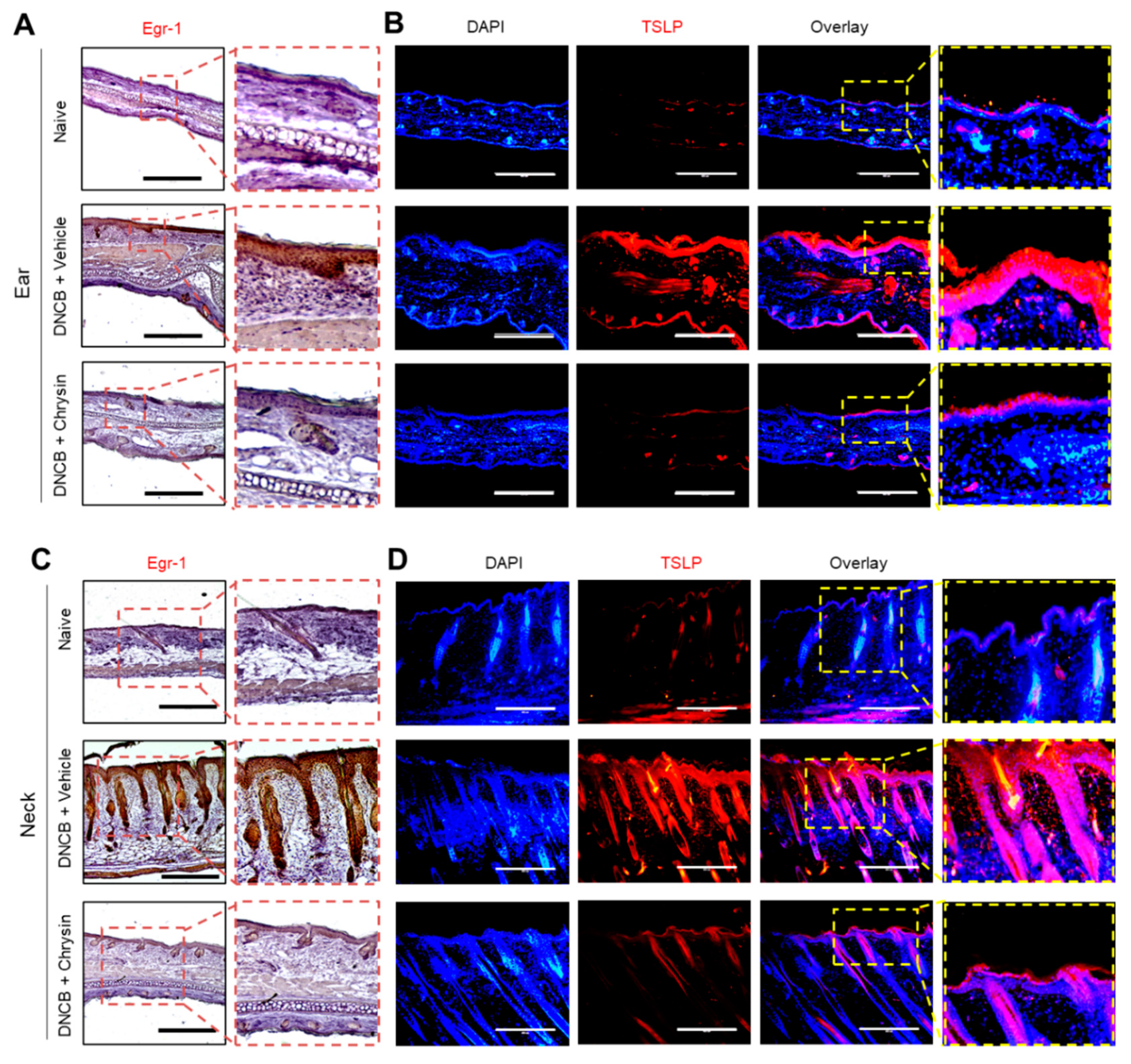
2.6. Oral Administration of Chrysin Reduces EGR1 and TSLP Expression in DNCB-Induced Skin Lesions in BALB/c Mice
2.7. Chrysin Inhibits the MAPK Pathways
2.8. MAPK Pathways Are Involved in TNFα-Induced EGR1 and TSLP Expression in HaCaT Keratinocytes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cells and Cell Culture
4.3. RT-PCR
- EGR1 forward, 5′-CAG CAG TCC CAT TTA CTC AG-3′;
- EGR1 reverse, 5′-GAC TGG TAG CTG GTA TTG-3;
- TSLP forward, 5′-TAG CAA TCG GCC ACA TTG CCT-3′;
- TSLP reverse, 5′-GAA GCG ACG CCA CAA TCC TTG-3;
- GAPDH forward, 5′-CCA AGG AGT AAG AAA CCC TGG AC-3′;
- GAPDH reverse, 5′-GGG CCG AGT TGG GAT AGG G-3′.
4.4. Quantitative Real-Time PCR (Q-PCR)
4.5. Western Blot Analysis
4.6. Construction of Human TSLP Promoter-Reporter Constructs
- −1338F: 5′-GGA CCA GAG CGA TGC AGG-3′
- −1214F: 5′-CAT GAG CCA AGC CAG GGA G-3′
- −1017F: 5′-AAA TCT GAG CCC GCC ATC TC-3′
- −369F: 5′-GGG ACA TAT GCA AGG ACT CC-3′
4.7. Luciferase Promoter–Reporter Assay
4.8. EMSA
4.9. Induction of AD-Like Skin Lesions in the Ear and Neck of Mice
4.10. Histological Analysis
4.11. Immunohistochemical and Immunofluorescence Analysis
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| CCL | C-C motif chemokine ligand |
| DNCB | 2,4-dinirochlorobenzene |
| EBS | EGR1-binding sequence |
| EGR1 | early rrowth response 1 |
| shEgr1 | EGR1 shRNA |
| EMSA | electrophoretic mobility shift assay |
| ERK | Extracellular signal-regulated kinases |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| H&E | hematoxylin and eosin |
| Ig | immunoglobulin |
| IKK | inhibitor of κB kinase |
| IL | interleukin |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinases |
| NF-κB | nuclear factor-κB |
| Q-PCR | quantitative real-time PCR |
| RT-PCR | reverse-transcription polymerase chain reaction |
| shCT | scrambled shRNA |
| TB | toluidine blue |
| Th2 | T helper cell 2 |
| TNFα | tumor necrosis factor-alpha |
| TSLP | thymic stromal lymphopoietin |
References
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Maarouf, M.; Vaughn, A.R.; Shi, V.Y. Topical micronutrients in atopic dermatitis—An evidence-based review. Dermatol. Ther. 2018, 31, e12659. [Google Scholar] [CrossRef]
- Bekic, S.; Martinek, V.; Talapko, J.; Majnaric, L.; Vasilj Mihaljevic, M.; Skrlec, I. Atopic dermatitis and comorbidity. Healthcare 2020, 8, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettis, E.; Ortoncelli, M.; Pellacani, G.; Foti, C.; Di Leo, E.; Patruno, C.; Rongioletti, F.; Argenziano, G.; Ferrucci, S.M.; Macchia, L.; et al. A multicenter study on the prevalence of clinical patterns and clinical phenotypes in adult atopic dermatitis. J. Investig. Allergol. Clin. Immunol. 2020, 30, 448–450. [Google Scholar] [CrossRef]
- Smith, C.H. New approaches to topical therapy. Clin. Exp. Dermatol. 2000, 25, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Sidbury, R.; Hanifin, J.M. Systemic therapy of atopic dermatitis. Clin. Exp. Dermatol. 2000, 25, 559–566. [Google Scholar] [CrossRef]
- Cline, A.; Bartos, G.J.; Strowd, L.C.; Feldman, S.R. Biologic Treatment options for pediatric psoriasis and atopic dermatitis. Children 2019, 6, 103. [Google Scholar] [CrossRef] [Green Version]
- Welsch, K.; Holstein, J.; Laurence, A.; Ghoreschi, K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur. J. Immunol. 2017, 47, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Newsom, M.; Bashyam, A.M.; Balogh, E.A.; Feldman, S.R.; Strowd, L.C. New and emerging systemic treatments for atopic dermatitis. Drugs 2020, 80, 1041–1052. [Google Scholar] [CrossRef]
- Ring, J.; Alomar, A.; Bieber, T.; Deleuran, M.; Fink-Wagner, A.; Gelmetti, C.; Gieler, U.; Lipozencic, J.; Luger, T.; Oranje, A.P.; et al. Guidelines for treatment of atopic eczema (atopic dermatitis) part I. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1045–1060. [Google Scholar] [CrossRef]
- Dattola, A.; Bennardo, L.; Silvestri, M.; Nistico, S.P. What′s new in the treatment of atopic dermatitis? Dermatol. Ther. 2019, 32, e12787. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.C. Epidemiology of atopic dermatitis. Clin. Exp. Dermatol. 2000, 25, 522–529. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trier, A.M.; Kim, B.S. Cytokine modulation of atopic itch. Curr. Opin. Immunol. 2018, 54, 7–12. [Google Scholar] [CrossRef]
- Girolomoni, G.; Sebastiani, S.; Albanesi, C.; Cavani, A. T-cell subpopulations in the development of atopic and contact allergy. Curr. Opin. Immunol. 2001, 13, 733–737. [Google Scholar] [CrossRef]
- Chan, L.S.; Robinson, N.; Xu, L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: An experimental animal model to study atopic dermatitis. J. Investig. Dermatol. 2001, 117, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Zheng, T.; Oh, M.H.; Oh, S.Y.; Schroeder, J.T.; Glick, A.B.; Zhu, Z. Transgenic expression of interleukin-13 in the skin induces a pruritic dermatitis and skin remodeling. J. Investig. Dermatol. 2009, 129, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Cevikbas, F.; Wang, X.; Akiyama, T.; Kempkes, C.; Savinko, T.; Antal, A.; Kukova, G.; Buhl, T.; Ikoma, A.; Buddenkotte, J.; et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: Involvement of TRPV1 and TRPA1. J. Allergy Clin. Immunol. 2014, 133, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.E.; Williams, D.E.; Morrissey, P.J.; Garka, K.; Foxworthe, D.; Price, V.; Friend, S.L.; Farr, A.; Bedell, M.A.; Jenkins, N.A.; et al. Molecular cloning and biological characterization of a novel murine lymphoid growth factor. J. Exp. Med. 2000, 192, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Friend, S.L.; Hosier, S.; Nelson, A.; Foxworthe, D.; Williams, D.E.; Farr, A. A thymic stromal cell line supports in vitro development of surface IgM + B cells and produces a novel growth factor affecting B and T lineage cells. Exp. Hematol. 1994, 22, 321–328. [Google Scholar]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Liu, Y.J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.H.; Malefyt Rde, W.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Ann. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef]
- Rochman, I.; Watanabe, N.; Arima, K.; Liu, Y.J.; Leonard, W.J. Cutting edge: Direct action of thymic stromal lymphopoietin on activated human CD4 + T cells. J. Immunol. 2007, 178, 6720–6724. [Google Scholar] [CrossRef] [Green Version]
- Rochman, Y.; Leonard, W.J. The role of thymic stromal lymphopoietin in CD8 + T cell homeostasis. J. Immunol. 2008, 181, 7699–7705. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J. Thymic stromal lymphopoietin: Master switch for allergic inflammation. J. Exp. Med. 2006, 203, 269–273. [Google Scholar] [CrossRef]
- Watanabe, N.; Hanabuchi, S.; Soumelis, V.; Yuan, W.; Ho, S.; de Waal Malefyt, R.; Liu, Y.J. Human thymic stromal lymphopoietin promotes dendritic cell-mediated CD4 + T cell homeostatic expansion. Nat. Immunol. 2004, 5, 426–434. [Google Scholar] [CrossRef]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J. Allergy Clin. Immunol. 2007, 120, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Allakhverdi, Z.; Comeau, M.R.; Jessup, H.K.; Yoon, B.R.; Brewer, A.; Chartier, S.; Paquette, N.; Ziegler, S.F.; Sarfati, M.; Delespesse, G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007, 204, 253–258. [Google Scholar] [CrossRef]
- Wilson, S.R.; The, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef]
- Adhikary, P.P.; Tan, Z.; Page, B.D.G.; Hedtrich, S. TSLP as druggable target—A silver-lining for atopic diseases? Pharmacol. Ther. 2021, 217, 107648. [Google Scholar] [CrossRef]
- Matera, M.G.; Rogliani, P.; Calzetta, L.; Cazzola, M. TSLP inhibitors for asthma: Current status and future prospects. Drugs 2020, 80, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Takai, T. TSLP expression: Cellular sources, triggers, and regulatory mechanisms. Allergol. Int. 2012, 61, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, R.; Natesan, V. Chrysin: Sources, beneficial pharmacological activities, and molecular mechanism of action. Phytochemistry 2018, 145, 187–196. [Google Scholar] [CrossRef]
- Naz, S.; Imran, M.; Rauf, A.; Orhan, I.E.; Shariati, M.A.; Iahtisham, U.H.; Iqra, Y.; Shahbaz, M.; Qaisrani, T.B.; Shah, Z.A.; et al. Chrysin: Pharmacological and therapeutic properties. Life Sci. 2019, 235, 116797. [Google Scholar] [CrossRef]
- Choi, J.K.; Jang, Y.H.; Lee, S.; Lee, S.R.; Choi, Y.A.; Jin, M.; Choi, J.H.; Park, J.H.; Park, P.H.; Choi, H.; et al. Chrysin attenuates atopic dermatitis by suppressing inflammation of keratinocytes. Food Chem. Toxicol. 2017, 110, 142–150. [Google Scholar] [CrossRef]
- Song, H.Y.; Kim, W.S.; Mushtaq, S.; Park, J.M.; Choi, S.H.; Cho, J.W.; Lim, S.T.; Byun, E.B. A novel chrysin derivative produced by γ irradiation attenuates 2,4-dinitrochlorobenzene-induced atopic dermatitis-like skin lesions in Balb/c mice. Food Chem. Toxicol. 2019, 128, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Lee, Y.H.; Koh, D.; Lim, Y.; Shin, S.Y. Chrysin inhibits NF-κB-dependent CCL5 Transcription by targeting IκB kinase in the atopic dermatitis-like inflammatory microenvironment. Int. J. Mol. Sci. 2020, 21, 7348. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schottelius, A.J.; Zugel, U.; Docke, W.D.; Zollner, T.M.; Rose, L.; Mengel, A.; Buchmann, B.; Becker, A.; Grutz, G.; Naundorf, S.; et al. The role of mitogen-activated protein kinase-activated protein kinase 2 in the p38/TNF-α pathway of systemic and cutaneous inflammation. J. Investig. Dermatol. 2010, 130, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Yeo, H.; Ahn, S.S.; Lee, J.Y.; Jung, E.; Jeong, M.; Kang, G.S.; Ahn, S.; Lee, Y.; Koh, D.; Lee, Y.H.; et al. Disrupting the DNA binding of EGR-1 with a small-molecule inhibitor ameliorates 2,4-dinitrochlorobenzene-induced skin inflammation. J. Investig. Dermatol. 2021, in press. [Google Scholar] [CrossRef]
- Redhu, N.S.; Saleh, A.; Halayko, A.J.; Ali, A.S.; Gounni, A.S. Essential role of NF-κB and AP-1 transcription factors in TNF-α-induced TSLP expression in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L479–L485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cultrone, A.; de Wouters, T.; Lakhdari, O.; Kelly, D.; Mulder, I.; Logan, E.; Lapaque, N.; Dore, J.; Blottiere, H.M. The NF-κB binding site located in the proximal region of the TSLP promoter is critical for TSLP modulation in human intestinal epithelial cells. Eur. J. Immunol. 2013, 43, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.I.; Lee, H.; Kim, J.H.; Bae, H.C.; Ryu, H.J.; Son, S.W. IL-33 induces Egr-1-dependent TSLP expression via the MAPK pathways in human keratinocytes. Exp. Dermatol. 2015, 24, 857–863. [Google Scholar] [CrossRef]
- Kitagaki, H.; Fujisawa, S.; Watanabe, K.; Hayakawa, K.; Shiohara, T. Immediate-type hypersensitivity response followed by a late reaction is induced by repeated epicutaneous application of contact sensitizing agents in mice. J. Investig. Dermatol. 1995, 105, 749–755. [Google Scholar] [CrossRef] [Green Version]
- Ando, T.; Matsumoto, K.; Namiranian, S.; Yamashita, H.; Glatthorn, H.; Kimura, M.; Dolan, B.R.; Lee, J.J.; Galli, S.J.; Kawakami, Y.; et al. Mast cells are required for full expression of allergen/SEB-induced skin inflammation. J. Investig. Dermatol. 2013, 133, 2695–2705. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, G.; Shankar, A.A. Toluidine blue: A review of its chemistry and clinical utility. J. Oral Maxillofac. Pathol. 2012, 16, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Utreras, E.; Futatsugi, A.; Rudrabhatla, P.; Keller, J.; Iadarola, M.J.; Pant, H.C.; Kulkarni, A.B. Tumor necrosis factor-α regulates cyclin-dependent kinase 5 activity during pain signaling through transcriptional activation of p35. J. Biol. Chem. 2009, 284, 2275–2284. [Google Scholar] [CrossRef] [Green Version]
- Mishra, J.P.; Mishra, S.; Gee, K.; Kumar, A. Differential involvement of calmodulin-dependent protein kinase II-activated AP-1 and c-Jun N-terminal kinase-activated EGR-1 signaling pathways in tumor necrosis factor-α and lipopolysaccharide-induced CD44 expression in human monocytic cells. J. Biol. Chem. 2005, 280, 26825–26837. [Google Scholar] [CrossRef] [Green Version]
- Son, S.W.; Min, B.W.; Lim, Y.; Lee, Y.H.; Shin, S.Y. Regulatory mechanism of TNFα autoregulation in HaCaT cells: The role of the transcription factor EGR-1. Biochem. Biophys. Res. Commun. 2008, 374, 777–782. [Google Scholar] [CrossRef]
- Li, M.; Hener, P.; Zhang, Z.; Kato, S.; Metzger, D.; Chambon, P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl. Acad. Sci. USA 2006, 103, 11736–11741. [Google Scholar] [CrossRef] [Green Version]
- Pavletich, N.P.; Pabo, C.O. Zinc finger-DNA recognition: Crystal structure of a Zif268-DNA complex at 2.1 A. Science 1991, 252, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Milbrandt, J. A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science 1987, 238, 797–799. [Google Scholar] [CrossRef]
- Gashler, A.; Sukhatme, V.P. Early growth response protein 1 (Egr-1): Prototype of a zinc-finger family of transcription factors. Prog. Nucleic Acid Res. Mol. Biol. 1995, 50, 191–224. [Google Scholar] [PubMed]
- Duclot, F.; Kabbaj, M. The role of early growth response 1 (EGR1) in brain plasticity and neuropsychiatric disorders. Front. Behav. Neurosci. 2017, 11, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, S.B.; Monroe, J.G. The role of early growth response gene 1 (egr-1) in regulation of the immune response. J. Leukoc. Biol. 1996, 60, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Liu, M.R.; Pei, D.S. Friend or foe, the role of EGR-1 in cancer. Med. Oncol. 2019, 37, 7. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Fang, F.; Tourtellotte, W.; Varga, J. Egr-1: New conductor for the tissue repair orchestra directs harmony (regeneration) or cacophony (fibrosis). J. Pathol. 2013, 229, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Bryant, M.; Drew, G.M.; Houston, P.; Hissey, P.; Campbell, C.J.; Braddock, M. Tissue repair with a therapeutic transcription factor. Hum. Gene Ther. 2000, 11, 2143–2158. [Google Scholar] [CrossRef]
- Jeong, S.H.; Kim, H.J.; Jang, Y.; Ryu, W.I.; Lee, H.; Kim, J.H.; Bae, H.C.; Choi, J.E.; Kye, Y.C.; Son, S.W. Egr-1 is a key regulator of IL-17A-induced psoriasin upregulation in psoriasis. Exp. Dermatol. 2014, 23, 890–895. [Google Scholar] [CrossRef]
- Yeo, H.; Ahn, S.S.; Lee, J.Y.; Shin, S.Y. EGR-1 acts as a transcriptional activator of KLK7 under IL-13 stimulation. Biochem. Biophys. Res. Commun. 2021, 534, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [Green Version]
- An, E.J.; Kim, Y.; Lee, S.H.; Choi, S.H.; Chung, W.S.; Jang, H.J. Ophiopogonin D ameliorates DNCB-induced atopic dermatitis-like lesions in BALB/c mice and TNF-α- inflamed HaCaT cell. Biochem. Biophys. Res. Commun. 2020, 522, 40–46. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, H.; Lee, Y.H.; Ahn, S.S.; Jung, E.; Lim, Y.; Shin, S.Y. Chrysin Inhibits TNFα-Induced TSLP Expression through Downregulation of EGR1 Expression in Keratinocytes. Int. J. Mol. Sci. 2021, 22, 4350. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094350
Yeo H, Lee YH, Ahn SS, Jung E, Lim Y, Shin SY. Chrysin Inhibits TNFα-Induced TSLP Expression through Downregulation of EGR1 Expression in Keratinocytes. International Journal of Molecular Sciences. 2021; 22(9):4350. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094350
Chicago/Turabian StyleYeo, Hyunjin, Young Han Lee, Sung Shin Ahn, Euitaek Jung, Yoongho Lim, and Soon Young Shin. 2021. "Chrysin Inhibits TNFα-Induced TSLP Expression through Downregulation of EGR1 Expression in Keratinocytes" International Journal of Molecular Sciences 22, no. 9: 4350. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094350