
Development of a Robust High-Throughput Screening Platform for Inhibitors of the Striatal-Enriched Tyrosine Phosphatase (STEP)

 ,
,
Abstract
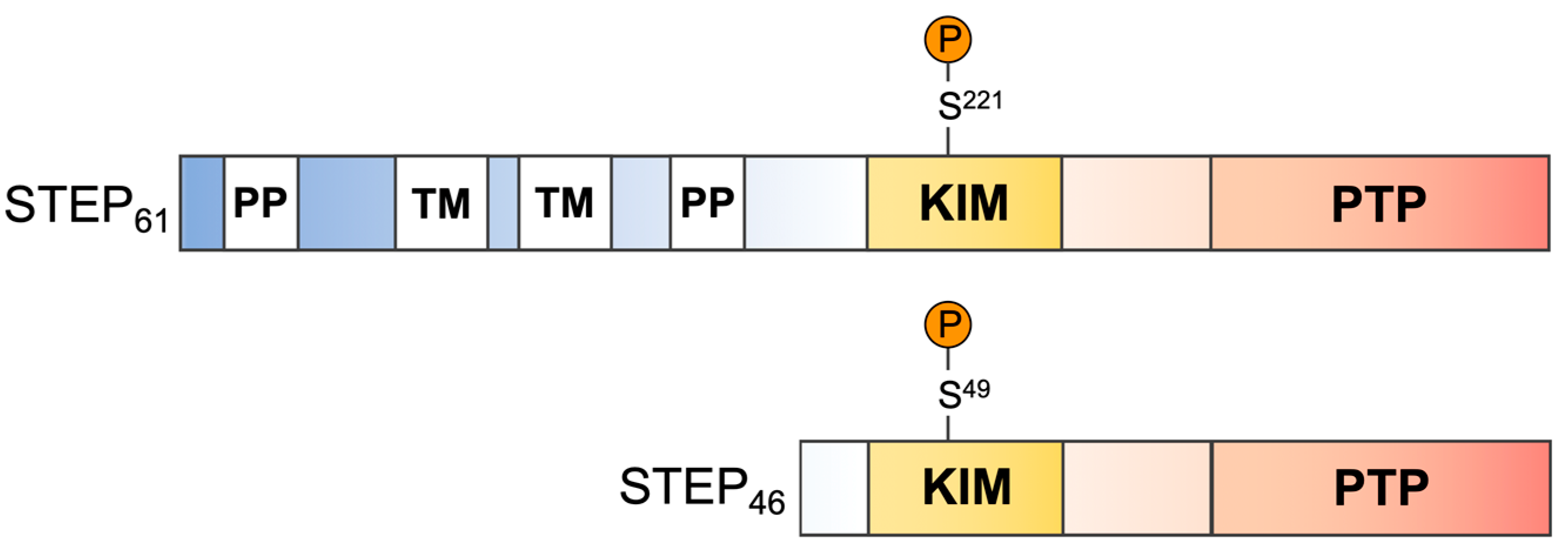
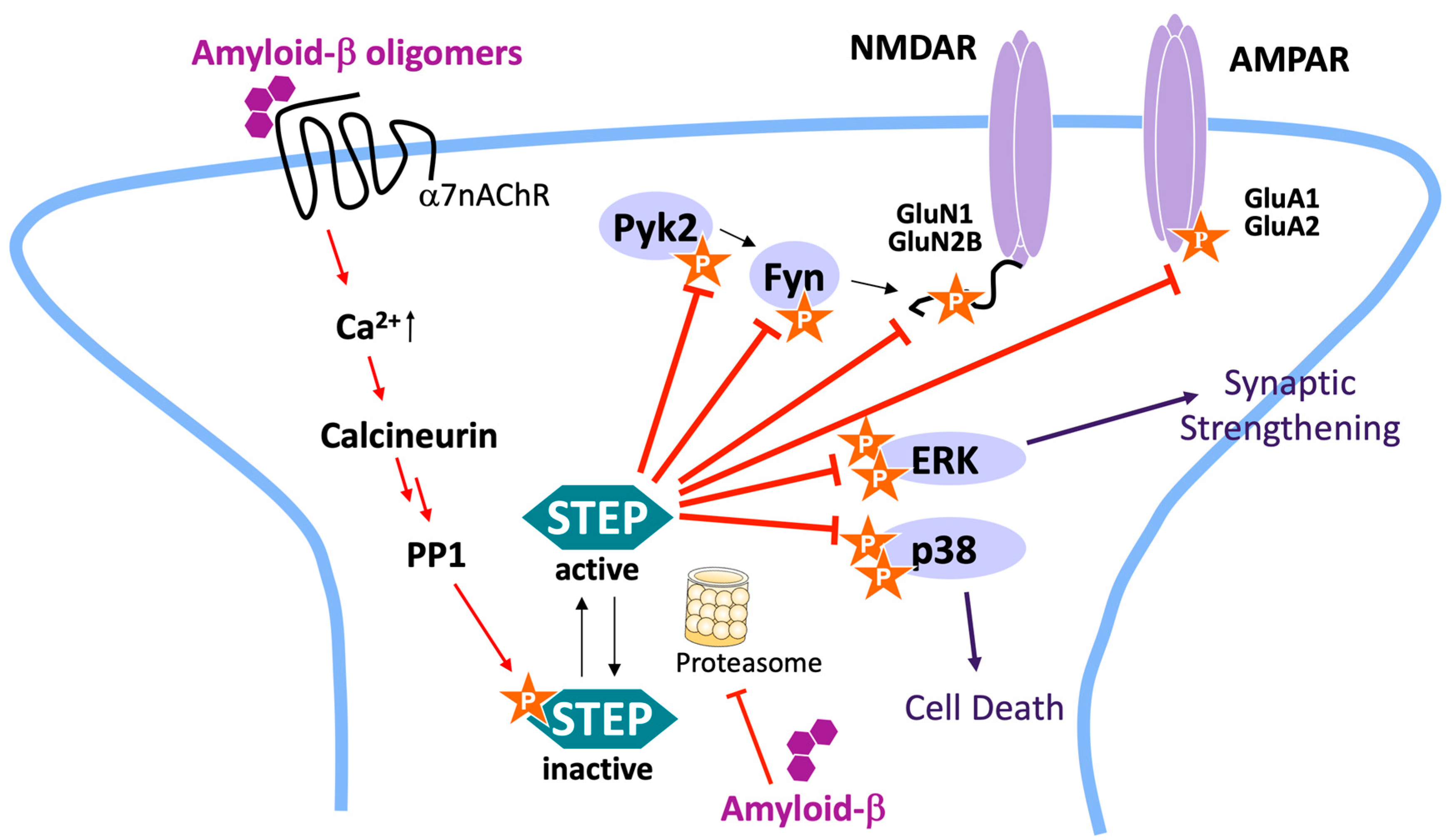
:1. Introduction
2. Results
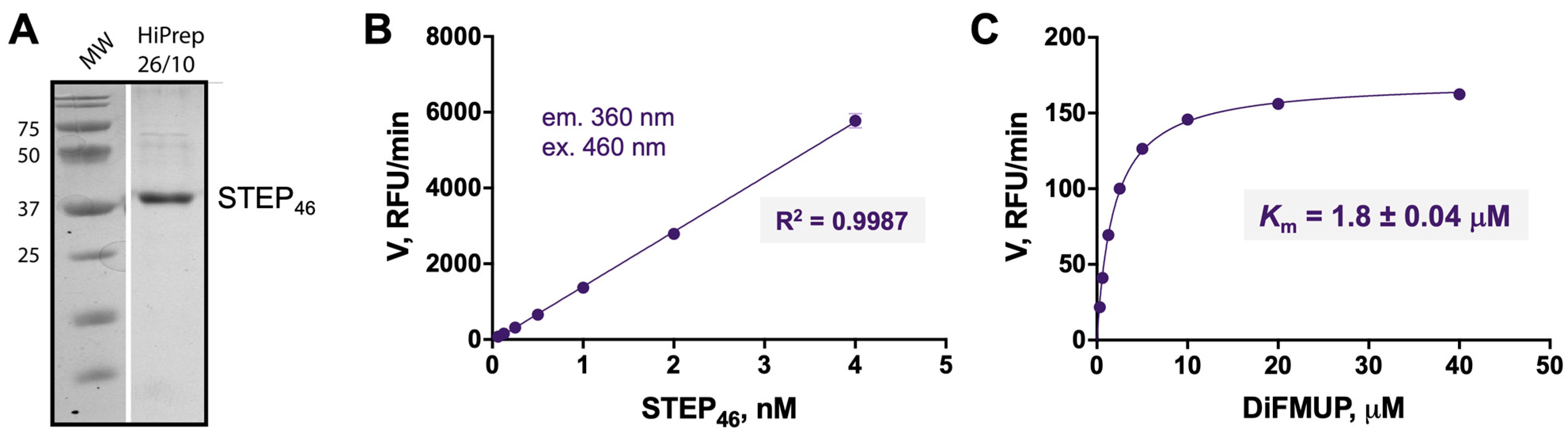
2.1. Production and Enzymatic Characterization of Recombinant Full-Length STEP46
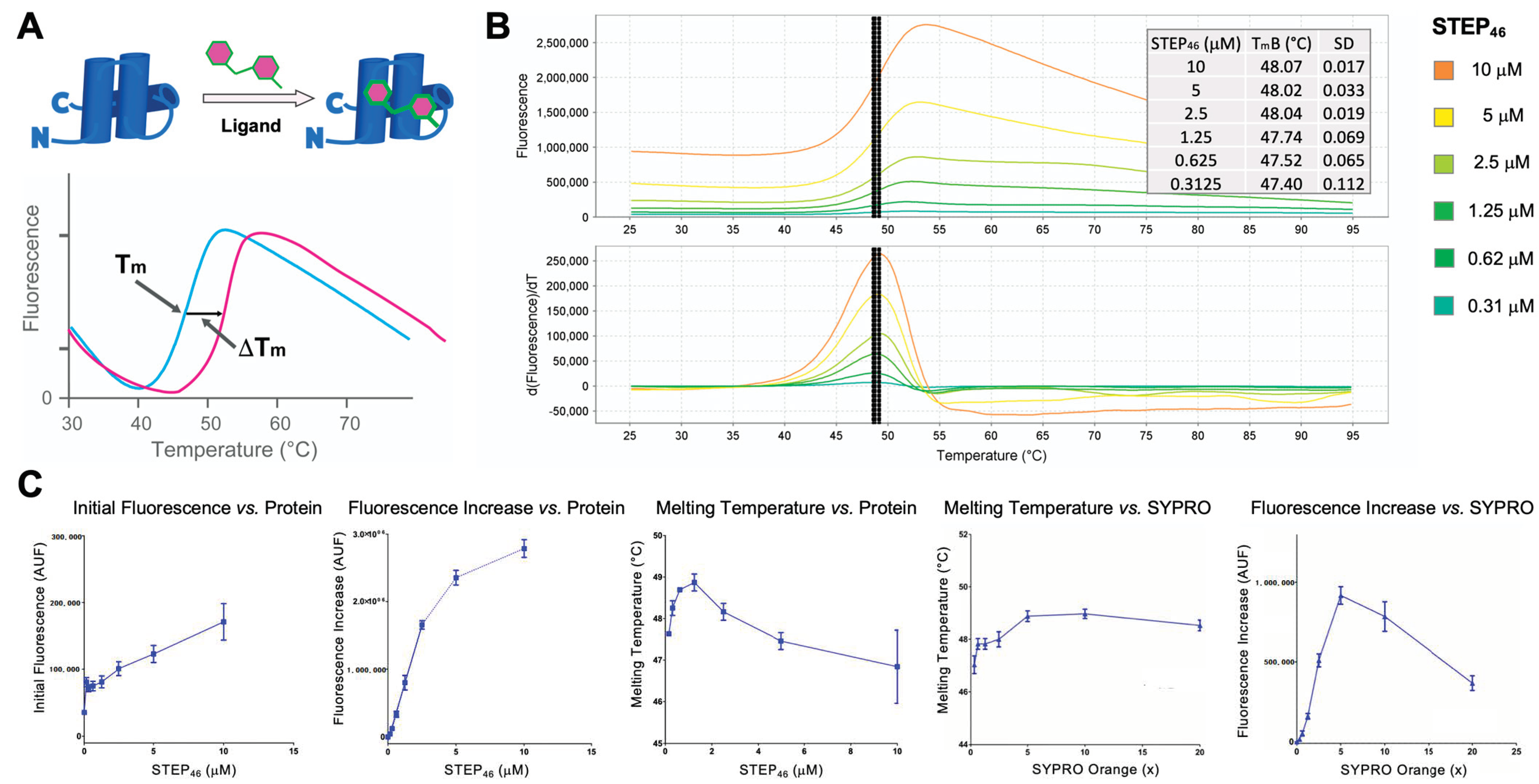
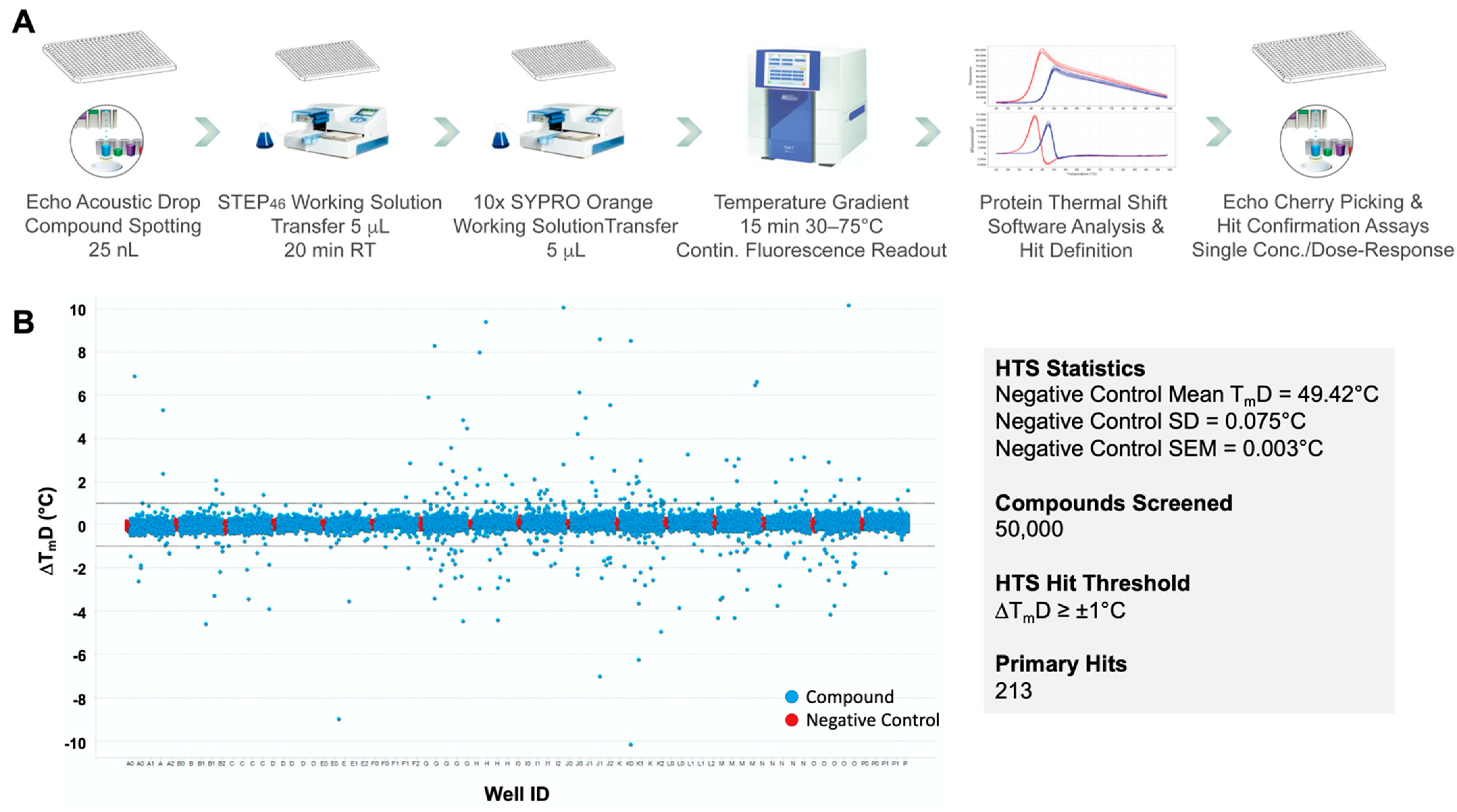
2.2. STEP46 Protein Thermal Shift (PTS) Assay Development
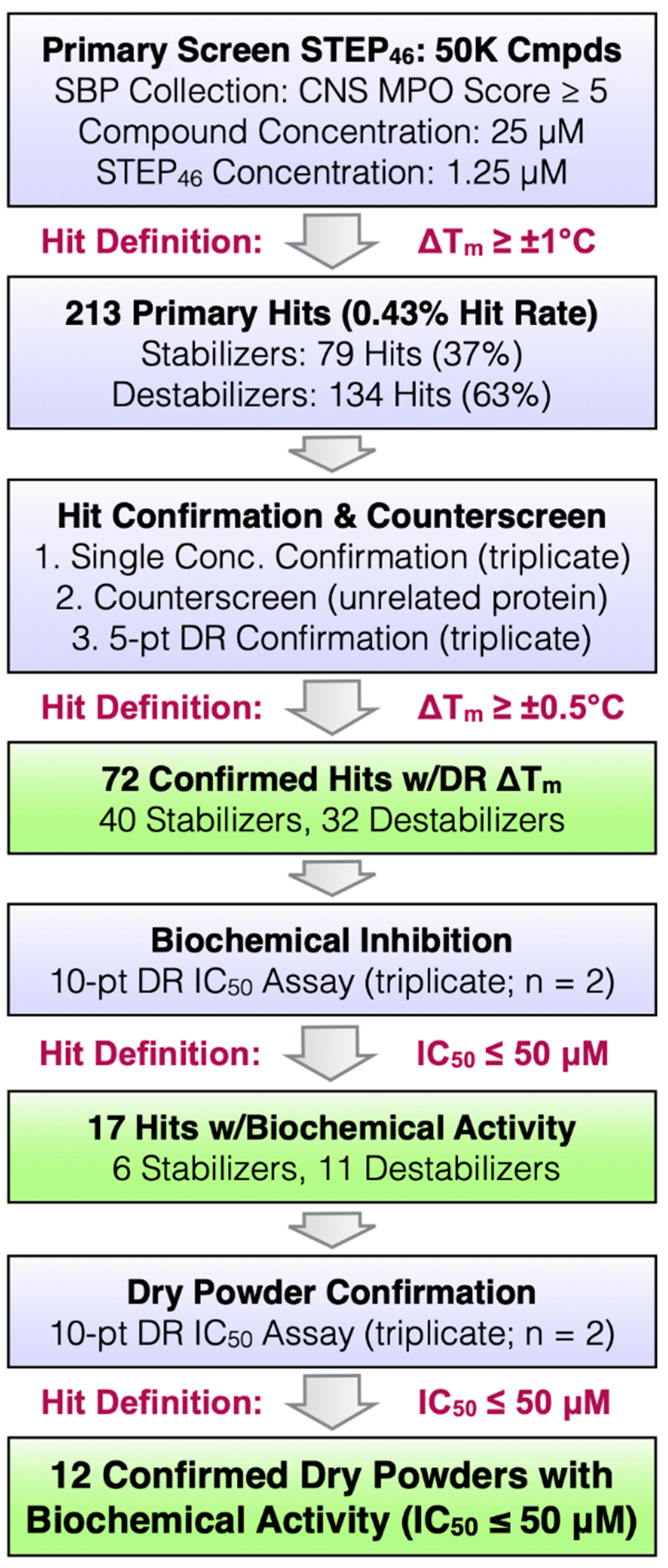
2.3. STEP46 HTS and Hit Confirmation Using the PTS Binding Assay
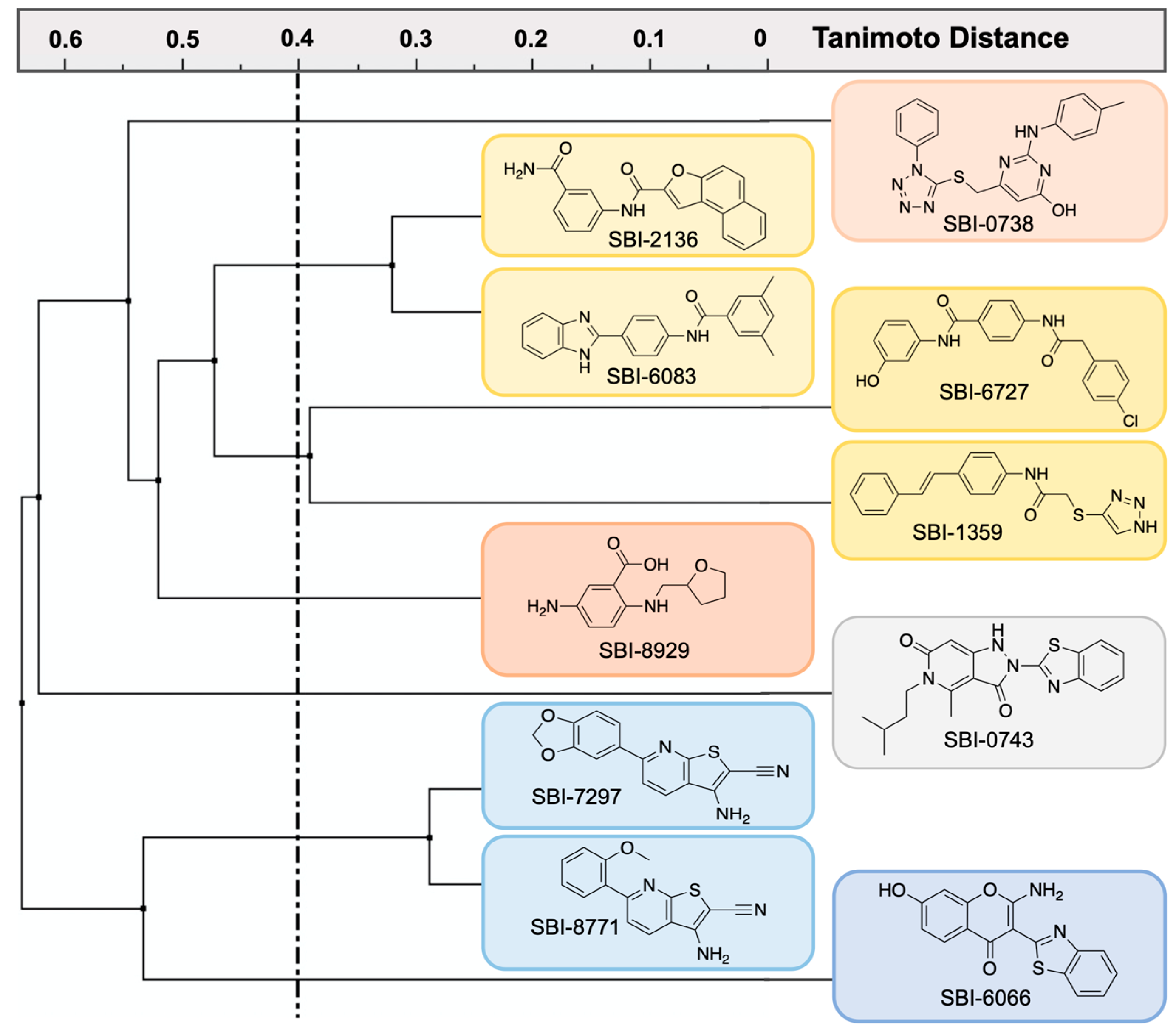
2.4. Biochemical Characterization of Confirmed STEP46 PTS Hits
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Protein Thermal Shift Assays (PTS)
4.3. Selection of 50K Small Molecules for HTS
4.4. STEP46 HTS of 50K Small Molecules
4.5. STEP46 and PTP1B Michaelis-Menten Kinetic Assays
4.6. STEP46 and PTP1B Biochemical Inhibition Assays
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β peptide |
| AD | Alzheimer’s disease |
| ADME | absorption, distribution, metabolism, and excretion |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic a |
| BSA | bovine serum albumin |
| CNS | central nervous system |
| DiFMUP | 6,8-difluoro-4-methylumbelliferyl phosphate |
| DMSO | dimethyl sulfoxide |
| DTT | dithiothreitol |
| ECFP | extended-connectivity FingerPrint |
| ERK | extracellular signal-regulated kinase |
| HTS | high-throughput screening |
| KIM | kinase-interaction motif |
| KO | knockout |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAPK | mitogen-activated protein kinase |
| Ni-NTA | nickel-nitrilotriacetic acid |
| NMDA | N-methyl-D-aspartate |
| PP1 | protein phosphatase 1 |
| PROTAC | proteolysis targeting chimeras |
| PSD | postsynaptic density |
| PTK | protein tyrosine kinase |
| PTP | protein tyrosine phosphatase |
| PTS | protein thermal shift |
| RT | room temperature |
| SAR | structure activity relationship |
| SD | standard deviation |
| SEM | standard error of the mean |
| STEP | striatal-enriched tyrosine phosphatase |
| TCEP | tris(2-carboxyethyl)phosphine |
| TKI | tyrosine kinase inhibitor |
| Tm | melting temperature |
References
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [Green Version]
- He, R.-J.; Yu, Z.-H.; Zhang, R.-Y.; Zhang, Z.-Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, S.M.; Bottini, N. Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci. 2017, 38, 524–540. [Google Scholar] [CrossRef]
- Cohen, P.; Alessi, D.R. Kinase drug discovery—What’s next in the field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialy, L.; Waldmann, H. Inhibitors of Protein Tyrosine Phosphatases: Next-Generation Drugs? Angew. Chem. Int. Ed. 2005, 44, 3814–3839. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Knapp, S.; Sundström, M. Recently targeted kinases and their inhibitors—The path to clinical trials. Curr. Opin. Pharmacol. 2014, 17, 58–63. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. FEBS J. 2012, 280, 346–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tautz, L.; Critton, D.; Grotegut, S. Protein tyrosine phosphatases: Structure, function, and implication in human disease. Methods Mol. Biol. 2013, 1053, 179–221. [Google Scholar]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer Disease. Dis. Mon. 2010, 56, 484–546. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Kirkitadze, M.D.; Bitan, G.; Teplow, D.B. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res. 2002, 69, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teich, A.F.; Arancio, O. Is the Amyloid Hypothesis of Alzheimer’s disease therapeutically relevant? Biochem. J. 2012, 446, 165–177. [Google Scholar] [CrossRef]
- Becker, R.E.; Greig, N.H.; Giacobini, E.; Schneider, L.S.; Ferrucci, L. A new roadmap for drug development for Alzheimer’s disease. Nat. Rev. Drug Discov. 2014, 13, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [Green Version]
- Lombroso, P.J.; Murdoch, G.; Lerner, M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc. Natl. Acad. Sci. USA 1991, 88, 7242–7246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombroso, P.J.; Ogren, M.; Kurup, P.; Nairn, A.C. Molecular underpinnings of neurodegenerative disorders: Striatal-enriched protein tyrosine phosphatase signaling and synaptic plasticity. F1000Research 2016, 5, 2932. [Google Scholar] [CrossRef]
- Goebel-Goody, S.M.; Baum, M.; Paspalas, C.D.; Fernandez, S.M.; Carty, N.C.; Kurup, P.; Lombroso, P.J. Therapeutic Implications for Striatal-Enriched Protein Tyrosine Phosphatase (STEP) in Neuropsychiatric Disorders. Pharmacol. Rev. 2011, 64, 65–87. [Google Scholar] [CrossRef]
- Paul, S.; Nairn, A.C.; Wang, P.; Lombroso, P.J. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 2002, 6, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.J.; Tarrega, C.; Blanco-Aparicio, C.; Pulido, R. Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38alpha is determined by a kinase specificity sequence and influenced by reducing agents. Biochem. J. 2003, 372, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.-H.; Liu, J.; Lombroso, P.J. Striatal Enriched Phosphatase 61 Dephosphorylates Fyn at Phosphotyrosine 420. J. Biol. Chem. 2002, 277, 24274–24279. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kurup, P.; Bartos, J.A.; Patriarchi, T.; Hell, J.W.; Lombroso, P.J. Striatal-enriched Protein-tyrosine Phosphatase (STEP) Regulates Pyk2 Kinase Activity. J. Biol. Chem. 2012, 287, 20942–20956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, E.; Nong, Y.; Almeida, C.; Paul, S.; Moran, T.; Choi, E. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Xu, J.; Kurup, P.; Zhang, Y.; Goebel-Goody, S.M.; Wu, P.H.; Hawasli, A.H.; Baum, M.L.; Bibb, J.A.; Lombroso, P.J. Extrasynaptic NMDA Receptors Couple Preferentially to Excitotoxicity via Calpain-Mediated Cleavage of STEP. J. Neurosci. 2009, 29, 9330–9343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurup, P.; Xu, J.; Carty, N.; Fernandez, S.M.; Nygaard, H.B.; Pittenger, C.; Greengard, P.; Strittmatter, S.M.; Nairn, A.C.; et al. Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 19014–19019. [Google Scholar] [CrossRef] [Green Version]
- Moult, P.R.; Gladding, C.M.; Sanderson, T.M.; Fitzjohn, S.M.; Bashir, Z.I.; Molnár, E.; Collingridge, G.L. Tyrosine Phosphatases Regulate AMPA Receptor Trafficking during Metabotropic Glutamate Receptor-Mediated Long-Term Depression. J. Neurosci. 2006, 26, 2544–2554. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.-X.; Zhang, Z.-Y. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008, 27, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Snyder, G.L.; Yokakura, H.; Picciotto, M.R.; Nairn, A.C.; Lombroso, P.J. The Dopamine/D1 Receptor Mediates the Phosphorylation and Inactivation of the Protein Tyrosine Phosphatase STEP via a PKA-Dependent Pathway. J. Neurosci. 2000, 20, 5630–5638. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kurup, P.; Nairn, A.C.; Lombroso, P.J. Striatal-enriched protein tyrosine phosphatase in Alzheimer’s disease. HIV-1 Mol. Boil. Pathog. 2012, 64, 303–325. [Google Scholar] [CrossRef] [Green Version]
- Kurup, P.K.; Xu, J.; Videira, R.A.; Ononenyi, C.; Baltazar, G.; Lombroso, P.J.; Nairn, A.C. STEP61is a substrate of the E3 ligase parkin and is upregulated in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, 1202–1207. [Google Scholar] [CrossRef] [Green Version]
- Carty, N.C.; Xu, J.; Kurup, P.; Brouillette, J.; Goebel-Goody, S.M.; Austin, D.R.; Yuan, P.; Chen, G.; Correa, P.R.; Haroutunian, V.; et al. The tyrosine phosphatase STEP: Implications in schizophrenia and the molecular mechanism underlying antipsychotic medications. Transl. Psychiatry 2012, 2, e137. [Google Scholar] [CrossRef] [Green Version]
- Goebel-Goody, S.M.; Wilson-Wallis, E.D.; Royston, S.; Tagliatela, S.M.; Naegele, J.R.; Lombroso, P.J. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 2012, 11, 586–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurup, P.; Zhang, Y.; Xu, J.; Venkitaramani, D.; Haroutunian, V.; Greengard, P. Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J. Neurosci. 2010, 30, 5948–5957. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kurup, P.; Xu, J.; Anderson, G.M.; Greengard, P.; Nairn, A.C.; Lombroso, P.J. Reduced levels of the tyrosine phosphatase STEP block beta amyloid-mediated GluA1/GluA2 receptor internalization. J. Neurochem. 2011, 119, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Venkitaramani, D.V.; Gladding, C.M.; Zhang, Y.; Kurup, P.; Molnar, E.; Collingridge, G.L.; Lombroso, P.J. The Tyrosine Phosphatase STEP Mediates AMPA Receptor Endocytosis after Metabotropic Glutamate Receptor Stimulation. J. Neurosci. 2008, 28, 10561–10566. [Google Scholar] [CrossRef]
- Venkitaramani, D.; Paul, S.; Zhang, Y.; Kurup, P.; Ding, L.; Tressler, L. Knockout of striatal enriched protein tyrosine phosphatase in mice results in increased ERK1/2 phosphorylation. Synapse 2009, 63, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkitaramani, D.V.; Moura, P.J.; Picciotto, M.R.; Lombroso, P.J. Striatal-enriched protein tyrosine phosphatase (STEP) knockout mice have enhanced hippocampal memory. Eur. J. Neurosci. 2011, 33, 2288–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Chatterjee, M.; Baguley, T.D.; Brouillette, J.; Kurup, P.; Ghosh, D.; Kanyo, J.; Zhang, Y.; Seyb, K.; Ononenyi, C.; et al. Inhibitor of the Tyrosine Phosphatase STEP Reverses Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. PLoS Biol. 2014, 12, e1001923. [Google Scholar] [CrossRef] [Green Version]
- Chatterji, T.; Gates, K.S. DNA cleavage by 7-methylbenzopentathiepin: A simple analog of the antitumor antibiotic varacin. Bioorg. Med. Chem. Lett. 1998, 8, 535–538. [Google Scholar] [CrossRef]
- Lee, A.H.F.; Chan, A.S.C.; Li, T. Acid-accelerated DNA-cleaving activities of antitumor antibiotic varacin. Chem. Commun. 2002, 2002, 2112–2113. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.F.; Chen, J.; Liu, N.; Leung, T.Y.C.; Chan, A.S.C.; Li, T. Acid-Promoted DNA-Cleaving Activities and Total Synthesis of Varacin, C.J. Am. Chem. Soc. 2002, 124, 13972–13973. [Google Scholar] [CrossRef] [PubMed]
- Greer, A. On the Origin of Cytotoxicity of the Natural Product Varacin. A Novel Example of a Pentathiepin Reaction That Provides Evidence for a Triatomic Sulfur Intermediate. J. Am. Chem. Soc. 2001, 123, 10379–10386. [Google Scholar] [CrossRef]
- Witten, M.R.; Wissler, L.; Snow, M.; Geschwindner, S.; Read, J.A.; Brandon, N.J.; Nairn, A.C.; Lombroso, P.J.; Käck, H.; Ellman, J.A. X-ray Characterization and Structure-Based Optimization of Striatal-Enriched Protein Tyrosine Phosphatase Inhibitors. J. Med. Chem. 2017, 60, 9299–9319. [Google Scholar] [CrossRef] [PubMed]
- Tautz, L.; Mustelin, T. Strategies for developing protein tyrosine phosphatase inhibitors. Methods 2007, 42, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Welte, S.; Baringhaus, K.-H.; Schmider, W.; Müller, G.; Petry, S.; Tennagels, N. 6,8-Difluoro-4-methylumbiliferyl phosphate: A fluorogenic substrate for protein tyrosine phosphatases. Anal. Biochem. 2005, 338, 32–38. [Google Scholar] [CrossRef]
- Ericsson, U.B.; Hallberg, B.M.; DeTitta, G.T.; Dekker, N.; Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006, 357, 289–298. [Google Scholar] [CrossRef]
- Cummings, M.D.; Farnum, M.A.; Nelen, M.I. Universal Screening Methods and Applications of ThermoFluor®. J. Biomol. Screen. 2006, 11, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach to Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nat. Cell Biol. 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Seetoh, W.G.; Abell, C. Disrupting the Constitutive, Homodimeric Protein-Protein Interface in CK2beta Using a Biophysical Fragment-Based Approach. J. Am. Chem. Soc. 2016, 138, 14303–14311. [Google Scholar] [CrossRef] [Green Version]
- Dai, R.; Wilson, D.J.; Geders, T.W.; Aldrich, C.C.; Finzel, B.C. Inhibition of My cobacterium tuberculosis Transaminase BioA by Aryl Hydrazines and Hydrazides. ChemBioChem 2014, 15, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Tautz, L.; Pellecchia, M.; Mustelin, T. Targeting the PTPome in human disease. Expert Opin. Ther. Targets 2006, 10, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Vintonyak, V.V.; Antonchick, A.P.; Rauh, D.; Waldmann, H. The therapeutic potential of phosphatase inhibitors. Curr. Opin. Chem. Biol. 2009, 13, 272–283. [Google Scholar] [CrossRef]
- Barr, A.J. Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Med. Chem. 2010, 2, 1563–1576. [Google Scholar] [CrossRef]
- He, R.; Zeng, L.-F.; He, Y.; Zhang, S.; Zhang, Z.-Y. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2012, 280, 731–750. [Google Scholar] [CrossRef]
- Köhn, M. Turn and Face the Strange: A New View on Phosphatases. ACS Cent. Sci. 2020, 6, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tautermann, C.S.; Binder, F.; Büttner, F.H.; Eickmeier, C.; Fiegen, D.; Gross, U.; Grundl, M.A.; Heilker, R.; Hobson, S.; Hoerer, S.; et al. Allosteric Activation of Striatal-Enriched Protein Tyrosine Phosphatase (STEP, PTPN5) by a Fragment-like Molecule. J. Med. Chem. 2018, 62, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.-M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-N.P.; Lamarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nat. Cell Biol. 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Romero, C.; Lambert, L.J.; Sheffler, D.J.; De Backer, L.J.; Raveendra-Panickar, D.; Celeridad, M.; Grotegut, S.; Rodiles, S.; Holleran, J.; Sergienko, E.; et al. A cellular target engagement assay for the characterization of SHP2 (PTPN11) phosphatase inhibitors. J. Biol. Chem. 2020, 295, 2601–2613. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance ID | Structure | STEP46 IC50, µM | STEP IC50 Curve, µM | PTP1B IC50, µM | PTP1B IC50 Curve, µM |
|---|---|---|---|---|---|
| SBI-6066 |  | 5.0 |  | >100 |  |
| SBI-2136 |  | 5.3 |  | 14 |  |
| SBI-6083 |  | 5.4 |  | >100 |  |
| SBI-0743 |  | 5.5 |  | 22 |  |
| SBI-0738 |  | 5.7 |  | 6.1 |  |
| SBI-1359 |  | 7.0 |  | 28 |  |
| SBI-8771 |  | 16 |  | 79 |  |
| SBI-6727 |  | 18 |  | 57 |  |
| SBI-7297 |  | 32 |  | >100 |  |
| SBI-8929 |  | 33 |  | 20 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert, L.J.; Grotegut, S.; Celeridad, M.; Gosalia, P.; Backer, L.J.D.; Bobkov, A.A.; Salaniwal, S.; Chung, T.D.; Zeng, F.-Y.; Pass, I.; et al. Development of a Robust High-Throughput Screening Platform for Inhibitors of the Striatal-Enriched Tyrosine Phosphatase (STEP). Int. J. Mol. Sci. 2021, 22, 4417. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094417
Lambert LJ, Grotegut S, Celeridad M, Gosalia P, Backer LJD, Bobkov AA, Salaniwal S, Chung TD, Zeng F-Y, Pass I, et al. Development of a Robust High-Throughput Screening Platform for Inhibitors of the Striatal-Enriched Tyrosine Phosphatase (STEP). International Journal of Molecular Sciences. 2021; 22(9):4417. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094417
Chicago/Turabian StyleLambert, Lester J, Stefan Grotegut, Maria Celeridad, Palak Gosalia, Laurent JS De Backer, Andrey A Bobkov, Sumeet Salaniwal, Thomas DY Chung, Fu-Yue Zeng, Ian Pass, and et al. 2021. "Development of a Robust High-Throughput Screening Platform for Inhibitors of the Striatal-Enriched Tyrosine Phosphatase (STEP)" International Journal of Molecular Sciences 22, no. 9: 4417. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094417