
NEIL1 and NEIL2 Are Recruited as Potential Backup for OGG1 upon OGG1 Depletion or Inhibition by TH5487


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
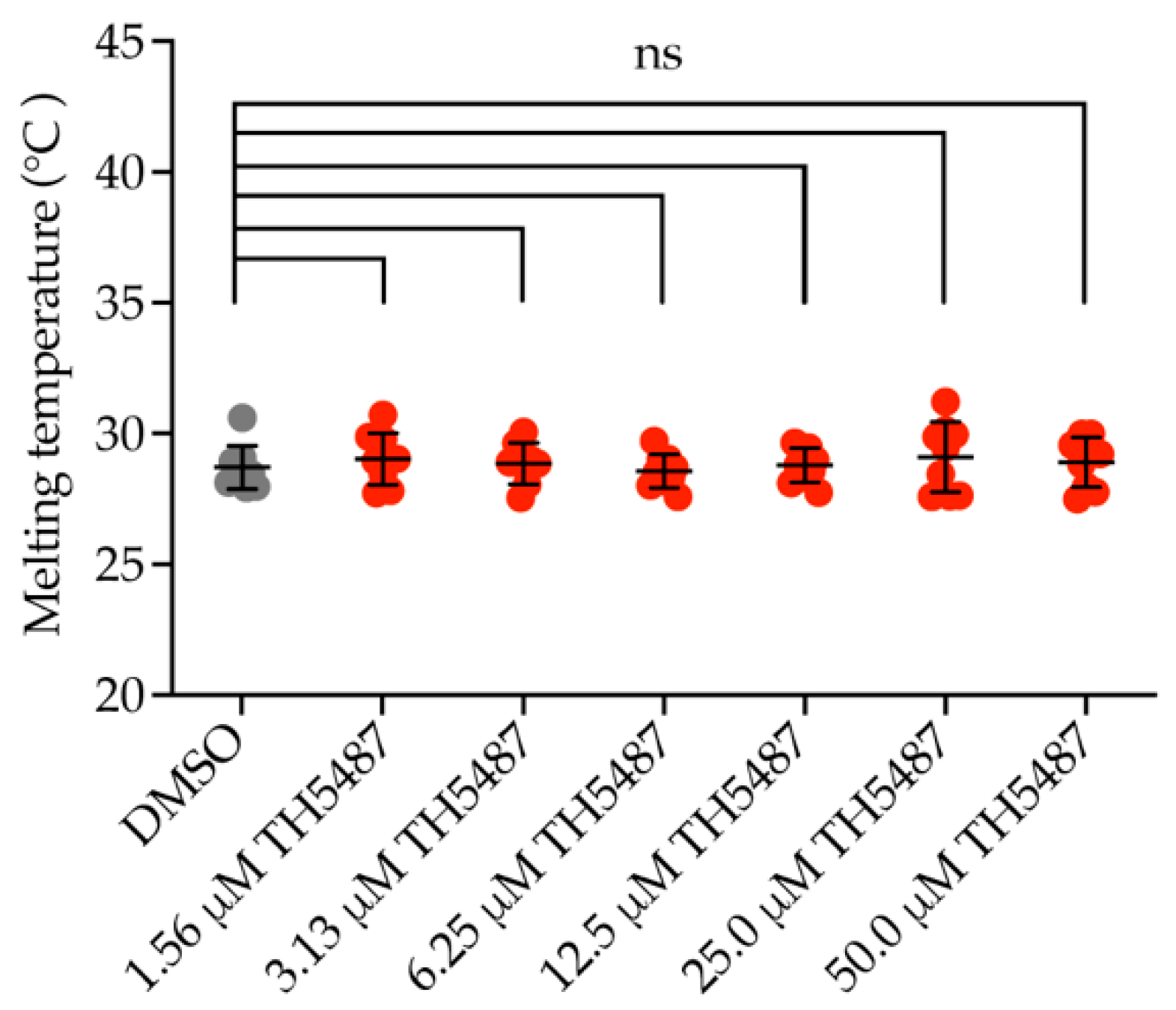
2.1. OGG1 Inhibitor TH5487 Does Not Bind to NEIL1
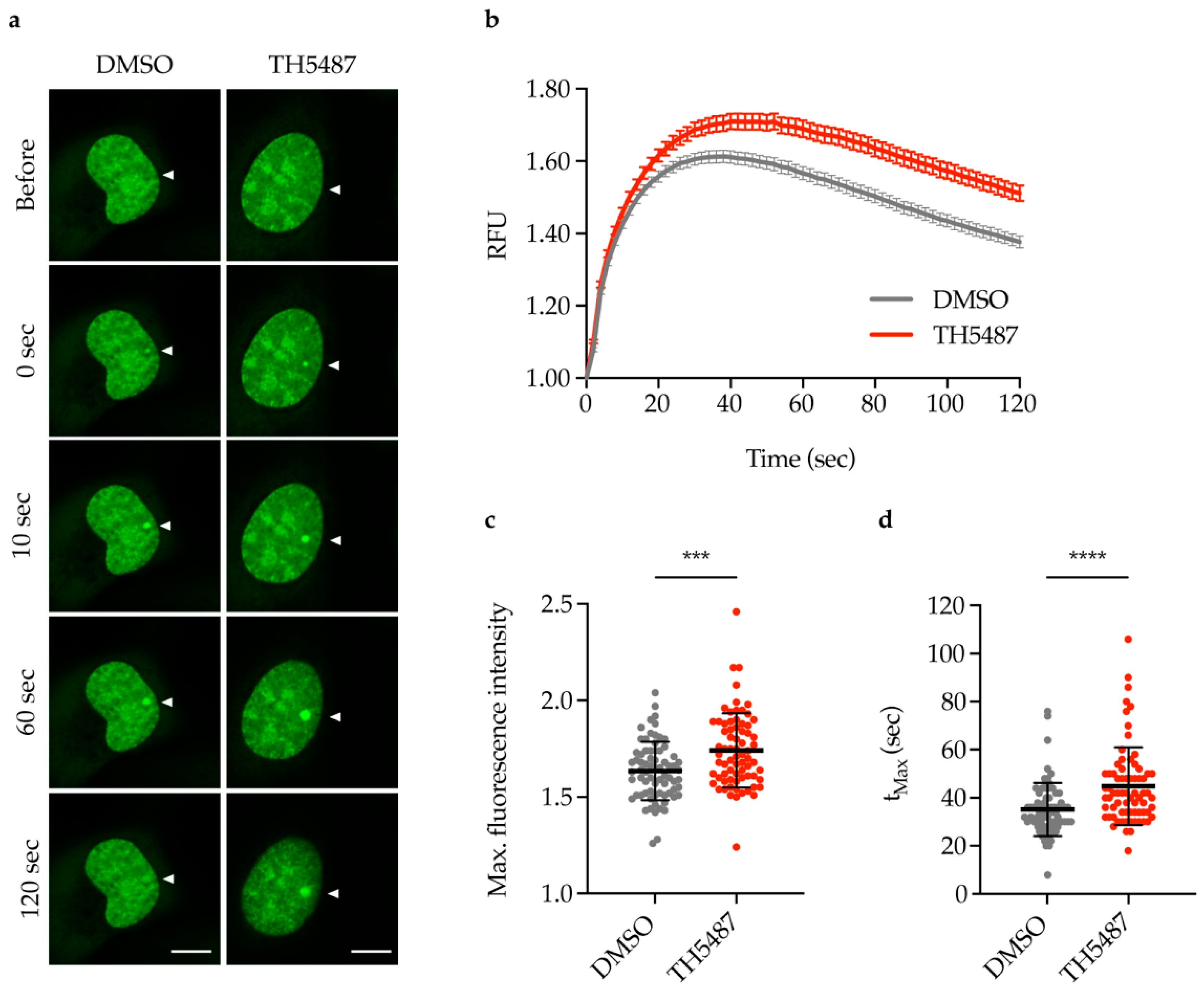
2.2. TH5487 Treatment Results in Increased Recruitment of NEIL1-GFP to DNA Damage Sites
2.3. NEIL1-Chromatin Binding Increases upon DNA Damage Induction and TH5487 Treatment
2.4. NEIL1 Retention Is Increased in a Dose-Dependent Manner upon DNA Damage Induction and TH5487 Treatment
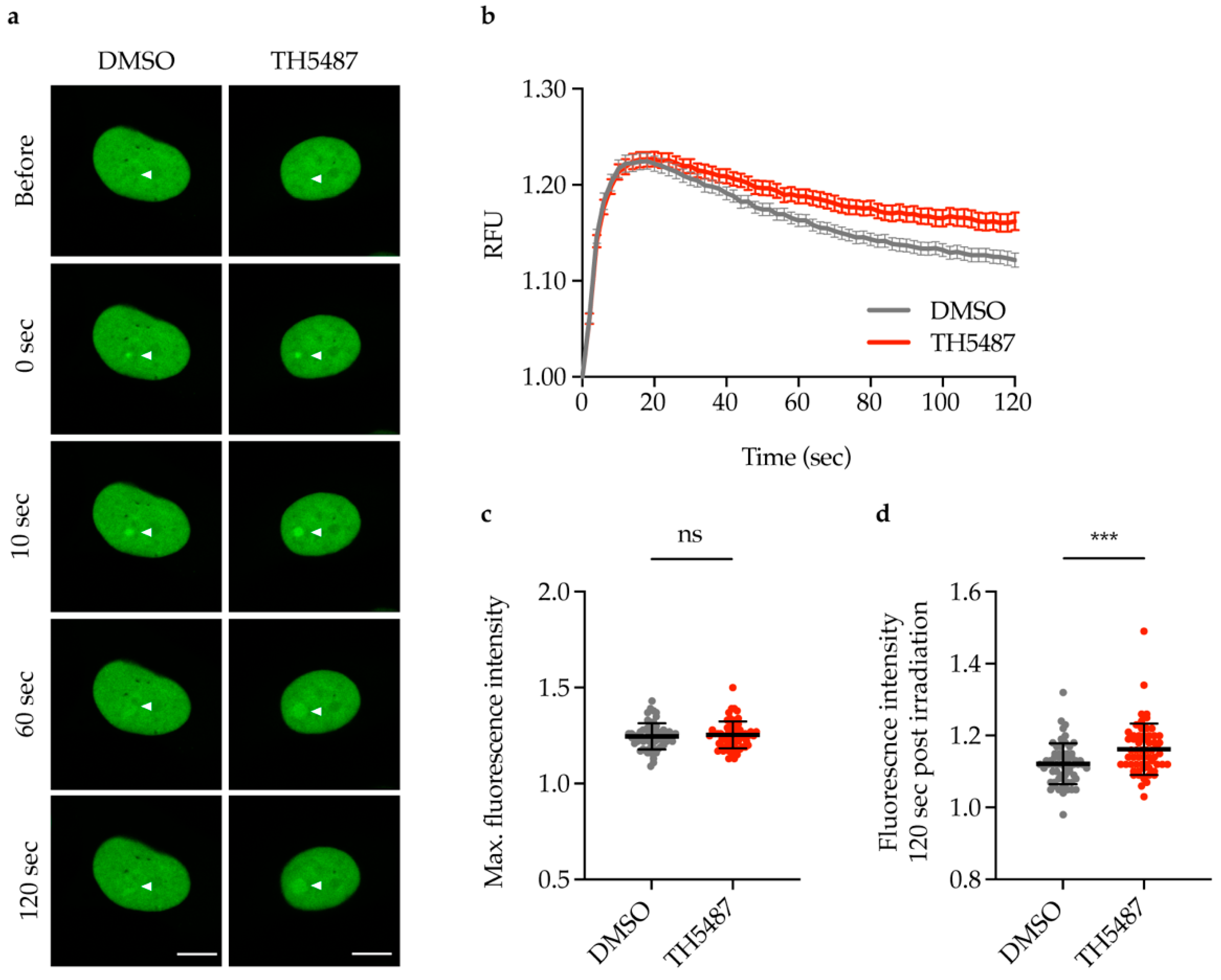
2.5. TH5487 Treatment Results in Prolonged Accumualtion of NEIL2-GFP at DNA Damage Sites
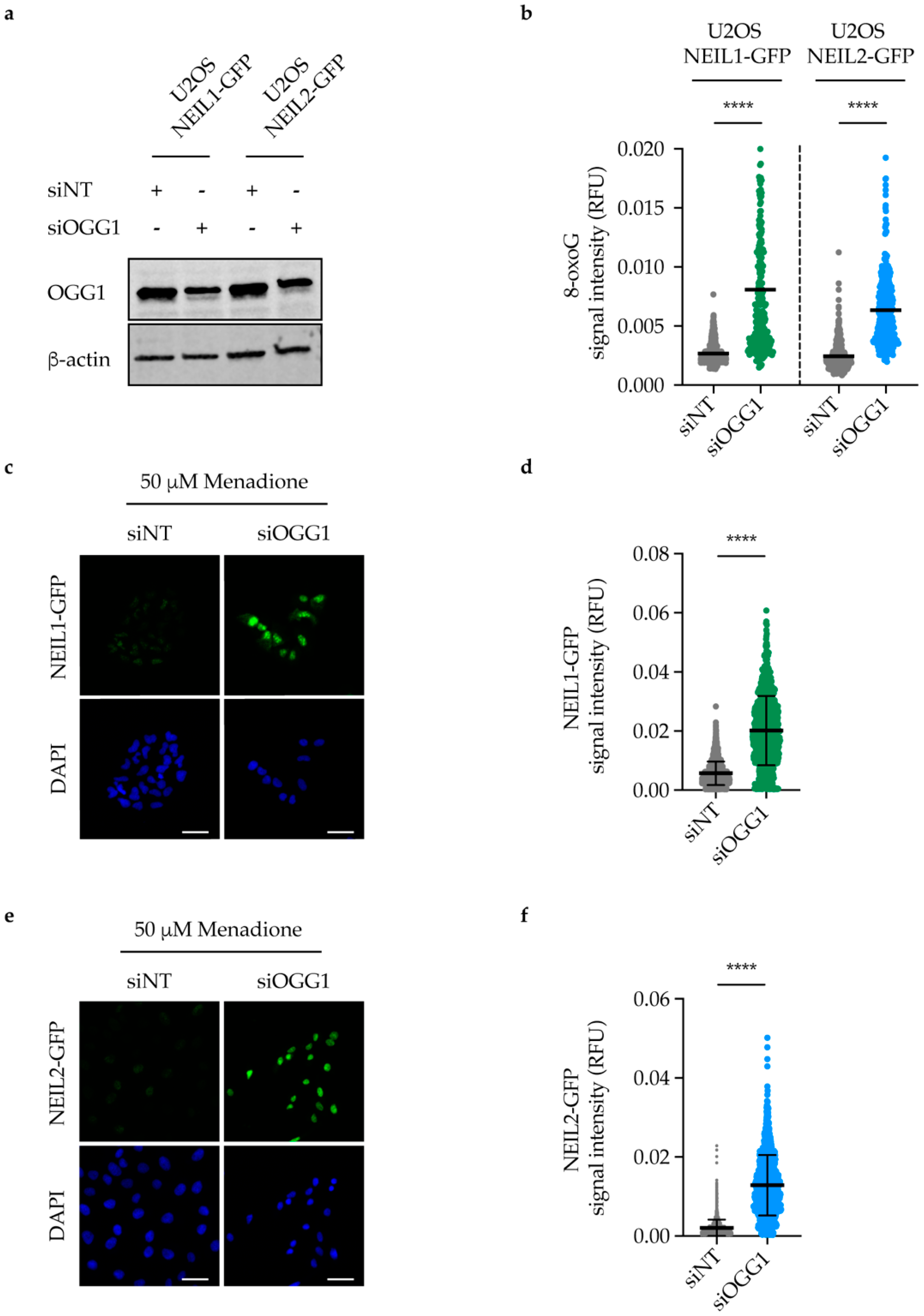
2.6. NEIL1 and NEIL2 Nuclear Retention Increases upon DNA Damage Induction in OGG1-Depleted Cells
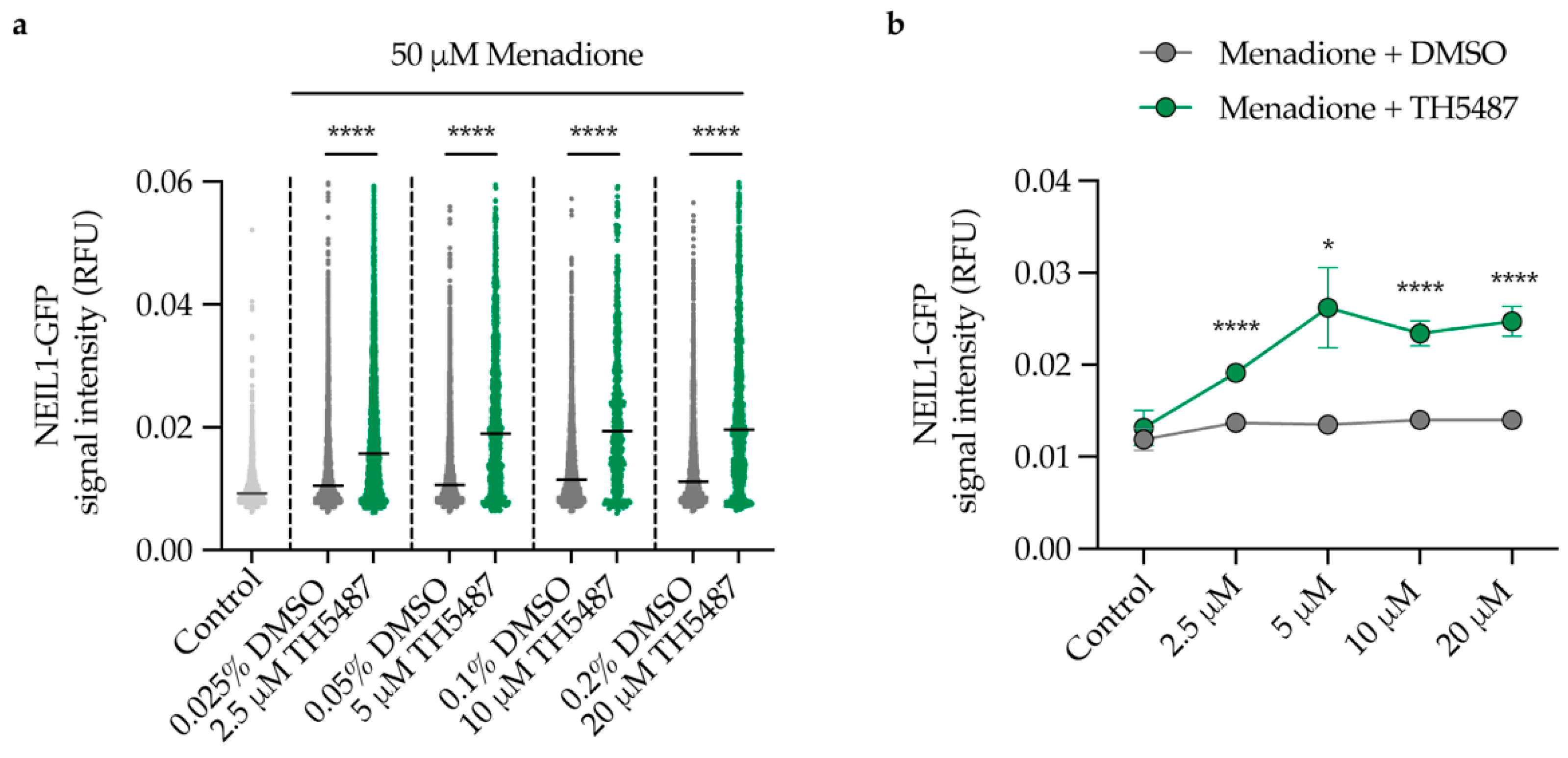
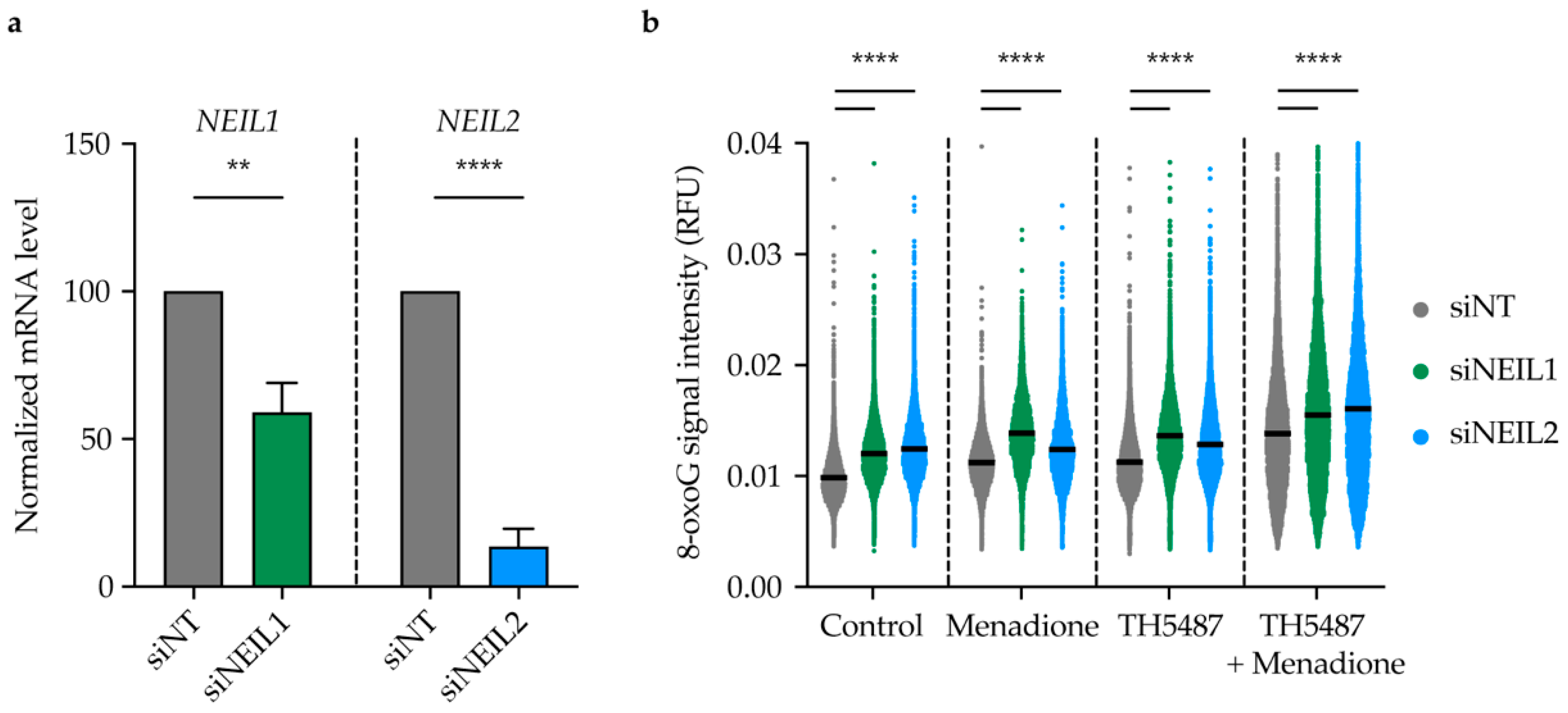
2.7. TH5487 Leads to 8-oxoG Lesion Accumulation upon Induction of Oxidative Stress in NEIL1-or NEIL2-Depleted Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Differential Scanning Fluorimetry (DSF)
4.3. NEIL1-GFP and NEIL2-GFP Plasmids Construction
4.4. Live Cell Microscopy, Laser Microirradiation and Fluorescence Recovery after Photobleaching
4.5. Quantitative Microscopy
4.6. OGG1 Knockdown and In Situ Extraction
4.7. Western Blot
4.8. NEIL1 and NEIL2 Knockdown
4.9. Cell Viability Assay
4.10. Real-Time PCR
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindahl, T.; Barnes, D.E. Repair of Endogenous DNA Damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Simic, M.G. One-Electron Redox Potentials of Purines and Pyrimidines. J. Phys. Chem. 1986, 90, 974–978. [Google Scholar] [CrossRef]
- Neeley, W.L.; Essigmann, J.M. Mechanisms of Formation, Genotoxicity, and Mutation of Guanine Oxidation Products. Chem. Res. Toxicol. 2006, 19, 491–505. [Google Scholar] [CrossRef]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-Excision Repair of Oxidative DNA Damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Karahalil, B.; Girard, P.M.; Boiteux, S.; Dizdaroglu, M. Substrate Specificity of the Ogg1 Protein of Saccharomyces Cerevisiae: Excision of Guanine Lesions Produced in DNA by Ionizing Radiation- or Hydrogen Peroxide/Metal Ion-Generated Free Radicals. Nucleic Acids Res. 1998, 26, 1228–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visnes, T.; Grube, M.; Hanna, B.M.F.; Benitez-Buelga, C.; Cázares-Körner, A.; Helleday, T. Targeting BER Enzymes in Cancer Therapy. DNA Repair 2018, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Bjørås, M.; Luna, L.; Johnson, B.; Hoff, E.; Haug, T.; Rognes, T.; Seeberg, E. Opposite Base-Dependent Reactions of a Human Base Excision Repair Enzyme on DNA Containing 7,8-Dihydro-8-Oxoguanine and Abasic Sites. EMBO J. 1997, 16, 6314–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zharkov, D.O.; Rosenquist, T.A.; Gerchman, S.E.; Grollman, A.P. Substrate Specificity and Reaction Mechanism of Murine 8-Oxoguanine-DNA Glycosylase. J. Biol. Chem. 2000, 275, 28607–28617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA Glycosylase 1: Beyond Repair of the Oxidatively Modified Base Lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hao, W.; Pan, L.; Boldogh, I.; Ba, X. The Roles of Base Excision Repair Enzyme OGG1 in Gene Expression. Cell. Mol. Life Sci. 2018, 75, 3741–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; Hao, W.; Zheng, X.; Zeng, X.; Ahmed Abbasi, A.; Boldogh, I.; Ba, X. OGG1-DNA Interactions Facilitate NF-kB Binding to DNA Targets. Sci. Rep. 2017, 7, 43297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-Molecule Inhibitor of OGG1 Suppresses Proinflammatory Gene Expression and Inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Muller, J.G.; Rachlin, E.M.; Burrows, C.J. Characterization of Spiroiminodihydantoin as a Product of One-Electron Oxidation of 8-Oxo-7,8-Dihydroguanosine. Org. Lett. 2000, 2, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S.; Jovanovic, S.V.; Bietti, M.; Bernhard, K. The Trap Depth (in DNA) of 8-Oxo-7,8-Dihydro-2′deoxyguanosine as Derived from Electron-Transfer Equilibria in Aqueous Solution. J. Am. Chem. Soc. 2000, 2373–2374. [Google Scholar] [CrossRef]
- Luo, W.; Muller, J.G.; Rachlin, E.M.; Burrows, C.J. Characterization of Hydantoin Products from One-Electron Oxidation of 8-Oxo-7,8-Dihydroguanosine in a Nucleoside Model. Chem. Res. Toxicol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Hailer, M.K.; Slade, P.G.; Martin, B.D.; Rosenquist, T.A.; Sugden, K.D. Recognition of the Oxidized Lesions Spiroiminodihydantoin and Guanidinohydantoin in DNA by the Mammalian Base Excision Repair Glycosylases NEIL1 and NEIL2. DNA Repair 2005, 4, 41–50. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Zhao, X.; Burrows, C.J.; David, S.S. Superior Removal of Hydantoin Lesions Relative to Other Oxidized Bases by the Human DNA Glycosylase HNEIL1. Biochemistry 2008, 47, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Doublié, S.; Wallace, S.S. Neil3, the Final Frontier for the DNA Glycosylases That Recognize Oxidative Damage. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2013, 743–744, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Takao, M.; Kanno, S.I.; Kobayashi, K.; Zhang, Q.M.; Yonei, S.; Van Der Horst, G.T.J.; Yasui, A. A Back-up Glycosylase in Nth1 Knock-out Mice Is a Functional Nei (Endonuclease VIII) Homologue. J. Biol. Chem. 2002, 277, 42205–42213. [Google Scholar] [CrossRef] [Green Version]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and Characterization of a Human DNA Glycosylase for Repair of Modified Bases in Oxidatively Damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katafuchi, A.; Nakano, T.; Masaoka, A.; Terato, H.; Iwai, S.; Hanaoka, F.; Ide, H. Differential Specificity of Human and Escherichia Coli Endonuclease III and VIII Homologues for Oxidative Base Lesions. J. Biol. Chem. 2004, 279, 14464–14471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.L.; Zharkov, D.O.; Dianov, G.L. NEIL1 Excises 3′ End Proximal Oxidative DNA Lesions Resistant to Cleavage by NTH1 and OGG1. Nucleic Acids Res. 2005, 33, 4849–4856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Krishnamurthy, N.; Burrows, C.J.; David, S.S. Mutation versus Repair: NEILl Removal of Hydantoin Lesions in Single-Stranded, Bulge, Bubble, and Duplex DNA Contexts. Biochemistry 2010, 49, 1658–1666. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative Repair of Oxidized Bases in the Human Genome Is Mediated by NEIL1 DNA Glycosylase Together with Replication Proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef] [Green Version]
- Rangaswamy, S.; Pandey, A.; Mitra, S.; Hegde, M.L. Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase. Genes 2017, 8, 175. [Google Scholar] [CrossRef] [Green Version]
- Albelazi, M.S.; Martin, P.R.; Mohammed, S.; Mutti, L.; Parsons, J.L.; Elder, R.H. The Biochemical Role of the Human NEIL1 and NEIL3 DNA Glycosylases on Model DNA Replication Forks. Genes 2019, 10, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvé, S.; Macé-Aimé, G.; Rosselli, F.; Saparbaev, M.K. The Human Oxidative DNA Glycosylase NEIL1 Excises Psoralen-Induced Interstrand DNA Cross-Links in a Three-Stranded DNA Structure. J. Biol. Chem. 2009, 284, 11963–11970. [Google Scholar] [CrossRef] [Green Version]
- McNeill, D.R.; Paramasivam, M.; Baldwin, J.; Huang, J.; Vyjayanti, V.N.; Seidman, M.M.; Wilson, D.M. NEIL1 Responds and Binds to Psoralen-Induced DNA Interstrand Crosslinks. J. Biol. Chem. 2013, 288, 12426–12436. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.R.; Couvé, S.; Zutterling, C.; Albelazi, M.S.; Groisman, R.; Matkarimov, B.T.; Parsons, J.L.; Elder, R.H.; Saparbaev, M.K. The Human DNA Glycosylases NEIL1 and NEIL3 Excise Psoralen-Induced DNA-DNA Cross-Links in a Four-Stranded DNA Structure. Sci. Rep. 2017, 7, 17438. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Fleming, A.M.; Averill, A.M.; Burrows, C.J.; Wallace, S.S. The NEIL Glycosylases Remove Oxidized Guanine Lesions from Telomeric and Promoter Quadruplex DNA Structures. Nucleic Acids Res. 2015, 43, 4039–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacolla, A.; Sengupta, S.; Ye, Z.; Yang, C.; Mitra, J.; De-Paula, R.B.; Hegde, M.L.; Ahmed, Z.; Mort, M.; Cooper, D.N.; et al. Heritable Pattern of Oxidized DNA Base Repair Coincides with Pre-Targeting of Repair Complexes to Open Chromatin. Nucleic Acids Res. 2021, 49, 221–243. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of Oxidized Bases in DNA Bubble Structures by Human DNA Glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential Repair of Oxidized Base Damage in the Transcribed Genes of Mammalian Cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.L.; Elder, R.H. DNA N-Glycosylase Deficient Mice: A Tale of Redundancy. In Mutation Research—Fundamental and Molecular Mechanisms of Mutagenesis; Elsevier: Amsterdam, The Netherlands, 2003; Volume 531, pp. 165–175. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA Glycosylases: In DNA Repair and Beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, E.C.; Meira, L.B. Database of Mouse Strains Carrying Targeted Mutations in Genes Affecting Biological Responses to DNA Damage Version 7. DNA Repair 2006, 5, 189–209. [Google Scholar] [CrossRef]
- Cortázar, D.; Kunz, C.; Selfridge, J.; Lettieri, T.; Saito, Y.; MacDougall, E.; Wirz, A.; Schuermann, D.; Jacobs, A.L.; Siegrist, F.; et al. Embryonic Lethal Phenotype Reveals a Function of TDG in Maintaining Epigenetic Stability. Nature 2011, 470, 419–423. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R.; et al. Deficiencies in Mouse Myh and Ogg1 Result in Tumor Predisposition and G to T Mutations in Codon 12 of the K-Ras Oncogene in Lung Tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef] [Green Version]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of Premutagenic DNA Lesions in Mice Defective in Removal of Oxidative Base Damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef] [Green Version]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 Gene Inactivation Results in Accumulation of 8-Hydroxyguanine in Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef] [Green Version]
- Hanna, B.M.F.; Helleday, T.; Mortusewicz, O. OGG1 Inhibitor TH5487 Alters OGG1 Chromatin Dynamics and Prevents Incisions. Biomolecules 2020, 10, 1483. [Google Scholar] [CrossRef]
- Visnes, T.; Benítez-Buelga, C.; Cázares-Körner, A.; Sanjiv, K.; Hanna, B.M.F.; Mortusewicz, O.; Rajagopal, V.; Albers, J.J.; Hagey, D.W.; Bekkhus, T.; et al. Targeting OGG1 Arrests Cancer Cell Proliferation by Inducing Replication Stress. Nucleic Acids Res. 2020, 48, 12234–12251. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ohno, M.; Tsuchimoto, D.; Sakumi, K.; Furuichi, M.; Nakabeppu, Y. Two Distinct Pathways of Cell Death Triggered by Oxidative Damage to Nuclear and Mitochondrial DNAs. EMBO J. 2008, 27, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Sakumi, K.; Tominaga, Y.; Furuichi, M.; Xu, P.; Tsuzuki, T.; Sekiguchi, M.; Nakabeppu, Y. Ogg1 Knockout-Associated Lung Tumorigenesis and Its Suppression by Mth1 Gene Disruption. Cancer Res. 2003, 63, 902–905. [Google Scholar]
- Michel, M.; Visnes, T.; Homan, E.J.; Seashore-Ludlow, B.; Hedenström, M.; Wiita, E.; Vallin, K.; Paulin, C.B.J.; Zhang, J.; Wallner, O.; et al. Computational and Experimental Druggability Assessment of Human DNA Glycosylases. ACS Omega 2019, 4, 11642–11656. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The Use of Differential Scanning Fluorimetry to Detect Ligand Interactions That Promote Protein Stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanna, B.M.F.; Michel, M.; Helleday, T.; Mortusewicz, O. NEIL1 and NEIL2 Are Recruited as Potential Backup for OGG1 upon OGG1 Depletion or Inhibition by TH5487. Int. J. Mol. Sci. 2021, 22, 4542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094542
Hanna BMF, Michel M, Helleday T, Mortusewicz O. NEIL1 and NEIL2 Are Recruited as Potential Backup for OGG1 upon OGG1 Depletion or Inhibition by TH5487. International Journal of Molecular Sciences. 2021; 22(9):4542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094542
Chicago/Turabian StyleHanna, Bishoy M. F., Maurice Michel, Thomas Helleday, and Oliver Mortusewicz. 2021. "NEIL1 and NEIL2 Are Recruited as Potential Backup for OGG1 upon OGG1 Depletion or Inhibition by TH5487" International Journal of Molecular Sciences 22, no. 9: 4542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094542