
Adrenoceptors Modulate Cholinergic Synaptic Transmission at the Neuromuscular Junction

Abstract
:1. Introduction
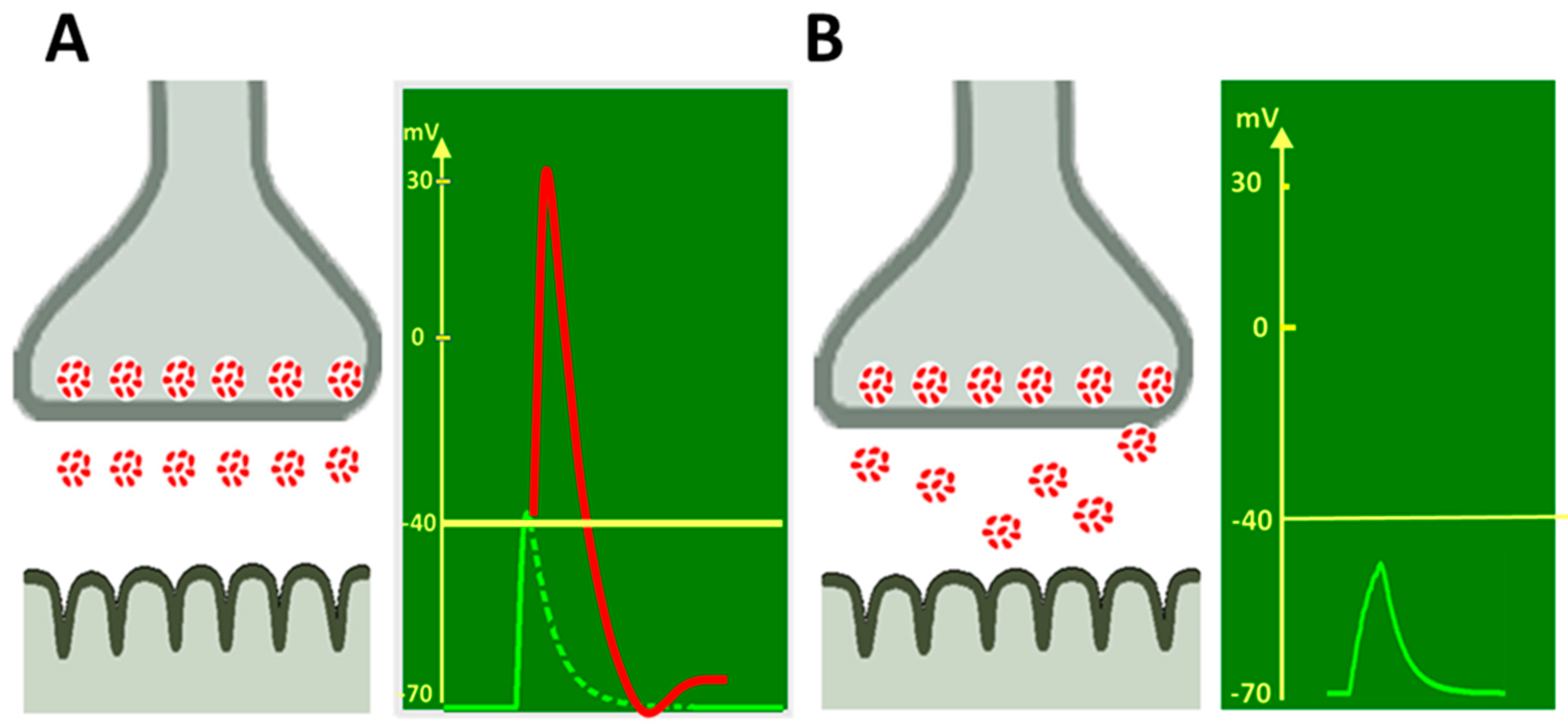
2. Activators and Blockers of Adrenergic Receptors Alter the Spontaneous ACh
Quantal Release at the NMJ
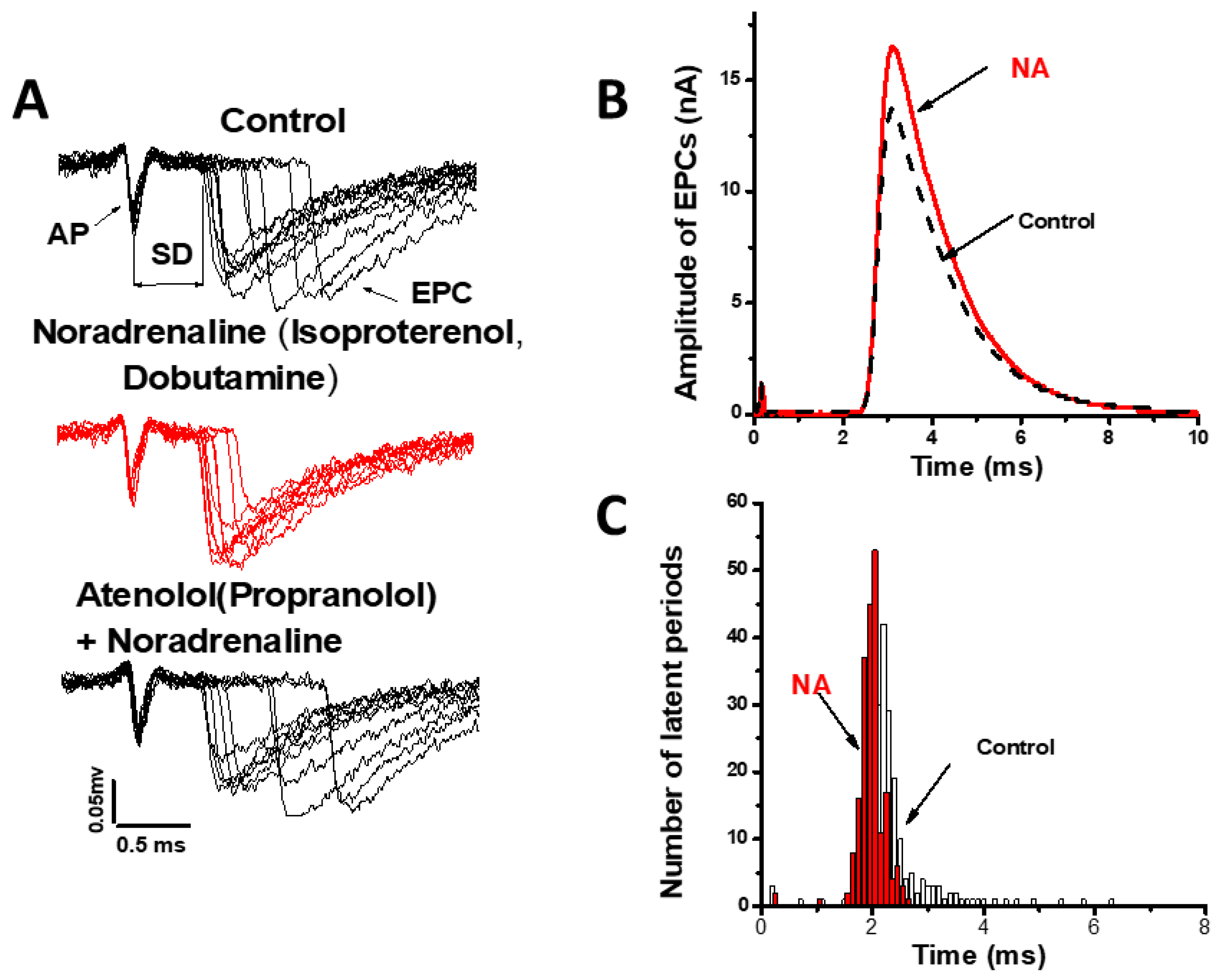
3. Sympathomimetic Effects on ACh Quantal Release Evoked by the Nerve Stimulus
3.1. Modulation of the Number of ACh Released Quanta
3.2. Modulation of the Kinetics of ACh Quantal Release
4. Effects of Adrenergic Drugs on the Postsynaptic Nicotinic Receptors at the NMJ
5. Morphological Evidence of the Existence of Adrenoceptors at the NMJ
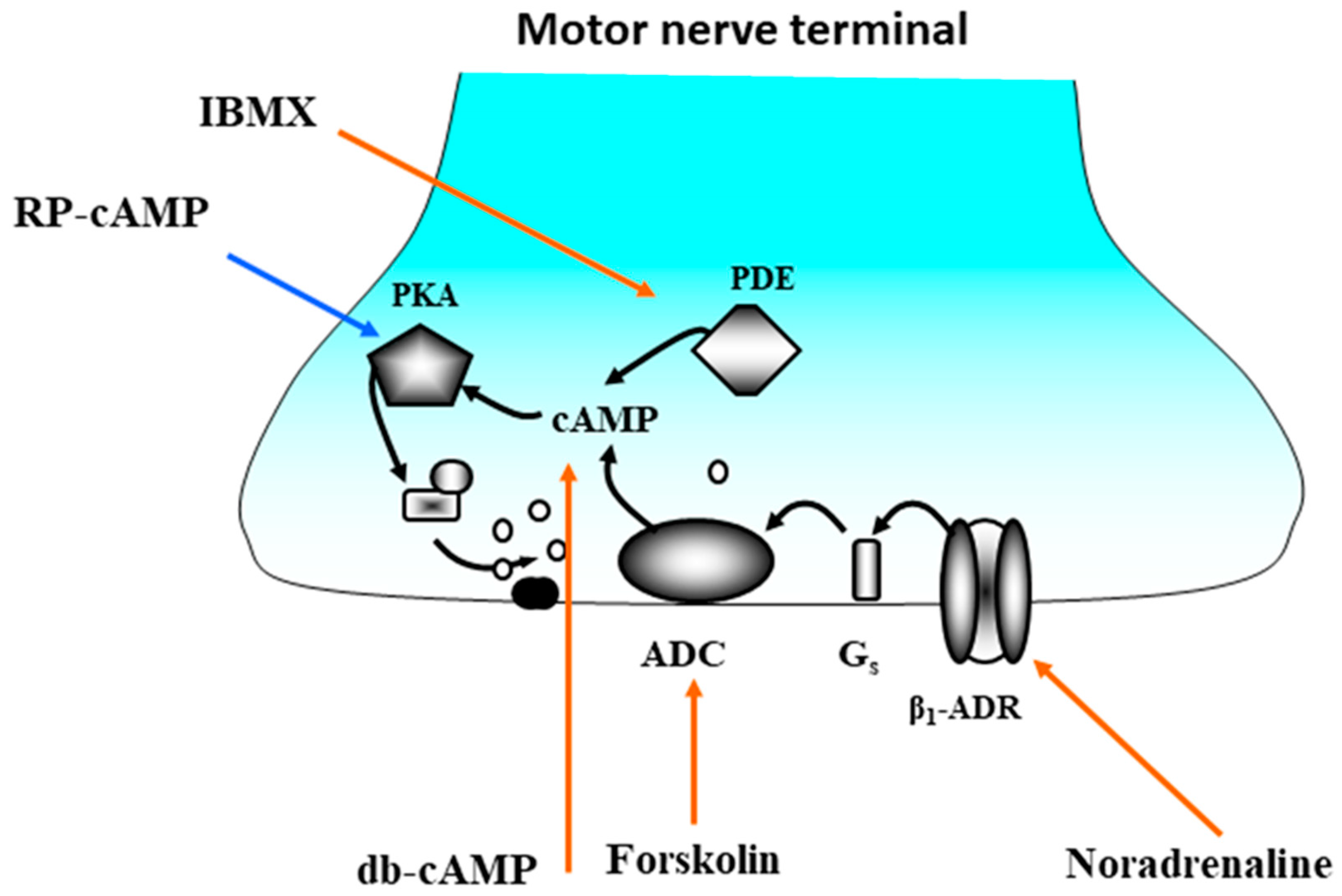
6. Effects of the Endogenous Catecholamines on Synaptic Transmission
7. Progress and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rudolf, R.; Khan, M.; Witzemann, V. Motor endplate—Anatomical, functional, and molecular concepts in the historical perspective. Cells 2019, 8, 387. [Google Scholar] [CrossRef] [Green Version]
- Lepore, E.; Casola, I.; Dobrowolny, G.; Musarò, A. Neuromuscular junction as an entity of nerve-muscle communication. Cells 2019, 8, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, P.R.; Cossins, J.; Beeson, D.; Vincent, A. The Neuromuscular Junction in Health and Disease: Molecular Mechanisms Governing Synaptic Formation and Homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.; Schäfer, E.A. The physiological effects of extracts of the suprarenal capsules. J. Physiol. 1895, 18, 230–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, S. On the general physiological effects of extracts of the suprarenal capsules. J. Physiol. 1897, 22, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orbeli, L.A. Die sympatetische Innervation der Skelettmuskeln. Bull. Inst. Sci. Leshaft. 1923, 6, 194–197. [Google Scholar]
- Ginetsinsky, A.G. The influence of the sympathetic nervous system on the function of the striated muscle. Russ. Fiziol. Zhurnal 1923, 6, 139. [Google Scholar]
- McMacken, G.M.; Spendiff, S.; Whittaker, R.G.; O’Connor, E.; Howarth, R.M.; Boczonadi, V.; Horvath, R.; Slater, C.R.; Lochmüller, H. Salbutamol modifies the neuromuscular junction in a mouse model of ColQ myasthenic syndrome. Hum. Mol. Genet. 2019, 28, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.Z.; Messi, M.L.; Wang, Z.M.; Abba, M.C.; Pereyra, A.; Birbrair, A.; Zhang, T.; O’Meara, M.; Kwan, P.; Lopez, E.I.S.; et al. The sympathetic nervous system regulates skeletal muscle motor innervation and acetylcholine receptor stability. Acta Physiol. 2019, 225, 13195. [Google Scholar] [CrossRef]
- Vanhaesebrouck, A.E.; Webster, R.; Maxwell, S.; Rodriguez Cruz, P.; Cossins, J.; Wickens, J.; Liu, W.-W.; Cetin, H.; Cheung, J.; Ramjattan, H.; et al. b2-Adrenergic receptor agonists ameliorate the adverse effect of long-term pyridostigmine on neuromuscular junction structure. BRAIN 2019, 142, 3713–3727. [Google Scholar] [CrossRef]
- Wang, Z.M.; Rodrigues, A.C.Z.; Messi, M.L.; Delbono, O. Aging Blunts Sympathetic Neuron Regulation of Motoneurons Synaptic Vesicle Release Mediated by β1– and α2B–Adrenergic Receptors in Geriatric Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.M.; Messi, M.L.; Grinevich, V.; Budygin, E.; Delbono, O. Postganglionic sympathetic neurons, but not locus coeruleus optostimulation, activates neuromuscular transmission in the adult mouse in vivo. Cell. Mol. Neurosci. 2020, 109, 103563. [Google Scholar] [CrossRef] [PubMed]
- Delbono, O.; Rodrigues, A.C.Z.; Bonilla, H.J.; Messi, M.L. The emerging role of the sympathetic nervous system in skeletal muscle motor innervation and sarcopenia. Age Res. Rev. 2021, 67, 101305. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Cruz, P.M.R.; Palace, J.; Beeson, D. The neuromuscular junction and wide heterogeneity of congenital myasthenic syndromes. Int. J. Mol. Sci. 2018, 19, 1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, G.; Hiscock, A.; Klein, A.; Niks, E.H.; Main, M.; Manzur, A.Y.; Ng, J.; de Vile, C.; Muntoni, F.; Beeson, D.; et al. Salbutamol benefits children with congenital myasthenic syndrome due to DOK7mutations. Neuromuscul. Disord. 2013, 23, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Webster, R.G.; Vanhaesebrouck, A.E.; Maxwell, S.E.; Cossins, J.A.; Liu, W.; Ueta, R.; Yamanashi, Y.; Beeson, D.M.W. Effect of salbutamol on neuromuscular junction function and structure in a mouse model of DOK7 congenital myasthenia. Hum. Mol. Genet. 2020, 29, 2325–2336. [Google Scholar] [CrossRef]
- Ghazanfari, N.; Morsch, M.; Tse, N.; Reddel, S.W.; Phillips, W.D. Effects of the ß2-adrenoceptor agonist, albuterol, in a mouse model of anti-MuSK myasthenia gravis. PLoS ONE 2014, 9, e87840. [Google Scholar] [CrossRef] [Green Version]
- Bartus, R.T.; Bétourné, A.; Basile, A.; Peterson, B.L.; Glass, J.; Boulis, N.M. β2-Adrenoceptor agonists as novel, safe and potentially effective therapies for Amyotrophic lateral sclerosis (ALS). Neurobiol. Dis. 2016, 85, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Takamori, M. Myasthenia Gravis: From the viewpoint of pathogenicity focusing on acetylcholine receptor clustering, trans–synaptic homeostasis and synaptic stability. Front. Mol. Neurosci. 2020, 13, 86. [Google Scholar] [CrossRef]
- Limanaqi, F.; Busceti, C.L.; Biagioni, F.; Cantini, F.; Lenzi, P.; Fornai, F. Cell-clearing systems bridging repeat expansion proteotoxicity and neuromuscular junction alterations in ALS and SBMA. Int. J. Mol. Sci. 2020, 21, 4021. [Google Scholar] [CrossRef] [PubMed]
- Fatt, P.; Katz, B. Spontaneous subthreshold activity at motor nerve endings. J. Physiol. 1952, 117, 109–128. [Google Scholar] [PubMed]
- Krnjevic, K.; Miledi, R. Some effects produced by adrenaline upon neuromuscular propagation in rats. J. Physiol. 1958, 141, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K. Effects of catecholamines on the neuromuscular junction in the rat diaphragm. J. Physiol. 1970, 211, 551–570. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Tomita, T. Noradrenaline action on nerve terminal in the rat diaphragm. J. Physiol. 1971, 217, 19–31. [Google Scholar]
- Bylund, D.B. Alpha- and beta-adrenergic receptors: Ahlquist’s landmark hypothesis of a single mediator with two receptors. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1479–E1481. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Dryden, W.F.; Singh, Y.N. Transduction of the modulatory effect of catecholamines at the mammalian motor neuron terminal. Synapse 1991, 7, 93–98. [Google Scholar] [CrossRef]
- Lim, S.P.; Muir, T.C. Microelectrode recording of the effects of agonists and antagonists on alpha-adrenoceptors on rat somatic nerve terminals. Br. J. Pharmacol. 1983, 80, 41–46. [Google Scholar]
- Rodrigues, A.Z.C.; Wang, Z.M.; Messi, M.L.; Delbono, O. Sympathomimetics regulate neuromuscular junction transmission through TRPV1, P/Q– and N–type Ca2+ channels. Mol. Cell. Neurosci. 2019, 95, 59–70. [Google Scholar] [CrossRef]
- Tsentsevitsky, A.; Nurullin, L.; Tyapkina, O.; Bukharaeva, E. Sympathomimetics regulate quantal acetylcholine release at neuromuscular junctions through various types of adrenoreceptors. Mol. Cell. Neurosci. 2020, 108, 103550. [Google Scholar] [CrossRef]
- Tsentsevitsky, A.; Kovyazina, I.; Bukharaeva, E. Diverse effects of noradrenaline and adrenaline on the quantal secretion of acetylcholine at the mouse neuromuscular junction. Neurosci 2019, 423, 162–171. [Google Scholar] [CrossRef]
- Bergman, H.; Glusman, S.; Harris-Warrick, R.M.; Kravitz, E.A.; Nussinovitch, I.; Rahamimoff, R. Noradrenaline augments tetanic potentiation of transmitter release by a calcium dependent process. Brain Res. 1981, 214, 200–204. [Google Scholar] [CrossRef]
- Bowman, W.C.; Raper, C. Effects of sympathomimetic amines on neuromuscular transmission. Br. J. Pharmacol. Chemother. 1966, 27, 313–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.J.; Harvey, A.L. Effects of the facilitatory compounds catechol, guanidine, noradrenaline and phencyclidine on presynaptic currents of mouse motor nerve terminals. Naunyn. Schmiedebergs. Arch. Pharmacol. 1988, 338, 133–1377. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I. Acetylcholine at motor nerves: Storage, release, and presynaptic modulation by autoreceptors and adrenoceptors. Int. Rev. Neurobiol. 1992, 4, 283–384. [Google Scholar]
- Starke, K.; Göthert, M.; Kilbinger, H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol. Rev. 1989, 69, 864–989. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Anschütz, S. β–Adrenoceptor stimulation enhances transmitter output from the rat phrenic nerve. Br. J. Pharmacol. 1988, 94, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Ladwein, E.; Szrama, E. Stimulation of alpha 1-adrenoceptors increases electrically evoked [3H]-acetylcholine release from the rat phrenic nerve. Eur. J. Pharmacol. 1989, 174, 77–83. [Google Scholar] [CrossRef]
- Wessler, I.; Holzer, G.; Künster, A. Stimulation of beta 1-adrenoceptors enhances electrically evoked [3H]-acetylcholine release from rat phrenic nerve. Clin. Exp. Pharmacol. Physiol. 1990, 17, 23–32. [Google Scholar] [CrossRef]
- Vizi, E.S. Evidence that catecholamines increase acetylcholine release from neuromuscular junction through stimulation of alpha–1 adrenoceptors. Naunyn. Schmiedebergs Arch. Pharmacol. 1991, 343, 435–438. [Google Scholar] [CrossRef]
- Snider, R.M.; Gerald, M.C. Noradrenergic-mediated potentiation of acetylcholine release from the phrenic nerve: Evidence for presynaptic alpha 1-adrenoceptor involvement. Life Sci. 1982, 31, 853–857. [Google Scholar] [CrossRef]
- Chiou, L.C.; Chang, C.C. Effect of clonidine on neuromuscular transmission and the nicotinic acetylcholine receptor. Proc. Natl. Sci. Counc. Repub. China. B 1984, 8, 148–154. [Google Scholar]
- Khuzakhmetova, V.; Bukharaeva, E. Adrenaline Facilitates Synaptic Transmission by Synchronizing Release of Acetylcholine Quanta from Motor Nerve Endings. Cell. Mol. Neurobiol. 2020, 41, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Eason, M.G.; Kurose, H.; Holt, B.D.; Raymond, J.R.; Liggett, S.B. Simultaneous coupling of alpha 2-adrenergic receptors to two G-proteins with opposing effects. Subtype-selective coupling of alpha 2C10, alpha 2C4, and alpha 2C2 adrenergic receptors to Gi and Gs. J. Biol. Chem. 1992, 267, 15795–15801. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, X.W.; Chang, A.M. Effects of Gi and beta2AR overexpression on the survival of rat cardiac myocytes injured by isoprenaline. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2007, 23, 274–275. [Google Scholar]
- Kobilka, B. Adrenergic receptors as models for G protein-coupled receptors. Annu. Rev. Neurosci. 1992, 15, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Tepper, J.M.; Young, S.J.; Groves, P.M. Neurophysiological consequences of presynaptic receptor activation: Changes in noradrenergic terminal excitability. Brain Res. 1981, 226, 155–170. [Google Scholar] [CrossRef]
- Williams, J.T.; North, R.A. Catecholamine inhibition of calcium action potentials in rat locus coeruleus neurones. Neuroscience 1985, 14, 103–109. [Google Scholar] [CrossRef]
- Horn, J.P.; McAfee, D.A. Alpha-drenergic inhibition of calcium-dependent potentials in rat sympathetic neurones. J. Physiol. 1980, 301, 191–204. [Google Scholar] [CrossRef]
- Canfield, D.R.; Dunlap, K. Pharmacological characterization of amine receptors on embryonic chick sensory neurones. Br. J. Pharmacol. 1984, 82, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Schoffelmeer, A.N.; Wierenga, E.A.; Mulder, A.H. Role of adenylate cyclase in presynaptic alpha 2-adrenoceptor- and mu-opioid receptor-mediated inhibition of [3H]noradrenaline release from rat brain cortex slices. J. Neurochem. 1986, 46, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.; Miledi, R. The measurement of synaptic delay, and the time course of acetylcholine release at the neuromuscular junction. Proc. R. Soc. Lond. B Biol. Sci. 1965, 161, 483–495. [Google Scholar] [PubMed]
- Barrett, E.F.; Stevens, C.F. The kinetics of transmitter release at the frog neuromuscular junction. J. Physiol. 1972, 227, 691–708. [Google Scholar] [CrossRef]
- Kaeser, P.S.; Regehr, W.G. Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu. Rev. Physiol. 2014, 76, 333–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukharaeva, E.A.; Samigullin, D.; Nikolsky, E.E.; Magazanik, L.G. Modulation of the kinetics of evoked quantal release at mouse neuromuscular junctions by calcium and strontium. J. Neurochem. 2007, 100, 939–949. [Google Scholar] [CrossRef]
- Van der Kloot, W. The kinetics of quantal releases during end-plate currents at the frog neuromuscular junction. J. Physiol. 1988, 402, 605–626. [Google Scholar] [CrossRef] [PubMed]
- Giniatullin, R.A.; Kheeroug, L.S.; Vyskocil, F. Modelling endplate currents: Dependence on quantum secretion probability and decay of miniature current. Eur. Biophys. J. 1995, 23, 443–446. [Google Scholar] [CrossRef]
- Bukharaeva, E.A.; Kim, K.K.; Nikol’skii, E.E.; Vyskochil, F. Synchronization of evoked secretion of quanta of mediator as a mechanism facilitating the action of sympathomimetics. Neurosci. Behav. Physiol. 2000, 30, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Bukcharaeva, E.A.; Kim, K.C.; Moravec, J.; Nikolsky, E.E.; Vyskocil, F. Noradrenaline synchronizes evoked quantal release at frog neuromuscular junctions. J. Physiol. 1999, 517, 1469–7793. [Google Scholar] [CrossRef]
- Bukharaeva, E.A.; Samigullin, D.; Nikolsky, E.; Vyskocil, F. Protein kinase A cascade regulates quantal release dispersion at frog muscle endplate. J. Physiol. 2002, 538, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Schneggenburger, R.; Neher, E. Presynaptic calcium and control of vesicle fusion. Curr. Opin. Neurobiol. 2005, 15, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Zhou, Z.Y.; Hemmings, H.C., Jr. α2-Adrenergic Receptor and Isoflurane Modulation of Presynaptic Ca2+ Influx and Exocytosis in Hippocampal Neurons. Anesthesiology 2016, 125, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milone, M.; Engel, A.G. Block of the endplate acetylcholine receptor channel by the sympathomimetic agents ephedrine, pseudoephedrine, and albuterol. Brain Res. 1996, 740, 346–352. [Google Scholar] [CrossRef]
- Sieb, J.P.; Engel, A.G. Ephedrine: Effects on neuromuscular transmission. Brain Res. 1993, 623, 167–171. [Google Scholar] [CrossRef]
- Webster, R.G.; Cossins, J.; Lashley, D.; Maxwell, S.; Liu, W.W.; Wickens, J.R.; Martinez-Martinez, P.; de Baets, M.; Beeson, D. A mouse model of the slow channel myasthenic syndrome: Neuromuscular physiology and effects of ephedrine treatment. Exp. Neurol. 2013, 248, 286–298. [Google Scholar] [CrossRef]
- Clausen, L.; Cossins, J.; Beeson, D. Beta-2 adrenergic receptor agonists enhance AChR clustering in C2C12 myotubes: Implications for therapy of myasthenic disorders. J. Neuromuscul. Dis. 2018, 5, 231–240. [Google Scholar] [CrossRef]
- Khan, M.M.; Lustrino, D.; Silveira, W.A.; Wild, F.; Straka, T.; Issop, Y.; O’Connor, E.; Cox, D.; Reischl, M.; Marquardt, T.; et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc. Natl. Acad. Sci. USA 2016, 113, 746–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Sainz, R.; Molenaar, P. Characterization of β1- and β2-adrenoreceptors in rat skeletal muscles. Biochem. Pharmacol. 1991, 42, 1783–1789. [Google Scholar] [CrossRef]
- Lynch, G.S.; Ryall, J.G. Role of beta-adrenoceptor signaling in skeletal muscle: Implications for muscle wasting and disease. Physiol. Rev. 2008, 88, 729–767. [Google Scholar] [CrossRef] [Green Version]
- Silveira, W.A.; Goncalves, D.A.; Graca, F.A.; Andrade–Lopes, A.L.; Bergantin, L.B.; Zanon, N.M.; Godinho, R.O.; Kettelhut, I.C.; Navegantes, L.C. Activating cAMP/PKA signaling in skeletal muscle suppresses the ubiquitin–proteasome–dependent proteolysis: Implications for sympathetic regulation. J. Appl. Physiol. 2014, 117, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Straka, T.; Vita, V.; Prokshi, K.; Hörner, S.J.; Khan, M.M.; Pirazzini, M.; Williams, M.P.I.; Hafner, M.; Zaglia, T.; Rudolf, R. Postnatal development and distribution of sympathetic innervation in mouse skeletal muscle. Int. J. Mol. Sci. 2018, 19, 1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Boeke, J. Die doppelte (motorische und sympathische) erente Innervation der quergestreiften Muskelfasern. Anat. Anz. 1913, 44, 343–356. [Google Scholar]
- Yasuda, T.; Sobue, G.; Mitsuma, T.; Takahashi, A. Peptidergic and adrenergic regulation of the intracellular 3′,5′-cyclic adenosine monophosphate content in cultured rat Schwann cells. J. Neurol. Sci. 1988, 88, 315–325. [Google Scholar] [CrossRef]
- Noronha-Matos, J.B.; Oliveira, L.; Peixoto, A.; Almeida, L.; Castellão-Santana, M.L.; Ambie, I.; Alves-do Prado, W.; Correia-de-Sá, P. Nicotinic α7 receptor-induced adenosine release from perisynaptic Schwann cells controls acetylcholine spillover from motor endplates. J. Neurochem. 2020, 154, 263–283. [Google Scholar] [CrossRef]
- Magnaghi, V.; Parducz, A.; Frasca, A.; Ballabio, M.; Procacci, P.; Racagni, G.; Bonanno, G.; Fumagalli, F. GABA synthesis in Schwann cells is induced by the neuroactive steroid allopregnanolone. J. Neurochem. 2010, 112, 980–990. [Google Scholar] [CrossRef]
- Yoon, B.E.; Lee, C.J. GABA as a rising gliotransmitter. Front. Neural Circuits 2014, 8, 141. [Google Scholar] [CrossRef] [Green Version]
- Petrov, K.A.; Girard, E.; Nikitashina, A.D.; Colasante, C.; Bernard, V.; Nurullin, L.; Leroy, J.; Samigullin, D.; Colak, O.; Nikolsky, E.; et al. Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by α7 nicotinic receptors and butyrylcholinesterase. J. Neurosci. 2014, 34, 11870–11883. [Google Scholar] [CrossRef] [Green Version]
- Fink, T.; Davey, D.F.; Ansselin, A.D. Glutaminergic and adrenergic receptors expressed on adult guinea pig Schwann cells in vitro. Can. J. Physiol. Pharmacol. 1999, 77, 204–210. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADR | Spontaneous ACh Release | Evoked ACh Release | ||||
|---|---|---|---|---|---|---|
| Quantal Content | Secretion Kinetics | |||||
| Inhibition | Facilitation | Inhibition | Facilitation | Synchronization | Desynchronization | |
| α | Adrenaline * Noradrenaline * Phenylephrine * Dexmedetomidine * Clonidine * [30,31,49] | Noradrenaline [23,24] Adrenaline [25] | Noradrenaline Adrenaline α-methylnor-adrenaline [32,33] | Adrenaline * [30,31] | Noradrenaline * [30,31] | |
| β | Noradrenaline [24] | Isoprenaline Noradrenaline [23,24,32,33,35,37] | ||||
| α1 | Phenylephrine [31] | Phenylephrine K [27,41] | Phenylephrine [33,38,40] | Adrenaline * [30,31] | ||
| α2 | Dexmedetomidine * Clonidine * [30] | Clonidine Xylazine [28] | Clonidine * Dexmedetomidine * [30] | Adrenaline * [31,43] | Dexmedetomidine [30] * | |
| β1 | Xamoterol * [30] | Isoproterenol [25] | Xamoterol * [30] | Isoprenaline [27,37,39] | Noradrenaline [59] | |
| β2 | Salbutamol Clenbuterol [9,29] | Salbutamol Clenbuterol Procaterol * [9,29] | Procaterol * [30] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukharaeva, E.; Khuzakhmetova, V.; Dmitrieva, S.; Tsentsevitsky, A. Adrenoceptors Modulate Cholinergic Synaptic Transmission at the Neuromuscular Junction. Int. J. Mol. Sci. 2021, 22, 4611. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094611
Bukharaeva E, Khuzakhmetova V, Dmitrieva S, Tsentsevitsky A. Adrenoceptors Modulate Cholinergic Synaptic Transmission at the Neuromuscular Junction. International Journal of Molecular Sciences. 2021; 22(9):4611. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094611
Chicago/Turabian StyleBukharaeva, Ellya, Venera Khuzakhmetova, Svetlana Dmitrieva, and Andrei Tsentsevitsky. 2021. "Adrenoceptors Modulate Cholinergic Synaptic Transmission at the Neuromuscular Junction" International Journal of Molecular Sciences 22, no. 9: 4611. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094611