
Blood–Brain Barrier and Neurovascular Unit In Vitro Models for Studying Mitochondria-Driven Molecular Mechanisms of Neurodegeneration


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
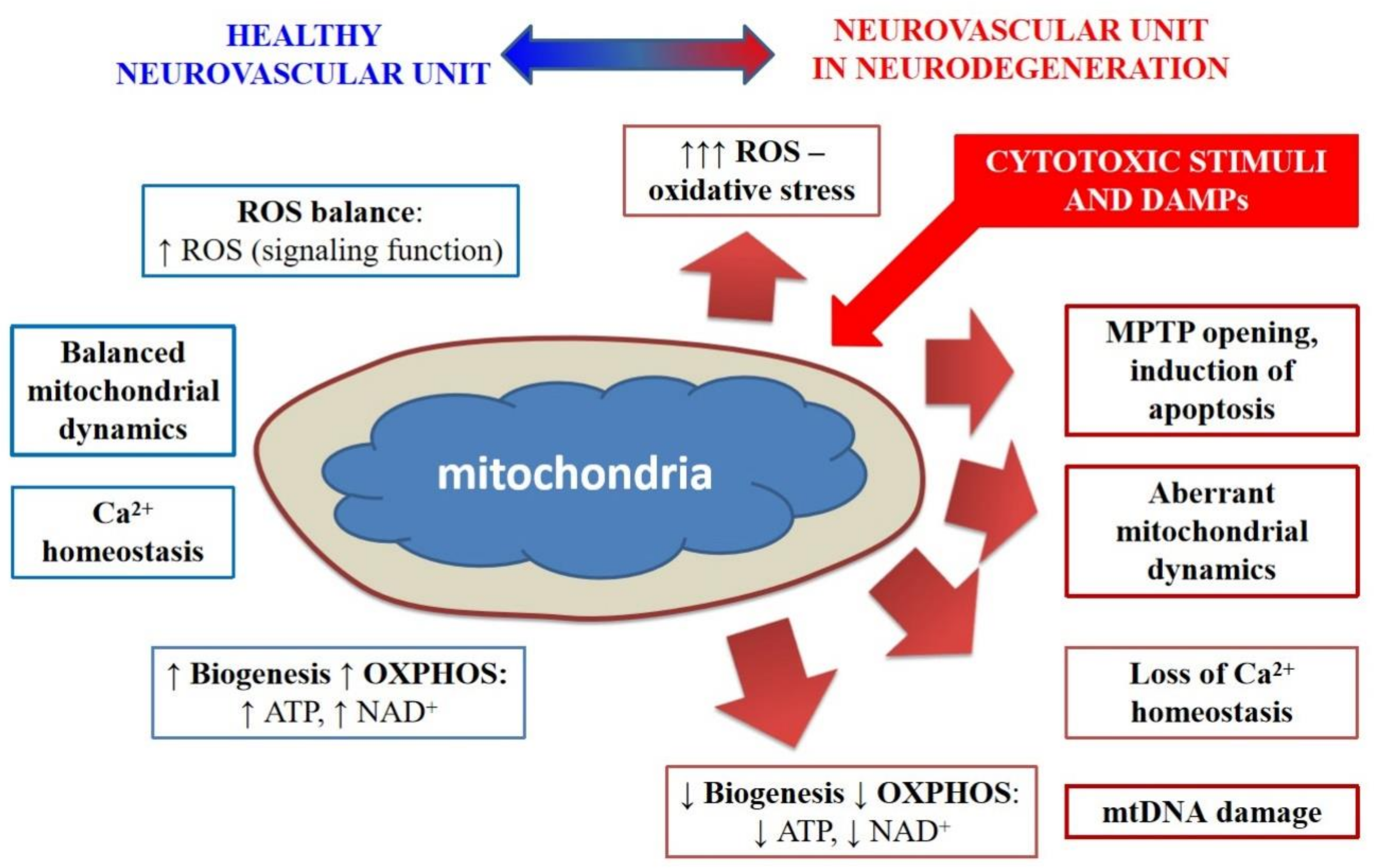
2. Mitochondrial Dysfunction and NVU/BBB Impairment in Alzheimer’s Type Neurodegeneration
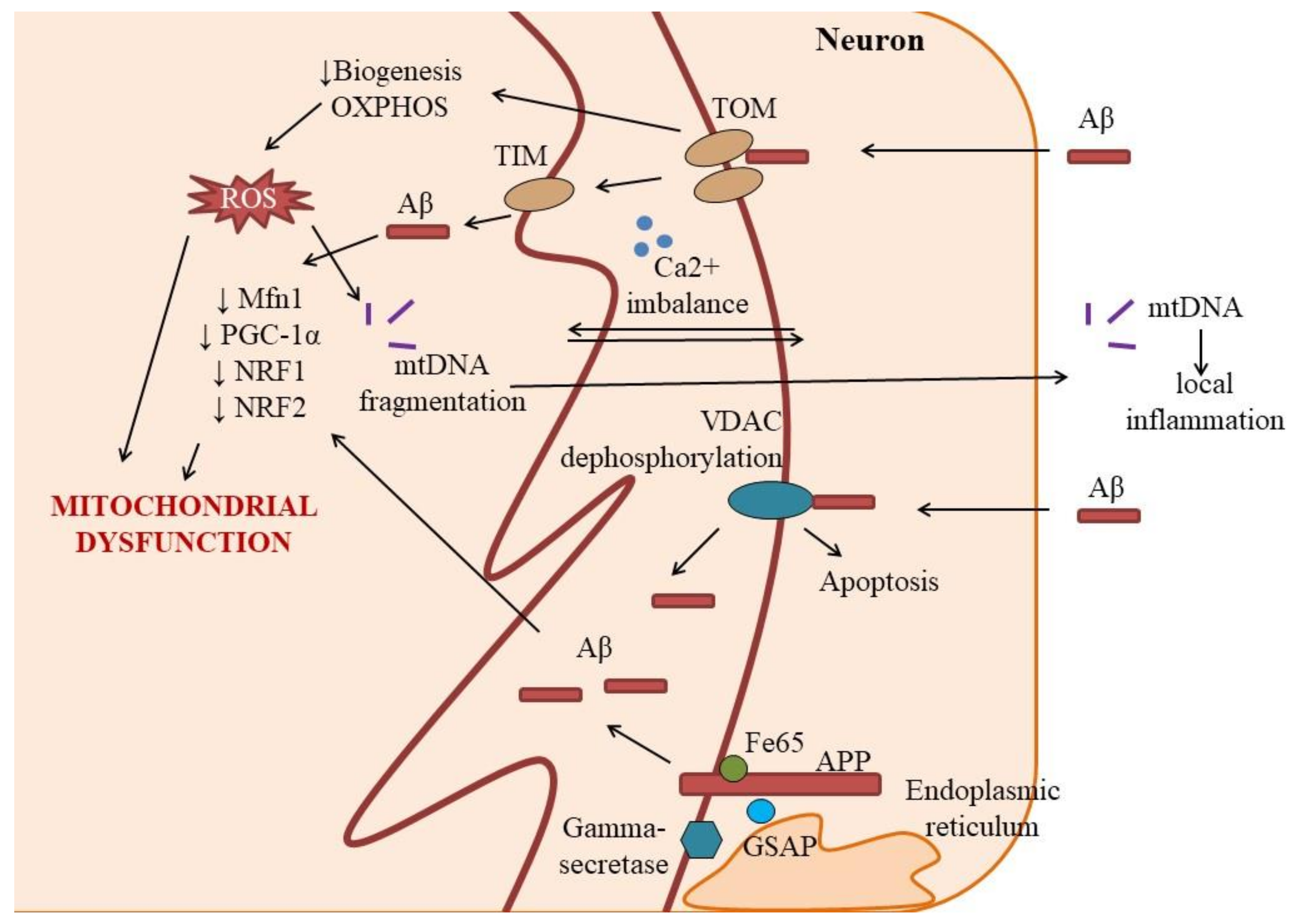
2.1. Aberrant Mitochondrial Function and Dynamics in Neurons
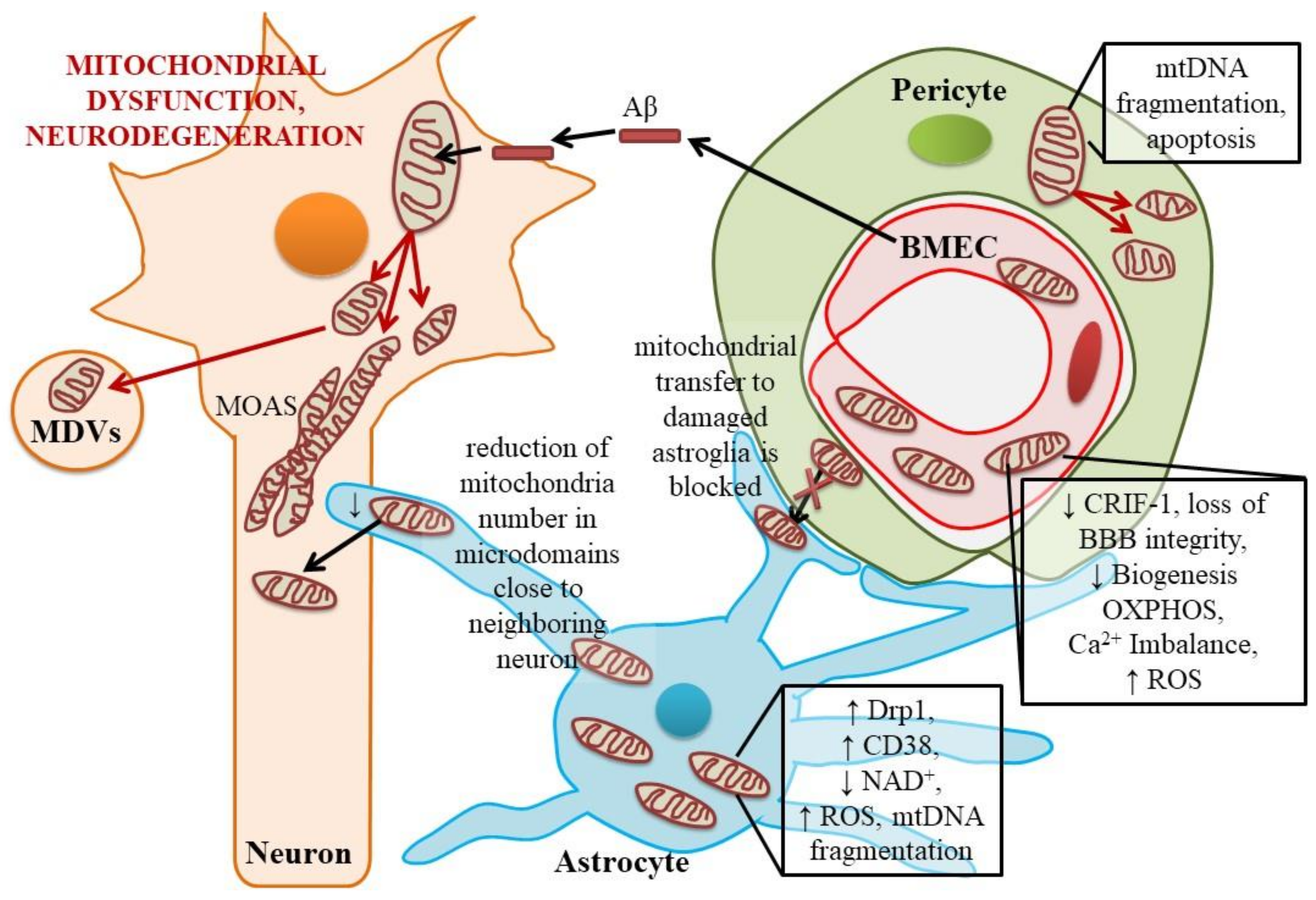
2.2. Dysfunctional Mitochondria in Astroglial Cells
2.3. Mitochondrial Dynamics and Metabolic Control in BMECs and Pericytes
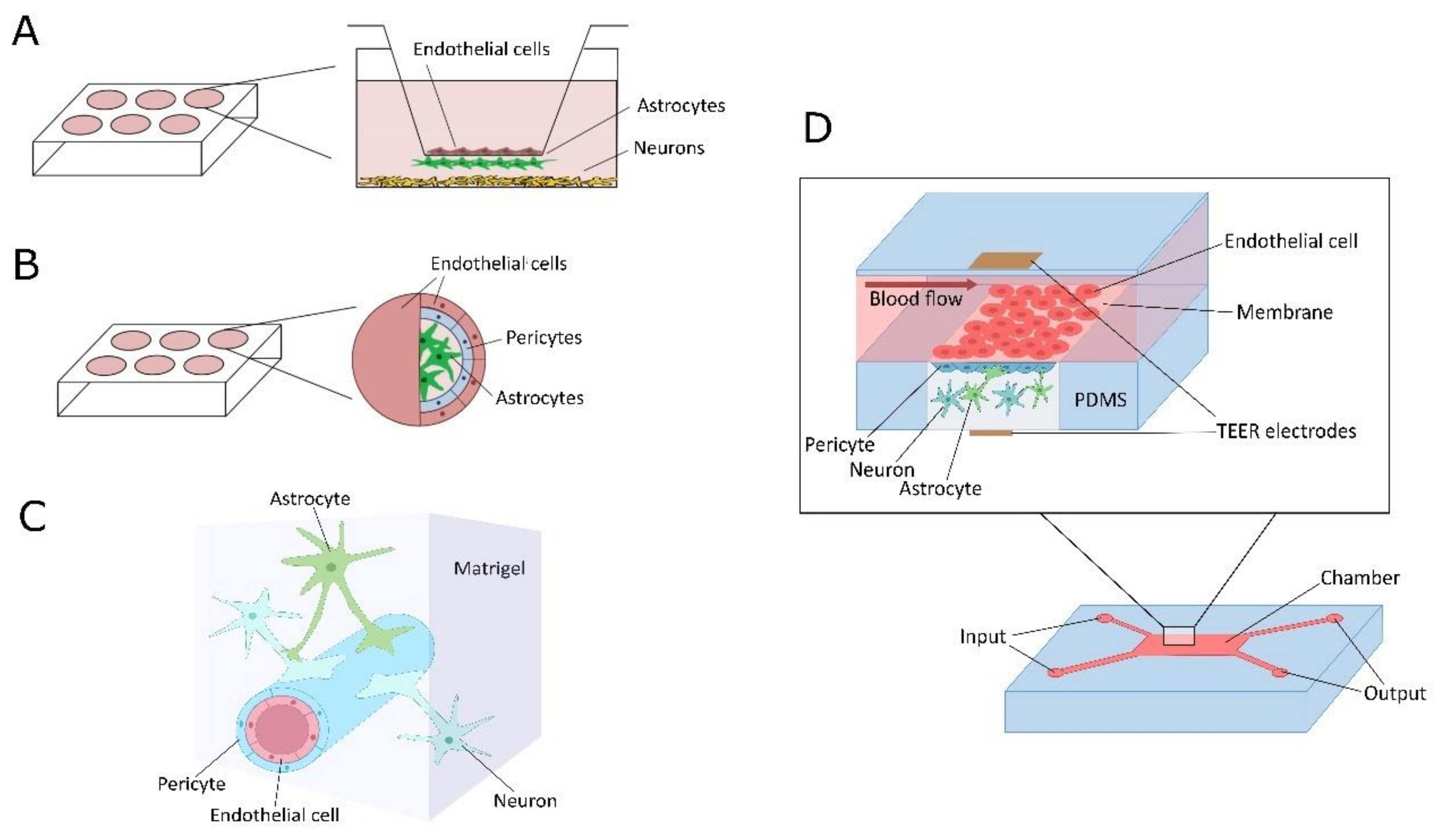
3. Application of In Vitro NVU/BBB Models for Studying Mitochondria-Related Changes in Cell Metabolism: Current Trends and Challenges
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Salmina, A.B.; Kuvacheva, N.V.; Morgun, A.V.; Komleva, Y.K.; Pozhilenkova, E.A.; Lopatina, O.L.; Gorina, Y.V.; Taranushenko, T.E.; Petrova, L.L. Glycolysis-mediated control of blood-brain barrier development and function. Int. J. Biochem. Cell Biol. 2015, 64, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Pozhilenkova, E.A.; Lopatina, O.L.; Komleva, Y.K.; Salmin, V.V.; Salmina, A.B. Blood-brain barrier-supported neurogenesis in healthy and diseased brain. Rev. Neurosci. 2017, 28, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langen, U.H.; Ayloo, S.; Gu, C. Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ustyantseva, E.I.; Medvedev, S.P.; Vetchinova, A.S.; Minina, J.M.; Illarioshkin, S.N.; Zakian, S.M. A Platform for Studying Neurodegeneration Mechanisms Using Genetically Encoded Biosensors. Biochemistry 2019, 84, 299–309. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Hasegawa, M. Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation. Biomolecules 2016, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Salmina, A.B.; Inzhutova, A.I.; Malinovskaya, N.A.; Petrova, M.M. Endothelial dysfunction and repair in Alzheimer-type neurodegeneration: Neuronal and glial control. J. Alzheimers Dis. 2010, 22, 17–36. [Google Scholar] [CrossRef]
- Salmina, A.B.; Komleva, Y.K.; Lopatina, O.L.; Birbrair, A. Pericytes in Alzheimer’s Disease: Novel Clues to Cerebral Amyloid Angiopathy Pathogenesis. Adv. Exp. Med. Biol. 2019, 1147, 147–166. [Google Scholar] [CrossRef]
- Salmina, A.B.; Komleva, Y.K.; Szijártó, I.A.; Gorina, Y.V.; Lopatina, O.L.; Gertsog, G.E.; Filipovic, M.R.; Gollasch, M. H2S- and NO-Signaling Pathways in Alzheimer’s Amyloid Vasculopathy: Synergism or Antagonism? Front. Physiol. 2015, 6, 361. [Google Scholar] [CrossRef] [Green Version]
- Komleva, Y.K.; Lopatina, O.L.; Gorina, Y.V.; Chernykh, A.I.; Trufanova, L.V.; Vais, E.F.; Kharitonova, E.V.; Zhukov, E.L.; Vahtina, L.Y.; Medvedeva, N.N.; et al. Expression of NLRP3 Inflammasomes in Neurogenic Niche Contributes to the Effect of Spatial Learning in Physiological Conditions but Not in Alzheimer’s Type Neurodegeneration. Cell Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, G. Mitochondria fragments fuel the fire of neuroinflammation. Sci. Transl. Med. 2019, 11, eaaz3714. [Google Scholar] [CrossRef]
- Wardelmann, K.; Blümel, S.; Rath, M.; Alfine, E.; Chudoba, C.; Schell, M.; Cai, W.; Hauffe, R.; Warnke, K.; Flore, T.; et al. Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 2019, 21, 68–81. [Google Scholar] [CrossRef]
- Qiu, J.; Tan, Y.-W.; Hagenston, A.M.; Martel, M.-A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.A.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Bischof, G.N.; Jessen, F.; Fliessbach, K.; Dronse, J.; Hammes, J.; Neumaier, B.; Onur, O.; Fink, G.R.; Kukolja, J.; Drzezga, A.; et al. Impact of tau and amyloid burden on glucose metabolism in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2016, 3, 934–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, C.; Santos, M.S.; Oliveira, C. Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cells. Neuroreport 1998, 9, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Kenney, P.M.; Bennett, J.P., Jr. Alzheimer’s Disease Frontal Cortex Mitochondria Show a Loss of Individual Respiratory Proteins but Preservation of Respiratory Supercomplexes. Int. J. Alzheimers Dis. 2019, 2019, 4814783. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Pickrell, A.M.; Fukui, H.; Moraes, C.T. Mitochondrial DNA damage in a mouse model of Alzheimer’s disease decreases amyloid beta plaque formation. Neurobiol. Aging 2013, 34, 2399–2407. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Hui, D.; Chalise, P.; Sharma, P.; Wang, X.; Andrews, S.J.; Pa, J.; Mahnken, J.D.; Morris, J.; Wilkins, H.M.; et al. Exploratory analysis of mtDNA haplogroups in two Alzheimer’s longitudinal cohorts. Alzheimers Dement. 2020, 16, 1164–1172. [Google Scholar] [CrossRef]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [Green Version]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Chai, Y.L.; Xing, H.; Chong, J.R.; Francis, P.T.; Ballard, C.G.; Chen, C.P.; Lai, M.K.P. Mitochondrial Translocase of the Outer Membrane Alterations May Underlie Dysfunctional Oxidative Phosphorylation in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 793–801. [Google Scholar] [CrossRef]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The Voltage-dependent Anion Channel 1 Mediates Amyloid β Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [Green Version]
- Martel, C.; Wang, Z.; Brenner, C. VDAC phosphorylation, a lipid sensor influencing the cell fate. Mitochondrion 2014, 19 Pt A, 69–77. [Google Scholar] [CrossRef]
- Fernandez-Echevarria, C.; Díaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Aβ promotes VDAC1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in Alzheimer’s disease. Neuroscience 2014, 278, 354–366. [Google Scholar] [CrossRef]
- Xu, P.; Chang, J.C.; Zhou, X.; Wang, W.; Bamkole, M.; Wong, E.; Bettayeb, K.; Jiang, L.-L.; Huang, T.; Luo, W.; et al. GSAP regulates mitochondrial function through the Mitochondria-associated ER membrane in the pathogenesis of Alzheimer’s disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Webster, M.T.; Pearce, B.R.; Bowen, D.M.; Francis, P.T. The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J. Neural Transm. 1998, 105, 839–853. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Panes, J.D.; Godoy, P.A.; Silva-Grecchi, T.; Celis, M.T.; Ramirez-Molina, O.; Gavilan, J.; Muñoz-Montecino, C.; Castro, P.A.; Moraga-Cid, G.; Yévenes, G.E.; et al. Changes in PGC-1α/SIRT1 Signaling Impact on Mitochondrial Homeostasis in Amyloid-Beta Peptide Toxicity Model. Front. Pharmacol. 2020, 11, 709. [Google Scholar] [CrossRef]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef] [Green Version]
- Weidling, I.; Swerdlow, R.H. Mitochondrial Dysfunction and Stress Responses in Alzheimer’s Disease. Biology 2019, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pépin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef]
- Norambuena, A.; Bloom, G.S. A Novel Communication Pathway Between Lysosomes and Mitochondria Is Disrupted in Alzheimer’s Disease. Contact 2019, 2, 2515256419865859. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Magistretti, P.J.; Pellerin, L. Alzheimer’s disease: The amyloid hypothesis and the Inverse Warburg effect. Front. Physiol. 2015, 5, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J. Neuron-glia metabolic coupling and plasticity. J. Exp. Biol. 2006, 209, 2304–2311. [Google Scholar] [CrossRef] [Green Version]
- Steinman, M.Q.; Gao, V.; Alberini, C.M. The Role of Lactate-Mediated Metabolic Coupling between Astrocytes and Neurons in Long-Term Memory Formation. Front. Integr. Neurosci. 2016, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate Produced by Glycogenolysis in Astrocytes Regulates Memory Processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109, 87–93. [Google Scholar] [CrossRef]
- Zhou, Z.; Okamoto, K.; Onodera, J.; Hiragi, T.; Andoh, M.; Ikawa, M.; Tanaka, K.F.; Ikegaya, Y.; Koyama, R. Astrocytic cAMP modulates memory via synaptic plasticity. Proc. Natl. Acad. Sci. USA 2021, 118, e2016584118. [Google Scholar] [CrossRef]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-dimensional Ca(2+) imaging advances understanding of astrocyte biology. Science 2017, 356. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Huang, J.-Z.; Chen, Y.; Hu, H.-J.; Tang, X.; Li, X. Effects and mechanism of amyloid β1-42 on mitochondria in astrocytes. Mol. Med. Rep. 2018, 17, 6997–7004. [Google Scholar] [CrossRef]
- Agarwal, A.; Wu, P.H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605. [Google Scholar] [CrossRef] [Green Version]
- Gollihue, J.L.; Norris, C.M. Astrocyte mitochondria: Central players and potential therapeutic targets for neurodegenerative diseases and injury. Ageing Res. Rev. 2020, 59, 101039. [Google Scholar] [CrossRef]
- Genda, E.N.; Jackson, J.G.; Sheldon, A.L.; Locke, S.F.; Greco, T.M.; O’Donnell, J.C.; Spruce, L.A.; Xiao, R.; Guo, W.; Putt, M.; et al. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J. Neurosci. 2011, 31, 18275–18288. [Google Scholar] [CrossRef] [Green Version]
- Voloboueva, L.A.; Suh, S.W.; Swanson, R.A.; Giffard, R.G. Inhibition of mitochondrial function in astrocytes: Implications for neuroprotection. J. Neurochem. 2007, 102, 1383–1394. [Google Scholar] [CrossRef]
- Kubik, L.L.; Philbert, M.A. The role of astrocyte mitochondria in differential regional susceptibility to environmental neurotoxicants: Tools for understanding neurodegeneration. Toxicol. Sci. 2015, 144, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.K.; Götz, M.; et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef] [Green Version]
- Ignatenko, O.; Chilov, D.; Paetau, I.; de Miguel, E.; Jackson, C.B.; Capin, G.; Paetau, A.; Terzioglu, M.; Euro, L.; Suomalainen, A. Loss of mtDNA activates astrocytes and leads to spongiotic encephalopathy. Nat. Commun. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.W.; Min, S.J.; Kim, J.E. CDK5 inhibitors prevent astroglial apoptosis and reactive astrogliosis by regulating PKA and DRP1 phosphorylations in the rat hippocampus. Neurosci. Res. 2017, 119, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liang, R.; Yang, B.; Zhou, Y.; Liu, M.; Fang, F.; Ding, J.; Fan, Y.; Hu, G. Aquaporin-4 mediates communication between astrocyte and microglia: Implications of neuroinflammation in experimental Parkinson’s disease. Neuroscience 2016, 317, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [Green Version]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife 2018, 7. [Google Scholar] [CrossRef]
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [Green Version]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef]
- Huang, Y.; Li, S.-n.; Zhou, X.-y.; Zhang, L.-x.; Chen, G.-x.; Wang, T.-h.; Xia, Q.-j.; Liang, N.; Zhang, X. The Dual Role of AQP4 in Cytotoxic and Vasogenic Edema Following Spinal Cord Contusion and Its Possible Association With Energy Metabolism via COX5A. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Amiry-Moghaddam, M.; Lindland, H.; Zelenin, S.; Roberg, B.Å.; Gundersen, B.B.; Petersen, P.; Rinvik, E.; Torgner, I.A.; Ottersen, O.P. Brain mitochondria contain aquaporin water channels: Evidence for the expression of a short AQP9 isoform in the inner mitochondrial membrane. FASEB J. 2005, 19, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef] [Green Version]
- Horenstein, A.L.; Faini, A.C.; Morandi, F.; Bracci, C.; Lanza, F.; Giuliani, N.; Paulus, A.; Malavasi, F. The Circular Life of Human CD38: From Basic Science to Clinics and Back. Molecules 2020, 25, 4844. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [Green Version]
- Higashida, H.; Egorova, A.; Higashida, C.; Zhong, Z.G.; Yokoyama, S.; Noda, M.; Zhang, J.S. Sympathetic potentiation of cyclic ADP-ribose formation in rat cardiac myocytes. J. Biol. Chem. 1999, 274, 33348–33354. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Higashida, H.; Yokoyama, S.; Kikuchi, M.; Munesue, T. CD38 and its role in oxytocin secretion and social behavior. Horm. Behav. 2012, 61, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Takaso, Y.; Noda, M.; Hattori, T.; Roboon, J.; Hatano, M.; Sugimoto, H.; Brenner, C.; Yamamoto, Y.; Okamoto, H.; Higashida, H.; et al. Deletion of CD38 and supplementation of NAD(+) attenuate axon degeneration in a mouse facial nerve axotomy model. Sci. Rep. 2020, 10, 17795. [Google Scholar] [CrossRef]
- Higashida, H.; Salmina, A.B.; Olovyannikova, R.Y.; Hashii, M.; Yokoyama, S.; Koizumi, K.; Jin, D.; Liu, H.X.; Lopatina, O.; Amina, S.; et al. Cyclic ADP-ribose as a universal calcium signal molecule in the nervous system. Neurochem. Int. 2007, 51, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Ceni, C.; Pochon, N.; Brun, V.; Muller-Steffner, H.; Andrieux, A.; Grunwald, D.; Schuber, F.; De Waard, M.; Lund, F.; Villaz, M.; et al. CD38-dependent ADP-ribosyl cyclase activity in developing and adult mouse brain. Biochem. J. 2003, 370, 175–183. [Google Scholar] [CrossRef]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.-J.; Lund, F.E.; Stein, R. Dual role of CD38 in microglial activation and activation-induced cell death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruzzone, S.; Verderio, C.; Schenk, U.; Fedele, E.; Zocchi, E.; Matteoli, M.; De Flora, A. Glutamate-mediated overexpression of CD38 in astrocytes cultured with neurones. J. Neurochem. 2004, 89, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Walseth, T.F.; Borgmann, K.; Wu, L.; Bidasee, K.R.; Kannan, M.S.; Ghorpade, A. CD38/cyclic ADP-ribose regulates astrocyte calcium signaling: Implications for neuroinflammation and HIV-1-associated dementia. J. Neuroimmune Pharmacol. 2008, 3, 154–164. [Google Scholar] [CrossRef]
- Liu, D.; Gao, Y.; Liu, J.; Huang, Y.; Yin, J.; Feng, Y.; Shi, L.; Meloni, B.P.; Zhang, C.; Zheng, M.; et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct. Target. Ther. 2021, 6, 65. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef]
- Bergau, N.; Maul, S.; Rujescu, D.; Simm, A.; Navarrete Santos, A. Reduction of Glycolysis Intermediate Concentrations in the Cerebrospinal Fluid of Alzheimer’s Disease Patients. Front. Neurosci. 2019, 13, 871. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Eide, P.K.; Hasan-Olive, M.M.; Hansson, H.A.; Enger, R. Increased occurrence of pathological mitochondria in astrocytic perivascular endfoot processes and neurons of idiopathic intracranial hypertension. J. Neurosci. Res. 2021, 99, 467–480. [Google Scholar] [CrossRef]
- Khilazheva, E.D.; Pisareva, N.V.; Morgun, A.V.; Boitsova, E.B.; Taranushenko, T.E.; Frolova, O.V.; Salmina, A.B. Activation of GPR81 lactate receptors stimulates mitochondrial biogenesis in cerebral microvessel endothelial cells. J. Clin. Exp. Neuropsychol. 2017, 11, 34–39. [Google Scholar]
- Subburaju, S.; Kaye, S.; Choi, Y.K.; Baruah, J.; Datta, D.; Ren, J.; Kumar, A.S.; Szabo, G.; Fukumura, D.; Jain, R.K.; et al. NAD+-mediated rescue of prenatal forebrain angiogenesis restores postnatal behavior. Sci. Adv. 2020, 6, eabb9766. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis revisited from a metabolic perspective: Role and therapeutic implications of endothelial cell metabolism. Biol. Open 2017, 7, 170219. [Google Scholar] [CrossRef] [Green Version]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517. [Google Scholar] [CrossRef]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Brandl, L.; Zhang, Y.; Kirstein, N.; Sendelhofert, A.; Boos, S.L.; Jung, P.; Greten, F.; Rad, R.; Menssen, A. Targeting c-MYC through Interference with NAMPT and SIRT1 and Their Association to Oncogenic Drivers in Murine Serrated Intestinal Tumorigenesis. Neoplasia 2019, 21, 974–988. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Florea, V.; Bhagavatula, N.; Simovic, G.; Macedo, F.Y.; Fock, R.A.; Rodrigues, C.O. c-Myc is essential to prevent endothelial pro-inflammatory senescent phenotype. PLoS ONE 2013, 8, e73146. [Google Scholar] [CrossRef] [Green Version]
- Malinovskaya, N.A.; Komleva, Y.K.; Salmin, V.V.; Morgun, A.V.; Shuvaev, A.N.; Panina, Y.A.; Boitsova, E.B.; Salmina, A.B. Endothelial Progenitor Cells Physiology and Metabolic Plasticity in Brain Angiogenesis and Blood-Brain Barrier Modeling. Front. Physiol. 2016, 7, 599. [Google Scholar] [CrossRef] [Green Version]
- Oikari, L.E.; Pandit, R.; Stewart, R.; Cuní-López, C.; Quek, H.; Sutharsan, R.; Rantanen, L.M.; Oksanen, M.; Lehtonen, S.; de Boer, C.M.; et al. Altered Brain Endothelial Cell Phenotype from a Familial Alzheimer Mutation and Its Potential Implications for Amyloid Clearance and Drug Delivery. Stem Cell Rep. 2020, 14, 924–939. [Google Scholar] [CrossRef]
- Caja, S.; Enríquez, J.A. Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox. Biol. 2017, 12, 821–827. [Google Scholar] [CrossRef]
- Falkenberg, K.D.; Rohlenova, K.; Luo, Y.; Carmeliet, P. The metabolic engine of endothelial cells. Nat. Metab. 2019, 1, 937–946. [Google Scholar] [CrossRef]
- Stapor, P.; Wang, X.; Goveia, J.; Moens, S.; Carmeliet, P. Angiogenesis revisited-role and therapeutic potential of targeting endothelial metabolism. J. Cell Sci. 2014, 127, 4331–4341. [Google Scholar] [CrossRef] [Green Version]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Jefferies, W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS ONE 2011, 6, e23789. [Google Scholar] [CrossRef] [Green Version]
- Hinca, S.B.; Salcedo, C.; Wagner, A.; Goldeman, C.; Sadat, E.; Aibar, M.M.D.; Maechler, P.; Brodin, B.; Aldana, B.I.; Helms, H.C.C. Brain endothelial cells metabolize glutamate via glutamate dehydrogenase to replenish TCA-intermediates and produce ATP under hypoglycemic conditions. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lecointre, M.; Hauchecorne, M.; Chaussivert, A.; Marret, S.; Leroux, P.; Jegou, S.; Leroux-Nicollet, I.; Gonzalez, B.J.; Henry, V.J. The efficiency of glutamate uptake differs between neonatal and adult cortical microvascular endothelial cells. J. Cereb. Blood Flow. Metab. 2014, 34, 764–767. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Kumar, T.P.; Joshee, S.; Kirschstein, T.; Subburaju, S.; Khalili, J.S.; Kloepper, J.; Du, C.; Elkhal, A.; Szabó, G.; et al. Endothelial cell-derived GABA signaling modulates neuronal migration and postnatal behavior. Cell Res. 2018, 28, 221–248. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.L.; Steuer, E.L.; Pochtarev, V.; Swain, R.A. Angiogenesis but not neurogenesis is critical for normal learning and memory acquisition. Neuroscience 2010, 171, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Jang, Y.; Han, J.; Kim, S.J.; Ju, X.; Lee, Y.L.; Cui, J.; Zhu, J.; Ryu, M.J.; Choi, S.Y.; et al. Endothelial-specific Crif1 deletion induces BBB maturation and disruption via the alteration of actin dynamics by impaired mitochondrial respiration. J. Cereb. Blood Flow Metab. 2020, 40, 1546–1561. [Google Scholar] [CrossRef]
- Kitazume, S.; Tachida, Y.; Kato, M.; Yamaguchi, Y.; Honda, T.; Hashimoto, Y.; Wada, Y.; Saito, T.; Iwata, N.; Saido, T.; et al. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 2010, 285, 40097–40103. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.A.; Santhanam, A.V.; Katusic, Z.S. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ. Res. 2010, 107, 1498–1502. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Katz, P.S.; Dutta, S.; Katakam, P.V.G.; Moreira, P.I.; Busija, D.W. Increased susceptibility to amyloid-β toxicity in rat brain microvascular endothelial cells under hyperglycemic conditions. J. Alzheimers Dis. 2014, 38, 75–83. [Google Scholar] [CrossRef]
- Quintana, D.D.; Garcia, J.A.; Anantula, Y.; Rellick, S.L.; Engler-Chiurazzi, E.B.; Sarkar, S.N.; Brown, C.M.; Simpkins, J.W. Amyloid-β Causes Mitochondrial Dysfunction via a Ca2+-Driven Upregulation of Oxidative Phosphorylation and Superoxide Production in Cerebrovascular Endothelial Cells. J. Alzheimers Dis. 2020, 75, 119–138. [Google Scholar] [CrossRef]
- Xu, J.; Chen, S.; Ku, G.; Ahmed, S.H.; Xu, J.; Chen, H.; Hsu, C.Y. Amyloid β Peptide–Induced Cerebral Endothelial Cell Death Involves Mitochondrial Dysfunction and Caspase Activation. J. Cereb. Blood Flow Metab. 2001, 21, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Durán-Prado, M.; Frontiñán, J.; Santiago-Mora, R.; Peinado, J.R.; Parrado-Fernández, C.; Gómez-Almagro, M.V.; Moreno, M.; López-Domínguez, J.A.; Villalba, J.M.; Alcaín, F.J. Coenzyme Q10 protects human endothelial cells from β-amyloid uptake and oxidative stress-induced injury. PLoS ONE 2014, 9, e109223. [Google Scholar] [CrossRef]
- Shapoval, N.S.; Malinovskaya, N.A.; Morgun, A.V.; Salmina, A.B.; Obolenskaya, O.N.; Medvedeva, N.A.; Medvedev, O.S. The effect of ubiquinol on cerebral endothelial cells in different regions of rat brain. Tsitologiya 2020, 62, 894–902. [Google Scholar] [CrossRef]
- Shapoval, N.S.; Medvedev, O.S.; Medvedeva, N.A.; Morgun, A.V.; Boytsova, E.B.; Osipova, E.D.; Salmina, A.B. Influence of the oxidized and reduced forms of coenzyme Q10 (ubiquinone and ubiquinol) to cerebral endothelial cells in the blood-brain barrier model. Tsitologiya 2020, 62, 428–436. [Google Scholar] [CrossRef]
- Haileselassie, B.; Joshi, A.U.; Minhas, P.S.; Mukherjee, R.; Andreasson, K.I.; Mochly-Rosen, D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J. Neuroinflammation 2020, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Boitsova, E.B.; Morgun, A.V.; Osipova, E.D.; Pozhilenkova, E.A.; Martinova, G.P.; Frolova, O.V.; Olovannikova, R.Y.; Tohidpour, A.; Gorina, Y.V.; Panina, Y.A.; et al. The inhibitory effect of LPS on the expression of GPR81 lactate receptor in blood-brain barrier model in vitro. J. Neuroinflammation 2018, 15, 196. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, K.; Molina, A.J.A.; Roy, S. High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8657–8664. [Google Scholar] [CrossRef] [Green Version]
- Castro, V.; Skowronska, M.; Lombardi, J.; He, J.; Seth, N.; Velichkovska, M.; Toborek, M. Occludin regulates glucose uptake and ATP production in pericytes by influencing AMP-activated protein kinase activity. J. Cereb. Blood. Flow Metab. 2018, 38, 317–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon-Martinez, L.; Villafranca-Baughman, D.; Quintero, H.; Kacerovsky, J.B.; Dotigny, F.; Murai, K.K.; Prat, A.; Drapeau, P.; Di Polo, A. Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 2020, 585, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, A.V.; Stamatovic, S.M.; Phillips, C.M.; Martinez-Revollar, G.; Keep, R.F. Modeling blood–brain barrier pathology in cerebrovascular disease in vitro: Current and future paradigms. Fluids Barriers CNS 2020, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Offeddu, G.S.; Shin, Y.; Kamm, R.D. Microphysiological models of neurological disorders for drug development. Curr. Opin. Biomed. Eng. 2020, 13, 119–126. [Google Scholar] [CrossRef]
- Gastfriend, B.D.; Palecek, S.P.; Shusta, E.V. Modeling the blood-brain barrier: Beyond the endothelial cells. Curr. Opin. Biomed. Eng. 2018, 5, 6–12. [Google Scholar] [CrossRef]
- Osipova, E.D.; Komleva, Y.K.; Morgun, A.V.; Lopatina, O.L.; Panina, Y.A.; Olovyannikova, R.Y.; Vais, E.F.; Salmin, V.V.; Salmina, A.B. Designing in vitro Blood-Brain Barrier Models Reproducing Alterations in Brain Aging. Front. Aging Neurosci. 2018, 10, 234. [Google Scholar] [CrossRef] [Green Version]
- Khilazheva, E.D.; Boytsova, E.B.; Pozhilenkova, E.A.; Solonchuk, Y.R.; Salmina, A.B. Obtaining a three-cell model of a neurovascular unit in vitro. Tissue Cell 2015, 9, 447–451. [Google Scholar] [CrossRef]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Venkataraman, L.; Fair, S.R.; McElroy, C.A.; Hester, M.E.; Fu, H. Modeling neurodegenerative diseases with cerebral organoids and other three-dimensional culture systems: Focus on Alzheimer’s disease. Stem Cell Rev. Rep. 2020, 1–22. [Google Scholar] [CrossRef]
- Wang, H. Modeling Neurological Diseases With Human Brain Organoids. Front. Synaptic. Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef]
- van der Helm, M.W.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L.I. Microfluidic organ-on-chip technology for blood-brain barrier research. Tissue Barriers 2016, 4, e1142493. [Google Scholar] [CrossRef] [Green Version]
- Winkelman, M.A.; Koppes, A.N.; Koppes, R.A.; Dai, G. Bioengineering the neurovascular niche to study the interaction of neural stem cells and endothelial cells. APL Bioeng. 2021, 5, 011507. [Google Scholar] [CrossRef]
- Kuvacheva, N.V.; Morgun, A.V.; Malinovskaya, N.A.; Gorina, Y.V.; Khilazheva, E.D.; Pozhilenkova, E.A.; Panina, Y.A.; Boytsova, E.B.; Ruzaeva, V.A.; Trufanova, L.V.; et al. Tight junction proteins of cerebral endothelial cells in early postnatal development. Tissue Cell 2016, 10, 372–377. [Google Scholar] [CrossRef]
- Ruzaeva, V.A.; Morgun, A.V.; Khilazheva, E.D.; Kuvacheva, N.V.; Pozhilenkova, E.A.; Boitsova, E.B.; Martynova, G.P.; Taranushenko, T.E.; Salmina, A.B. Development of blood-brain barrier under the modulation of HIF activity in astroglialand neuronal cells in vitro. Biomed. Khim. 2016, 62, 664–669. [Google Scholar] [CrossRef] [Green Version]
- Malinovskaya, N.A.; Morgun, A.V.; Pisareva, N.V.; Osipova, E.D.; Boytsova, E.B.; Panina, Y.A.; Zhukov, E.L.; Medvedeva, N.N.; Salmina, A.B. Changes in the Permeability and Expression of Markers of the Structural and Functional Integrity of the Blood–Brain Barrier under Early Postnatal Hypoxia in vivo. Neurochem. Res. 2018, 12, 228–240. [Google Scholar] [CrossRef]
- Khilazheva, E.D.; Morgun, A.V.; Boytsova, E.B.; Mosiagina, A.I.; Shuvaev, A.N.; Malinovskaya, N.A.; Uspenskaya, Y.A.; Pozhilenkova, E.A.; Salmina, A.B. Features of the in vitro expression profile of hippocampal neurogenic niche cells during optogenetic stimulation. Biochemistry 2021, 67, 34–41. [Google Scholar] [CrossRef]
- Morgun, A.V.; Osipova, E.D.; Boytsova, E.B.; Shuvaev, A.N.; Komleva, Y.K.; Trufanova, L.V.; Vais, E.F.; Salmina, A.B. Astroglia-mediated regulation of cell development in the model of neurogenic niche in vitro treated with Aβ1-42. Biomed. Khim. 2019, 65, 366–373. [Google Scholar] [CrossRef]
- Salmin, V.V.; Komleva, Y.K.; Kuvacheva, N.V.; Morgun, A.V.; Khilazheva, E.D.; Lopatina, O.L.; Pozhilenkova, E.A.; Shapovalov, K.A.; Uspenskaya, Y.A.; Salmina, A.B. Differential Roles of Environmental Enrichment in Alzheimer’s Type of Neurodegeneration and Physiological Aging. Front. Aging Neurosci. 2017, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Pérez Del Palacio, J.; Díaz, C.; de la Cruz, M.; Annang, F.; Martín, J.; Pérez-Victoria, I.; González-Menéndez, V.; de Pedro, N.; Tormo, J.R.; Algieri, F.; et al. High-Throughput Screening Platform for the Discovery of New Immunomodulator Molecules from Natural Product Extract Libraries. J. Biomol. Screen 2016, 21, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Salvador, E.; Förster, C. In silico, in vitro, and in vivo methods to analyse drug permeation across the blood-brain barrier: A critical review. Anaesth. Intensive Care 2013, 1, 13. [Google Scholar] [CrossRef]
- Cho, C.-F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Nzou, G.; Wicks, R.T.; Wicks, E.E.; Seale, S.A.; Sane, C.H.; Chen, A.; Murphy, S.V.; Jackson, J.D.; Atala, A.J. Human Cortex Spheroid with a Functional Blood Brain Barrier for High-Throughput Neurotoxicity Screening and Disease Modeling. Sci. Rep. 2018, 8, 7413. [Google Scholar] [CrossRef] [Green Version]
- Qian, T.; Maguire, S.E.; Canfield, S.G.; Bao, X.; Olson, W.R.; Shusta, E.V.; Palecek, S.P. Directed differentiation of human pluripotent stem cells to blood-brain barrier endothelial cells. Sci. Adv. 2017, 3, e1701679. [Google Scholar] [CrossRef] [Green Version]
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Canfield, S.G.; Stebbins, M.J.; Faubion, M.G.; Gastfriend, B.D.; Palecek, S.P.; Shusta, E.V. An isogenic neurovascular unit model comprised of human induced pluripotent stem cell-derived brain microvascular endothelial cells, pericytes, astrocytes, and neurons. Fluids Barriers CNS 2019, 16, 25. [Google Scholar] [CrossRef]
- Jagadeesan, S.; Workman, M.J.; Herland, A.; Svendsen, C.N.; Vatine, G.D. Generation of a Human iPSC-Based Blood-Brain Barrier Chip. J. Vis. Exp. 2020. [Google Scholar] [CrossRef]
- Hartlaub, A.M.; McElroy, C.A.; Maitre, N.L.; Hester, M.E. Modeling Human Brain Circuitry Using Pluripotent Stem Cell Platforms. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Nickels, S.L.; Modamio, J.; Mendes-Pinheiro, B.; Monzel, A.S.; Betsou, F.; Schwamborn, J.C. Reproducible generation of human midbrain organoids for in vitro modeling of Parkinson’s disease. Stem Cell Res. 2020, 46, 101870. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, S.N.; Xu, T.Y.; Miao, Z.W.; Su, D.F.; Miao, C.Y. Organoid technology for brain and therapeutics research. CNS Neurosci. Ther. 2017, 23, 771–778. [Google Scholar] [CrossRef]
- Hämäläinen, R.H. Mitochondrial DNA mutations in iPS cells: mtDNA integrity as standard iPSC selection criteria? EMBO J. 2016, 35, 1960–1962. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ryan, S.K.; Deboer, E.; Cook, K.; Fitzgerald, S.; Lachman, H.M.; Wallace, D.C.; Goldberg, E.M.; Anderson, S.A. Mitochondrial deficits in human iPSC-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl. Psychiatry 2019, 9, 302. [Google Scholar] [CrossRef] [Green Version]
- Duong, A.; Evstratova, A.; Sivitilli, A.; Hernandez, J.J.; Gosio, J.; Wahedi, A.; Sondheimer, N.; Wrana, J.L.; Beaulieu, J.-M.; Attisano, L.; et al. Characterization of mitochondrial health from human peripheral blood mononuclear cells to cerebral organoids derived from induced pluripotent stem cells. Sci. Rep. 2021, 11, 4523. [Google Scholar] [CrossRef]
- Papaspyropoulos, A.; Tsolaki, M.; Foroglou, N.; Pantazaki, A.A. Modeling and Targeting Alzheimer’s Disease With Organoids. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Workman, M.J.; Svendsen, C.N. Recent advances in human iPSC-derived models of the blood–brain barrier. Fluids Barriers CNS 2020, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.H.; Marinelli, N.A.; Shi, Y.; McClatchey, P.M.; Balotin, K.M.; Gullett, D.R.; Hagerla, K.A.; Bowman, A.B.; Ess, K.C.; Wikswo, J.P.; et al. A Simplified, Fully Defined Differentiation Scheme for Producing Blood-Brain Barrier Endothelial Cells from Human iPSCs. Stem Cell Rep. 2019, 12, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Faley, S.L.; Neal, E.H.; Wang, J.X.; Bosworth, A.M.; Weber, C.M.; Balotin, K.M.; Lippmann, E.S.; Bellan, L.M. iPSC-Derived Brain Endothelium Exhibits Stable, Long-Term Barrier Function in Perfused Hydrogel Scaffolds. Stem Cell Rep. 2019, 12, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, H.; Gastfriend, B.D.; Soldati, S.; Perriot, S.; Mathias, A.; Sano, Y.; Shimizu, F.; Gosselet, F.; Kanda, T.; Palecek, S.P.; et al. Advancing human induced pluripotent stem cell-derived blood-brain barrier models for studying immune cell interactions. FASEB J. 2020, 34, 16693–16715. [Google Scholar] [CrossRef]
- DeStefano, J.G.; Jamieson, J.J.; Linville, R.M.; Searson, P.C. Benchmarking in vitro tissue-engineered blood–brain barrier models. Fluids Barriers CNS 2018, 15, 32. [Google Scholar] [CrossRef] [Green Version]
- Berger, E.; Magliaro, C.; Paczia, N.; Monzel, A.S.; Antony, P.; Linster, C.L.; Bolognin, S.; Ahluwalia, A.; Schwamborn, J.C. Millifluidic culture improves human midbrain organoid vitality and differentiation. Lab.Chip 2018, 18, 3172–3183. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Sun, L.; Wang, M.; Liu, J.; Zhong, S.; Li, R.; Li, P.; Guo, L.; Fang, A.; Chen, R.; et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 2020, 18, e3000705. [Google Scholar] [CrossRef]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef]
- Ham, O.; Jin, Y.B.; Kim, J.; Lee, M.O. Blood vessel formation in cerebral organoids formed from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2020, 521, 84–90. [Google Scholar] [CrossRef]
- Matsui, T.K.; Tsuru, Y.; Hasegawa, K.; Kuwako, K.-I. Vascularization of human brain organoids. Stem Cells 2021, 1–8. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmina, A.B.; Kharitonova, E.V.; Gorina, Y.V.; Teplyashina, E.A.; Malinovskaya, N.A.; Khilazheva, E.D.; Mosyagina, A.I.; Morgun, A.V.; Shuvaev, A.N.; Salmin, V.V.; et al. Blood–Brain Barrier and Neurovascular Unit In Vitro Models for Studying Mitochondria-Driven Molecular Mechanisms of Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 4661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094661
Salmina AB, Kharitonova EV, Gorina YV, Teplyashina EA, Malinovskaya NA, Khilazheva ED, Mosyagina AI, Morgun AV, Shuvaev AN, Salmin VV, et al. Blood–Brain Barrier and Neurovascular Unit In Vitro Models for Studying Mitochondria-Driven Molecular Mechanisms of Neurodegeneration. International Journal of Molecular Sciences. 2021; 22(9):4661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094661
Chicago/Turabian StyleSalmina, Alla B., Ekaterina V. Kharitonova, Yana V. Gorina, Elena A. Teplyashina, Natalia A. Malinovskaya, Elena D. Khilazheva, Angelina I. Mosyagina, Andrey V. Morgun, Anton N. Shuvaev, Vladimir V. Salmin, and et al. 2021. "Blood–Brain Barrier and Neurovascular Unit In Vitro Models for Studying Mitochondria-Driven Molecular Mechanisms of Neurodegeneration" International Journal of Molecular Sciences 22, no. 9: 4661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094661