
The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease

 , , ,
, , ,
Abstract
:1. Introduction
1.1. Parkinson’s Disease
1.2. Microglia
2. Human Post-Mortem Tissue Studies
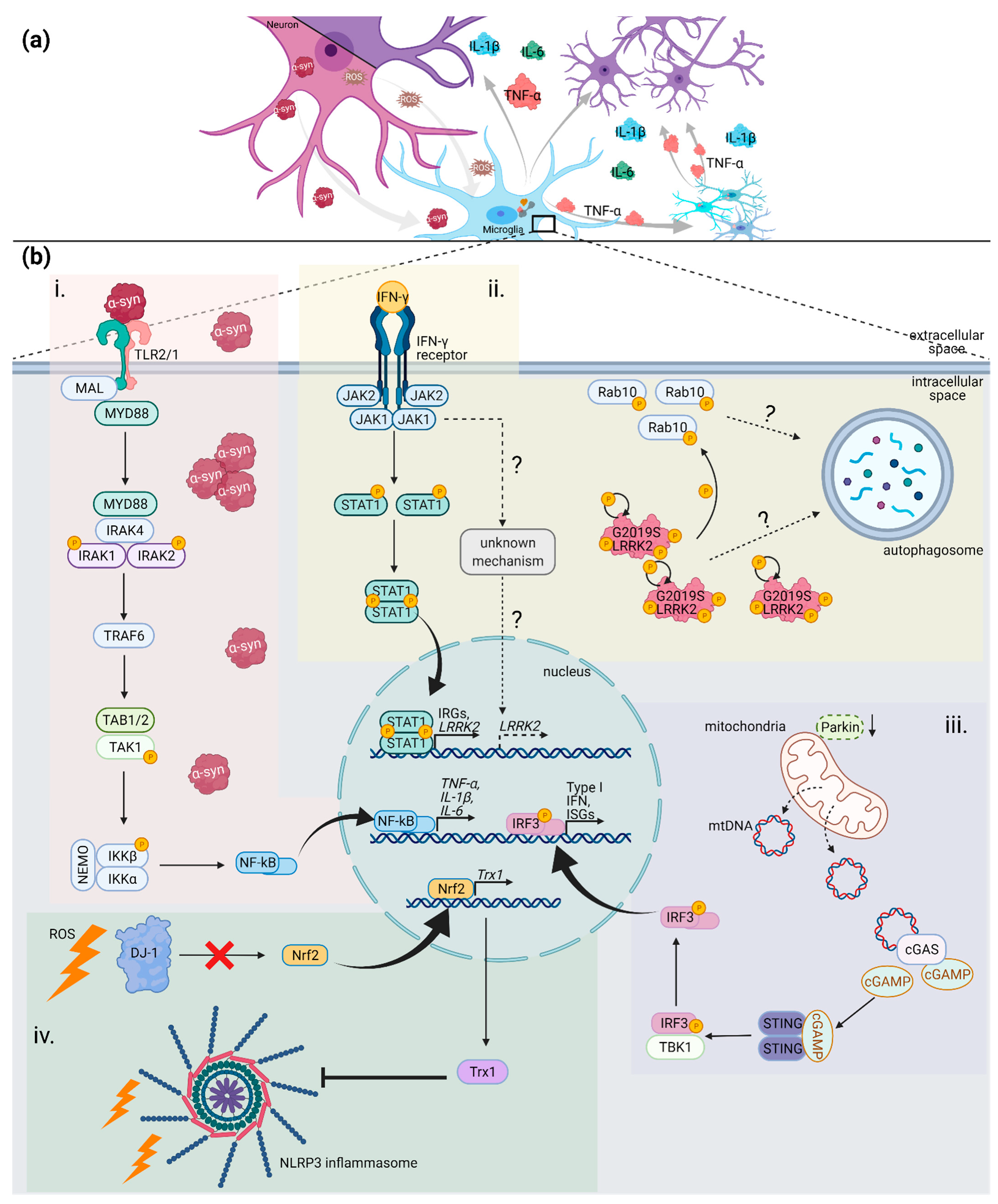
3. (Neuro) Inflammation in PD
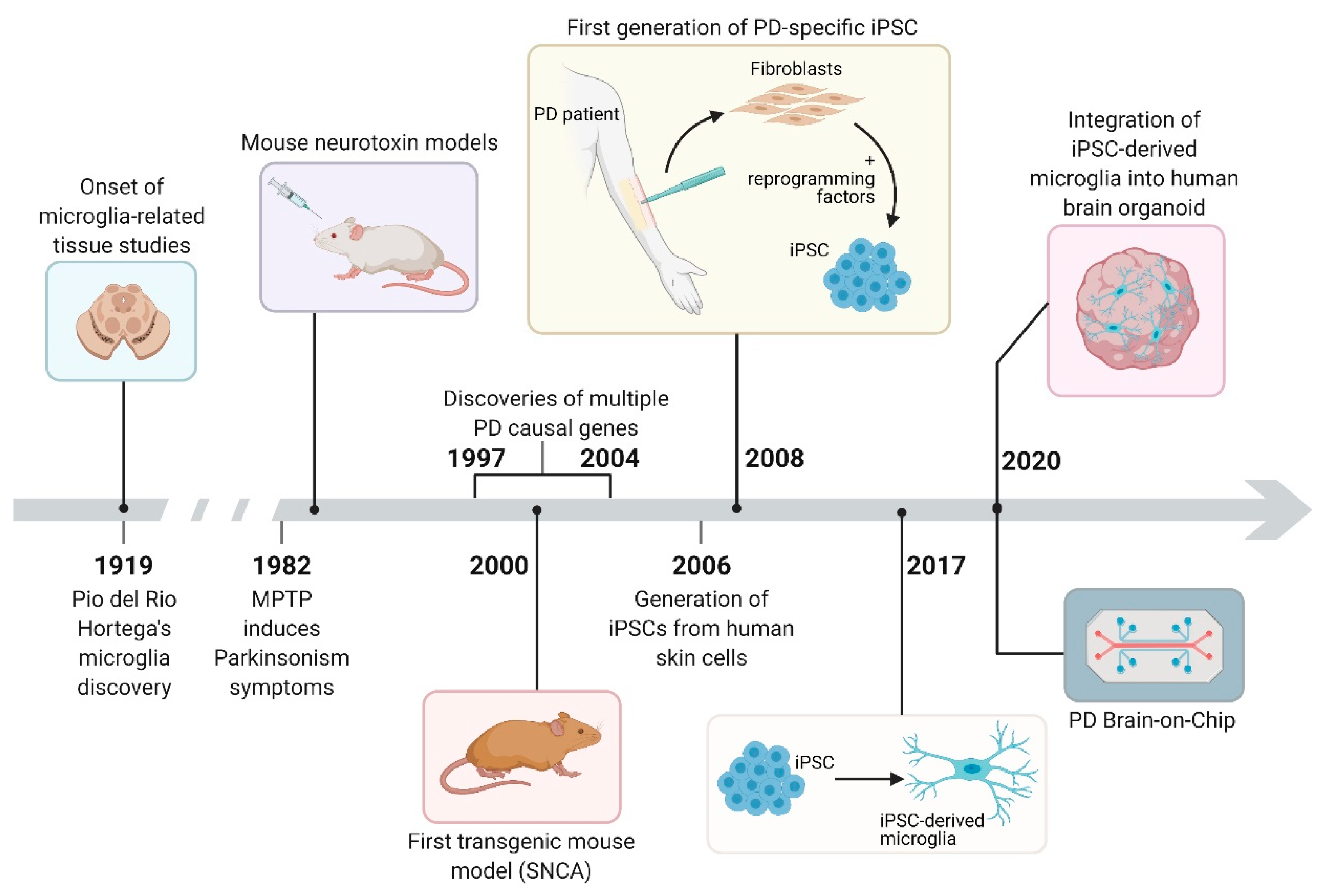
4. Disease Modeling: Animal Models, Patient-Derived iPSC Models, and Cell-Based 3D Models and Platforms
4.1. Animal Models
4.1.1. Neurotoxin Model
4.1.2. SNCA
4.1.3. LRRK2
4.1.4. PRKN
4.1.5. DJ-1
4.2. Human Induced Pluripotent Stem Cell (iPSC) Studies
4.2.1. SNCA
4.2.2. LRRK2
4.3. Human Cell-Based 3D Models and Platforms
5. Immunotherapies in Parkinson’s Disease
5.1. Anti-Inflammatory Treatments in PD
5.2. Anti-α-Synuclein Immunotherapies
6. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Toulouse, A.; Sullivan, A.M. Progress in Parkinson’s Disease-Where Do We Stand? Prog. Neurobiol. 2008, 85, 376–392. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s Disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s Disease Genes pink1 and Parkin Promote Mitochondrial Fission And/or Inhibit Fusion in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Del Rey, N.L.-G.; Quiroga-Varela, A.; Garbayo, E.; Carballo-Carbajal, I.; Fernández-Santiago, R.; Monje, M.H.G.; Trigo-Damas, I.; Blanco-Prieto, M.J.; Blesa, J. Advances in Parkinson’s Disease: 200 Years Later. Front. Neuroanat. 2018, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Hindle, J.V. Ageing, Neurodegeneration and Parkinson’s Disease. Age Ageing 2010, 39, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a Primary Risk Factor for Parkinson’s Disease: Evidence from Studies of Non-Human Primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Chade, A.R.; Kasten, M.; Tanner, C.M. Nongenetic Causes of Parkinson’s Disease. In Parkinson’s Disease and Related Disorders; Springer: Vienna, Austria, 2006; pp. 147–151. [Google Scholar]
- Braak, H.; Rüb, U.; Del Tredici, K. Cognitive Decline Correlates with Neuropathological Stage in Parkinson’s Disease. J. Neurol. Sci. 2006, 248, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-Motor Features of Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of Active Nerve Cell Degeneration in the Substantia Nigra of Humans Years after 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New Insights into the Complex Role of Mitochondria in Parkinson’s Disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Hauser, D.N.; Hastings, T.G. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease and Monogenic Parkinsonism. Neurobiol. Dis. 2013, 51, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.E.; Lozano, A.M. Parkinson’s Disease. First of Two Parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why Neurodegenerative Diseases Are Progressive: Uncontrolled Inflammation Drives Disease Progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Ho, M.S.; Zorec, R.; Parpura, V. (Eds.) Neuroglia in Neurodegenerative Diseases; Springer: Singapore, 2019. [Google Scholar]
- Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Matute, C. Pío Del Río Hortega and the Discovery of the Oligodendrocytes. Front. Neuroanat. 2015, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; de Castro, F.; Del Río-Hortega, J.; Rafael Iglesias-Rozas, J.; Garrosa, M.; Kettenmann, H. The “Big-Bang” for Modern Glial Biology: Translation and Comments on Pío Del Río-Hortega 1919 Series of Papers on Microglia. Glia 2016, 64, 1801–1840. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Priller, J.; Flügel, A.; Wehner, T.; Boentert, M.; Haas, C.A.; Prinz, M.; Fernández-Klett, F.; Prass, K.; Bechmann, I.; de Boer, B.A.; et al. Targeting Gene-Modified Hematopoietic Cells to the Central Nervous System: Use of Green Fluorescent Protein Uncovers Microglial Engraftment. Nat. Med. 2001, 7, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Bruttger, J.; Karram, K.; Wörtge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 2015, 43, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V Cortical Neurons Require Microglial Support for Survival during Postnatal Development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New Tools for Studying Microglia in the Mouse and Human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.G.; Tang, T.M.; Mendsaikhan, A.; Tooyama, I.; Serrano, G.E.; Sue, L.I.; Beach, T.G.; Lue, L.-F. Patterns of Expression of Purinergic Receptor P2RY12, a Putative Marker for Non-Activated Microglia, in Aged and Alzheimer’s Disease Brains. Int. J. Mol. Sci. 2020, 21, 678. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.G.; Mrak, R.E.; Griffin, W.S. Enlarged and Phagocytic, but Not Primed, Interleukin-1 Alpha-Immunoreactive Microglia Increase with Age in Normal Human Brain. Acta Neuropathol. 1998, 95, 229–234. [Google Scholar] [CrossRef]
- Conde, J.R.; Streit, W.J. Microglia in the Aging Brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wang, X.; Xiong, W.; Chen, J. In Vivo Reprogramming Reactive Glia into iPSCs to Produce New Neurons in the Cortex Following Traumatic Brain Injury. Sci. Rep. 2016, 6, 22490. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of Major Histocompatibility Complex Class II-Positive Microglia and Cytokine Profile of Parkinson’s Disease Brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Mastroeni, D.; Nolz, J.; Sekar, S.; Delvaux, E.; Serrano, G.; Cuyugan, L.; Liang, W.S.; Beach, T.G.; Rogers, J.; Coleman, P.D. Laser-Captured Microglia in the Alzheimer’s and Parkinson’s Brain Reveal Unique Regional Expression Profiles and Suggest a Potential Role for Hepatitis B in the Alzheimer’s Brain. Neurobiol. Aging 2018, 63, 12–21. [Google Scholar] [CrossRef]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.-A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 2019, 179, 1609–1622.e16. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A Single-Cell Atlas of the Human Substantia Nigra Reveals Cell-Specific Pathways Associated with Neurological Disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef] [PubMed]
- Thrupp, N.; Sala Frigerio, C.; Wolfs, L.; Skene, N.G.; Poovathingal, S.; Fourne, Y.; Matthews, P.M.; Theys, T.; Mancuso, R.; de Strooper, B.; et al. Single Nucleus Sequencing Fails to Detect Microglial Activation in Human Tissue. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Smajić, S.; Prada-Medina, C.A.; Landoulsi, Z.; Dietrich, C.; Spielmann, M. Single-Cell Sequencing of the Human Midbrain Reveals Glial Activation and a Neuronal State Specific to Parkinson’s Disease. medRxiv 2020. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sammler, E. LRRK2 Kinase in Parkinson’s Disease. Science 2018, 360, 36–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Gao, H.-M.; Hong, J.-S. Parkinson’s Disease and Exposure to Infectious Agents and Pesticides and the Occurrence of Brain Injuries: Role of Neuroinflammation. Environ. Health Perspect. 2003, 111, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Dobbs, R.J.; Charlett, A.; Purkiss, A.G.; Dobbs, S.M.; Weller, C.; Peterson, D.W. Association of Circulating TNF-Alpha and IL-6 with Ageing and Parkinsonism. Acta Neurol. Scand. 1999, 100, 34–41. [Google Scholar] [CrossRef]
- Scalzo, P.; Kümmer, A.; Cardoso, F.; Teixeira, A.L. Increased Serum Levels of Soluble Tumor Necrosis Factor-Alpha Receptor-1 in Patients with Parkinson’s Disease. J. Neuroimmunol. 2009, 216, 122–125. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 Mitigate STING-Induced Inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; König, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Brüggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial Damage-Associated Inflammation Highlights Biomarkers in PRKN/PINK1 Parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of Interferon-Gamma in Microglial-Mediated Loss of Dopaminergic Neurons. J. Neurosci. 2007, 27, 3328–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menza, M.; Dobkin, R.D.; Marin, H.; Mark, M.H.; Gara, M.; Bienfait, K.; Dicke, A.; Kusnekov, A. The Role of Inflammatory Cytokines in Cognition and Other Non-Motor Symptoms of Parkinson’s Disease. Psychosomatics 2010, 51, 474–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-Motor Symptoms in Patients with Parkinson’s Disease—Correlations with Inflammatory Cytokines in Serum. PLoS ONE 2012, 7, e47387. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Le, W.-D.; Appel, S.H. Role of Fcgamma Receptors in Nigral Cell Injury Induced by Parkinson Disease Immunoglobulin Injection into Mouse Substantia Nigra. Exp. Neurol. 2002, 176, 322–327. [Google Scholar] [CrossRef]
- Benkler, M.; Agmon-Levin, N.; Hassin-Baer, S.; Cohen, O.S.; Ortega-Hernandez, O.-D.; Levy, A.; Moscavitch, S.-D.; Szyper-Kravitz, M.; Damianovich, M.; Blank, M.; et al. Immunology, Autoimmunity, and Autoantibodies in Parkinson’s Disease. Clin. Rev. Allergy Immunol. 2012, 42, 164–171. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Papachroni, K.K.; Ninkina, N.; Papapanagiotou, A.; Hadjigeorgiou, G.M.; Xiromerisiou, G.; Papadimitriou, A.; Kalofoutis, A.; Buchman, V.L. Autoantibodies to Alpha-Synuclein in Inherited Parkinson’s Disease. J. Neurochem. 2007, 101, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Yanamandra, K.; Gruden, M.A.; Casaite, V.; Meskys, R.; Forsgren, L.; Morozova-Roche, L.A. α-Synuclein Reactive Antibodies as Diagnostic Biomarkers in Blood Sera of Parkinson’s Disease Patients. PLoS ONE 2011, 6, e18513. [Google Scholar] [CrossRef]
- Double, K.L.; Rowe, D.B.; Carew-Jones, F.M.; Hayes, M.; Chan, D.K.Y.; Blackie, J.; Corbett, A.; Joffe, R.; Fung, V.S.; Morris, J.; et al. Anti-Melanin Antibodies Are Increased in Sera in Parkinson’s Disease. Exp. Neurol. 2009, 217, 297–301. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Sommer, A.; Marxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s Disease Patients Have a Complex Phenotypic and Functional Th1 Bias: Cross-Sectional Studies of CD4+ Th1/Th2/T17 and Treg in Drug-Naïve and Drug-Treated Patients. J. Neuroinflammation 2018, 15, 205. [Google Scholar] [CrossRef]
- Karaaslan, Z.; Kahraman, Ö.T.; Şanlı, E.; Ergen, H.A.; Ulusoy, C.; Bilgiç, B.; Yılmaz, V.; Tüzün, E.; Hanağası, H.A.; Küçükali, C.İ. Inflammation and Regulatory T Cell Genes Are Differentially Expressed in Peripheral Blood Mononuclear Cells of Parkinson’s Disease Patients. Sci. Rep. 2021, 11, 2316. [Google Scholar] [CrossRef]
- Subbarayan, M.S.; Hudson, C.; Moss, L.D.; Nash, K.R.; Bickford, P.C. T Cell Infiltration and Upregulation of MHCII in Microglia Leads to Accelerated Neuronal Loss in an α-Synuclein Rat Model of Parkinson’s Disease. J. Neuroinflammation 2020, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, T.; Habu, S. Humanized Mice; Springer: Berlin/Heidelberg, Germany, 2008; ISBN 9783540756460. [Google Scholar]
- Członkowska, A.; Kohutnicka, M.; Kurkowska-Jastrzebska, I.; Członkowski, A. Microglial Reaction in MPTP (1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Induced Parkinson’s Disease Mice Model. Neurodegeneration 1996, 5, 137–143. [Google Scholar] [CrossRef]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.-K.; Ischiropoulos, H.; Przedborski, S. Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Giuliani, F.; Hader, W.; Yong, V.W. Minocycline Attenuates T Cell and Microglia Activity to Impair Cytokine Production in T Cell-Microglia Interaction. J. Leukoc. Biol. 2005, 78, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice Deficient in TNF Receptors Are Protected against Dopaminergic Neurotoxicity: Implications for Parkinson’s Disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [Green Version]
- Barcia, C.; Ros, C.M.; Annese, V.; Gómez, A.; Ros-Bernal, F.; Aguado-Yera, D.; Martínez-Pagán, M.E.; de Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. IFN-γ Signaling, with the Synergistic Contribution of TNF-α, Mediates Cell Specific Microglial and Astroglial Activation in Experimental Models of Parkinson’s Disease. Cell Death Dis. 2011, 2, e142. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.-M.; Hong, J.-S.; Zhang, W.; Liu, B. Synergistic Dopaminergic Neurotoxicity of the Pesticide Rotenone and Inflammogen Lipopolysaccharide: Relevance to the Etiology of Parkinson’s Disease. J. Neurosci. 2003, 23, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.-M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.-S. HMGB1 Acts on Microglia Mac1 to Mediate Chronic Neuroinflammation That Drives Progressive Neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Klintworth, H.; Garden, G.; Xia, Z. Rotenone and Paraquat Do Not Directly Activate Microglia or Induce Inflammatory Cytokine Release. Neurosci. Lett. 2009, 462, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Chen, D.; Hu, Q.; Wang, G. Rotenone Directly Induces BV2 Cell Activation via the p38 MAPK Pathway. PLoS ONE 2013, 8, e72046. [Google Scholar] [CrossRef]
- Athanassiadou, A.; Voutsinas, G.; Psiouri, L.; Leroy, E.; Polymeropoulos, M.H.; Ilias, A.; Maniatis, G.M.; Papapetropoulos, T. Genetic Analysis of Families with Parkinson Disease That Carry the Ala53Thr Mutation in the Gene Encoding Alpha-Synuclein. Am. J. Hum. Genet. 1999, 65, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the Alpha-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-Synuclein Mutation Causes a Novel Parkinsonian-Pyramidal Syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic Investigation of Alpha-Synuclein Multiplication and Parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro Mutation in the Gene Encoding Alpha-Synuclein in Parkinson’s Disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Kim, W.S.; Kågedal, K.; Halliday, G.M. Alpha-Synuclein Biology in Lewy Body Diseases. Alzheimers. Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.D.; Stone, D.K.; Mosley, R.L.; Gendelman, H.E. Nitrated {alpha}-Synuclein-Induced Alterations in Microglial Immunity Are Regulated by CD4+ T Cell Subsets. J. Immunol. 2009, 182, 4137–4149. [Google Scholar] [CrossRef] [Green Version]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated Alpha-Synuclein Immunity Accelerates Degeneration of Nigral Dopaminergic Neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated Alpha-Synuclein Activates Microglia: A Process Leading to Disease Progression in Parkinson’s Disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.A.; Anthony, D.C. The Acute Inflammatory Response to Intranigral α-Synuclein Differs Significantly from Intranigral Lipopolysaccharide and Is Exacerbated by Peripheral Inflammation. J. Neuroinflammation 2011, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein Activates Microglia in a Model of Parkinson’s Disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.-F. Regionally-Specific Microglial Activation in Young Mice over-Expressing Human Wildtype Alpha-Synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Béraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Ton, V.; Neitzke, K.; Chasovskikh, S.; Mhyre, T.R.; Maguire-Zeiss, K.A. α-Synuclein Alters Toll-Like Receptor Expression. Front. Neurosci. 2011, 5, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-Dependent TLR1/2 Signaling by Misfolded α-Synuclein, a Protein Linked to Neurodegenerative Disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome Inhibition Prevents α-Synuclein Pathology and Dopaminergic Neurodegeneration in Mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Pike, A.F.; Varanita, T.; Herrebout, M.A.C.; Plug, B.C.; Kole, J.; Musters, R.J.P.; Teunissen, C.E.; Hoozemans, J.J.M.; Bubacco, L.; Veerhuis, R. α-Synuclein Evokes NLRP3 Inflammasome-Mediated IL-1β Secretion from Primary Human Microglia. Glia 2021. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, K.; Maguire-Zeiss, K. MMP13 Expression Is Increased Following Mutant α-Synuclein Exposure and Promotes Inflammatory Responses in Microglia. Front. Neurosci. 2020, 14, 585544. [Google Scholar] [CrossRef]
- Zhang, W.; Dallas, S.; Zhang, D.; Guo, J.-P.; Pang, H.; Wilson, B.; Miller, D.S.; Chen, B.; Zhang, W.; McGeer, P.L.; et al. Microglial PHOX and Mac-1 Are Essential to the Enhanced Dopaminergic Neurodegeneration Elicited by A30P and A53T Mutant Alpha-Synuclein. Glia 2007, 55, 1178–1188. [Google Scholar] [CrossRef]
- Gasser, T. Usefulness of Genetic Testing in PD and PD Trials: A Balanced Review. J. Parkinsons. Dis. 2015, 5, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 Is Involved in the IFN-Gamma Response and Host Response to Pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, M.; Selvanantham, T.; Swinton, E.; Padmore, R.F.; Tong, Y.; Kabbach, G.; Venderova, K.; Girardin, S.E.; Bulman, D.E.; Scherzer, C.R.; et al. Parkinson’s Disease-Linked LRRK2 Is Expressed in Circulating and Tissue Immune Cells and Upregulated Following Recognition of Microbial Structures. J. Neural Transm. 2011, 118, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s Disease-Linked Leucine-Rich Repeat Kinase 2(R1441G) Mutation Increases Proinflammatory Cytokine Release from Activated Primary Microglial Cells and Resultant Neurotoxicity. Neuroscience 2012, 208, 41–48. [Google Scholar] [CrossRef]
- Zhang, F.-R.; Huang, W.; Chen, S.-M.; Sun, L.-D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.-X.; Yang, H.-T.; Yang, R.-D.; et al. Genomewide Association Study of Leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeno, J.; Asano, K.; Matsushita, T.; Matsumoto, T.; Kiyohara, Y.; Iida, M.; Nakamura, Y.; Kamatani, N.; Kubo, M. Meta-Analysis of Published Studies Identified Eight Additional Common Susceptibility Loci for Crohn’s Disease and Ulcerative Colitis. Inflamm. Bowel Dis. 2011, 17, 2407–2415. [Google Scholar] [CrossRef] [PubMed]
- Van Limbergen, J.; Wilson, D.C.; Satsangi, J. The Genetics of Crohn’s Disease. Annu. Rev. Genomics Hum. Genet. 2009, 10, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Bell, S.L.; Huntington, T.E.; Vail, K.J.; Srinivasan, R.; Patrick, K.L.; Watson, R.O. LRRK2 Regulates Innate Immune Responses and Neuroinflammation during Mycobacterium Tuberculosis Infection. bioRxiv 2019, 699066. [Google Scholar] [CrossRef] [Green Version]
- Goldwurm, S.; Zini, M.; Mariani, L.; Tesei, S.; Miceli, R.; Sironi, F.; Clementi, M.; Bonifati, V.; Pezzoli, G. Evaluation of LRRK2 G2019S Penetrance: Relevance for Genetic Counseling in Parkinson Disease. Neurology 2007, 68, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J., 3rd; Shen, J. Loss of Leucine-Rich Repeat Kinase 2 Causes Impairment of Protein Degradation Pathways, Accumulation of Alpha-Synuclein, and Apoptotic Cell Death in Aged Mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Patel, J.C.; Wang, J.; Avshalumov, M.V.; Nicholson, C.; Buxbaum, J.D.; Elder, G.A.; Rice, M.E.; Yue, Z. Enhanced Striatal Dopamine Transmission and Motor Performance with LRRK2 Overexpression in Mice Is Eliminated by Familial Parkinson’s Disease Mutation G2019S. J. Neurosci. 2010, 30, 1788–1797. [Google Scholar] [CrossRef]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; DeSilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef]
- Kim, B.; Yang, M.-S.; Choi, D.; Kim, J.-H.; Kim, H.-S.; Seol, W.; Choi, S.; Jou, I.; Kim, E.-Y.; Joe, E.-H. Impaired Inflammatory Responses in Murine Lrrk2-Knockdown Brain Microglia. PLoS ONE 2012, 7, e34693. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.; Inesta-Vaquera, F.; Zhang, J.; Xie, C.; Cai, H.; Arthur, S.; Tan, L.; Choi, H.; Gray, N.; Cohen, P.; et al. The IkappaB Kinase Family Phosphorylates the Parkinson’s Disease Kinase LRRK2 at Ser935 and Ser910 during Toll-like Receptor Signaling. PLoS ONE 2012, 7, e39132. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.B.; Hanss, Z.; Krüger, R. The Genetic Architecture of Mitochondrial Dysfunction in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Djarmati, A.; Hedrich, K.; Schäfer, N.; Scaglione, C.; Marchese, R.; Kock, N.; Schüle, B.; Hiller, A.; Lohnau, T.; et al. PINK1, Parkin, and DJ-1 Mutations in Italian Patients with Early-Onset Parkinsonism. Eur. J. Hum. Genet. 2005, 13, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Dela Cruz, C.S.; Kang, M.-J. Mitochondrial Dysfunction and Damage Associated Molecular Patterns (DAMPs) in Chronic Inflammatory Diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Cannon, T.C.; Tran, T.; Ruhn, K.A.; Martinez, T.N.; Hong, J.; Marvin, M.; Hartley, M.; Treviño, I.; O’Brien, D.E.; Casey, B.; et al. Parkin Deficiency Increases Vulnerability to Inflammation-Related Nigral Degeneration. J. Neurosci. 2008, 28, 10825–10834. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.-M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.-C.; et al. Parkin Deficiency Modulates NLRP3 Inflammasome Activation by Attenuating an A20-Dependent Negative Feedback Loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.A.; Nguyen, A.D.; Chang, J.; Goldberg, M.S.; Lee, J.-K.; Tansey, M.G. Lipopolysaccharide and Tumor Necrosis Factor Regulate Parkin Expression via Nuclear Factor-Kappa B. PLoS ONE 2011, 6, e23660. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. DJ-1, a Cancer- and Parkinson’s Disease-Associated Protein, Stabilizes the Antioxidant Transcriptional Master Regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.-H.; Lee, M.-J.; Liou, H.-C.; Liou, H.-H.; Fu, W.-M. Microglia-Derived Cytokines/chemokines Are Involved in the Enhancement of LPS-Induced Loss of Nigrostriatal Dopaminergic Neurons in DJ-1 Knockout Mice. PLoS ONE 2016, 11, e0151569. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Choi, D.-J.; Jeong, H.-K.; Kim, J.; Kim, D.W.; Choi, S.Y.; Park, S.-M.; Suh, Y.H.; Jou, I.; Joe, E.-H. DJ-1 Facilitates the Interaction between STAT1 and Its Phosphatase, SHP-1, in Brain Microglia and Astrocytes: A Novel Anti-Inflammatory Function of DJ-1. Neurobiol. Dis. 2013, 60, 1–10. [Google Scholar] [CrossRef]
- Trudler, D.; Weinreb, O.; Mandel, S.A.; Youdim, M.B.H.; Frenkel, D. DJ-1 Deficiency Triggers Microglia Sensitivity to Dopamine toward a pro-Inflammatory Phenotype That Is Attenuated by Rasagiline. J. Neurochem. 2014, 129, 434–447. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 Deficiency Impairs Autophagy and Reduces Alpha-Synuclein Phagocytosis by Microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.-J.; Wang, H.-L.; Yin, B.-L.; Ren, X.-Y. Down-Regulation of DJ-1 Augments Neuroinflammation via Nrf2/Trx1/NLRP3 Axis in MPTP-Induced Parkinson’s Disease Mouse Model. Neuroscience 2020, 442, 253–263. [Google Scholar] [CrossRef]
- Volpato, V.; Webber, C. Addressing Variability in iPSC-Derived Models of Human Disease: Guidelines to Promote Reproducibility. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Haenseler, W.; Zambon, F.; Lee, H.; Vowles, J.; Rinaldi, F.; Duggal, G.; Houlden, H.; Gwinn, K.; Wray, S.; Luk, K.C.; et al. Excess α-Synuclein Compromises Phagocytosis in iPSC-Derived Macrophages. Sci. Rep. 2017, 7, 9003. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-Synuclein-Antibody Complexes Activate the NLRP3 Inflammasome in hiPSC-Derived Microglia. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Flynn, R.; Sharma, I.; Haberman, E.; Carling, P.J.; Nicholls, F.J.; Stegmann, M.; Vowles, J.; Haenseler, W.; Wade-Martins, R.; et al. LRRK2 Is Recruited to Phagosomes and Co-Recruits RAB8 and RAB10 in Human Pluripotent Stem Cell-Derived Macrophages. Stem Cell Rep. 2020, 14, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakopoulou, V.; Ivanyuk, D.; De Cicco, S.; Haq, W.; Arsić, A.; Yu, C.; Messelodi, D.; Oldrati, M.; Schöndorf, D.C.; Perez, M.-J.; et al. Interferon-γ Signaling Synergizes with LRRK2 in Neurons and Microglia Derived from Human Induced Pluripotent Stem Cells. Nat. Commun. 2020, 11, 5163. [Google Scholar] [CrossRef]
- Speidel, A.; Felk, S.; Reinhardt, P.; Sterneckert, J.; Gillardon, F. Leucine-Rich Repeat Kinase 2 Influences Fate Decision of Human Monocytes Differentiated from Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0165949. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-Organization of Axial Polarity, inside-out Layer Pattern, and Species-Specific Progenitor Dynamics in Human ES Cell–derived Neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of Function of the Mitochondrial Peptidase PITRM1 Induces Proteotoxic Stress and Alzheimer’s Disease-like Pathology in Human Cerebral Organoids. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Nickels, S.L.; Modamio, J.; Mendes-Pinheiro, B.; Monzel, A.S.; Betsou, F.; Schwamborn, J.C. Reproducible Generation of Human Midbrain Organoids for in Vitro Modeling of Parkinson’s Disease. Stem Cell Res. 2020, 46, 101870. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Takanashi, M.; Oyama, G.; Yoritaka, A.; Hatano, T.; Shiba-Fukushima, K.; Nagai, M.; Nishiyama, K.; Hasegawa, K.; Inoshita, T.; et al. Reduced Astrocytic Reactivity in Human Brains and Midbrain Organoids with PRKN Mutations. NPJ Parkinsons Dis. 2020, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Rõlova, T.; Lehtonen, Š.; Goldsteins, G.; Kettunen, P.; Koistinaho, J. Metabolic and Immune Dysfunction of Glia in Neurodegenerative Disorders: Focus on iPSC Models. Stem Cells 2021, 39, 256–265. [Google Scholar] [CrossRef]
- Brownjohn, P.W.; Smith, J.; Solanki, R.; Lohmann, E.; Houlden, H.; Hardy, J.; Dietmann, S.; Livesey, F.J. Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem Cell Rep. 2018, 10, 1294–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, C.M.; Gama, L.; Krasemann, S.; Chesnut, M.; Odwin-Dacosta, S.; Hogberg, H.T.; Hartung, T.; Pamies, D. Microglia Increase Inflammatory Responses in iPSC-Derived Human BrainSpheres. Front. Microbiol. 2018, 9, 2766. [Google Scholar] [CrossRef]
- Fagerlund, I.; Dougalis, A.; Shakirzyanova, A.; Gómez-Budia, M.; Konttinen, H.; Ohtonen, S.; Feroze, F.; Koskuvi, M.; Kuusisto, J.; Hernández, D.; et al. Microglia Orchestrate Neuronal Activity in Brain Organoids. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sá, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia Innately Develop within Cerebral Organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Hosmane, S.; Tegenge, M.A.; Rajbhandari, L.; Uapinyoying, P.; Ganesh Kumar, N.; Thakor, N.; Venkatesan, A. Toll/interleukin-1 Receptor Domain-Containing Adapter Inducing Interferon-β Mediates Microglial Phagocytosis of Degenerating Axons. J. Neurosci. 2012, 32, 7745–7757. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D Human Triculture System Modeling Neurodegeneration and Neuroinflammation in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Kodella, K.R.; Manatakis, D.V.; Hinojosa, C.D.; Manolakos, E.S.; Rubin, L.L.; Hamilton, G.A.; Karalis, K. Modeling Alpha-Synuclein Pathology in a Human Brain-Chip to Assess Blood-Brain Barrier Disruption in Parkinson’s Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ribecco-Lutkiewicz, M.; Sodja, C.; Haukenfrers, J.; Haqqani, A.S.; Ly, D.; Zachar, P.; Baumann, E.; Ball, M.; Huang, J.; Rukhlova, M.; et al. A Novel Human Induced Pluripotent Stem Cell Blood-Brain Barrier Model: Applicability to Study Antibody-Triggered Receptor-Mediated Transcytosis. Sci. Rep. 2018, 8, 1873. [Google Scholar] [CrossRef]
- Bergmann, S.; Lawler, S.E.; Qu, Y.; Fadzen, C.M.; Wolfe, J.M.; Regan, M.S.; Pentelute, B.L.; Agar, N.Y.R.; Cho, C.-F. Blood-Brain-Barrier Organoids for Investigating the Permeability of CNS Therapeutics. Nat. Protoc. 2018, 13, 2827–2843. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Teismann, P.; Ferger, B. Inhibition of the Cyclooxygenase Isoenzymes COX-1 and COX-2 Provide Neuroprotection in the MPTP-Mouse Model of Parkinson’s Disease. Synapse 2001, 39, 167–174. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.M.; Hernán, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal Anti-Inflammatory Drugs and the Risk of Parkinson Disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Yi, J.; Yang, J.; Li, P.; Cheng, X.; Mao, P. Nonsteroidal Anti-Inflammatory Drugs Use and Risk of Parkinson Disease: A Dose-Response Meta-Analysis. Medicine 2018, 97, e12172. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Ruhn, K.A.; Martinez, T.N.; McAlpine, F.E.; Blesch, A.; Tansey, M.G. Intranigral Lentiviral Delivery of Dominant-Negative TNF Attenuates Neurodegeneration and Behavioral Deficits in Hemiparkinsonian Rats. Mol. Ther. 2008, 16, 1572–1579. [Google Scholar] [CrossRef]
- García, M.C.; Cinquina, V.; Palomo-Garo, C.; Rábano, A.; Fernández-Ruiz, J. Identification of CB₂ Receptors in Human Nigral Neurons That Degenerate in Parkinson’s Disease. Neurosci. Lett. 2015, 587, 1–4. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [Green Version]
- Schwenkgrub, J.; Joniec-Maciejak, I.; Sznejder-Pachołek, A.; Wawer, A.; Ciesielska, A.; Bankiewicz, K.; Członkowska, A.; Członkowski, A. Effect of Human Interleukin-10 on the Expression of Nitric Oxide Synthases in the MPTP-Based Model of Parkinson’s Disease. Pharmacol. Rep. 2013, 65, 44–49. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Wang, J.; Yang, B.; Weng, Q.; He, Q. Targeting Microglia and Macrophages: A Potential Treatment Strategy for Multiple Sclerosis. Front. Pharmacol. 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, M.J.; Cantu, M.A.; Kasanga, E.A.; Moore, C.; Salvatore, M.F.; Meshul, C.K. Glatiramer Acetate Reverses Motor Dysfunction and the Decrease in Tyrosine Hydroxylase Levels in a Mouse Model of Parkinson’s Disease. Neuroscience 2019, 414, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Zella, M.A.S.; Metzdorf, J.; Ostendorf, F.; Maass, F.; Muhlack, S.; Gold, R.; Haghikia, A.; Tönges, L. Novel Immunotherapeutic Approaches to Target Alpha-Synuclein and Related Neuroinflammation in Parkinson’s Disease. Cells 2019, 8, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-Terminal-Truncated Alpha-Synuclein by Immunotherapy Attenuates Neurodegeneration and Propagation in Parkinson’s Disease-like Models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-J.; Lee, H.-J.; Rockenstein, E.; Ho, D.-H.; Park, E.-B.; Yang, N.-Y.; Desplats, P.; Masliah, E.; Lee, S.-J. Antibody-Aided Clearance of Extracellular α-Synuclein Prevents Cell-to-Cell Aggregate Transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- El-Agnaf, O.; Overk, C.; Rockenstein, E.; Mante, M.; Florio, J.; Adame, A.; Vaikath, N.; Majbour, N.; Lee, S.-J.; Kim, C.; et al. Differential Effects of Immunotherapy with Antibodies Targeting α-Synuclein Oligomers and Fibrils in a Transgenic Model of Synucleinopathy. Neurobiol. Dis. 2017, 104, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Kim, H.-J.; Jeon, B. Immunotherapy Targeting Neurodegenerative Proteinopathies: α-Synucleinopathies and Tauopathies. J. Mov. Disord. 2020, 13, 11–19. [Google Scholar] [CrossRef]
- Alon, S.; Goodwin, D.R.; Sinha, A.; Wassie, A.T.; Chen, F.; Daugharthy, E.R.; Bando, Y.; Kajita, A.; Xue, A.G.; Marrett, K.; et al. Expansion Sequencing: Spatially Precise in Situ Transcriptomics in Intact Biological Systems. Science 2021, 371. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-Definition Spatial Transcriptomics for in Situ Tissue Profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque Associated Microglia Hyper-Secrete Extracellular Vesicles and Accelerate Tau Propagation in a Humanized APP Mouse Model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Condition | Samples | Brain Region | Gender | Age (Years) | Tissue State | Method | Analyzed Microglia | Major Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| Mastroeni et al. 2018 [35] | Age-matched control subjects (absence of PD pathology), PD patients | Controls: n = 6, PD patients: n = 6 | SN, HPC (CA1) | All M | controls: 73.6 ± 6 PD patients: 74.6 ± 15.6 | Post-mortem frozen unfixed | RT-PCR of immunolabeled (LN3) laser-captured microdissected microglia | Control SN n = 3600 microglia cells, control CA1 n = 3600 microglia cells, PD SN n = 3600 microglia cells, PD CA1 n = 3600 microglia cells | Regional heterogeneity (inter- and intra-condition), PD-specific active pathways including inflammation-related aldosterone and reactive oxygen species metabolism |
| Geirsdottir et al. 2019 [36] | Control subjects (absence of neuropathology) | controls: n = 6 | Various | 3 F, 3 M | 9–55 | Fresh (surgically removed excess tissue surrounding epileptic focal) | scRNAseq (MARS-seq2.0) | n = 1069 microglia cells | Complex human microglia heterogeneity, Neurodegeneration-linked pathways and PD risk factors most profoundly expressed in human microglia |
| Agarwal et al. 2020 [37] | Age-matched control subjects (no neurological disease) | Controls: n = 5 | Cortex (middle frontal gyrus), SN (central portion at the level of the third nerve encompassing both ventral and dorsal tiers) | Cortex: 1 F, 3 M; SN: 2 F, 4M | 55–70 | Post-mortem frozen unfixed | snRNAseq (10X Genomics Chromium Single Cell Kit [v2 chemistry]) | Cortex n = 500 microglia nuclei, SN n = 325 microglia nuclei | PD risk variants in neurons but not in microglia |
| Smajic et al. 2021 [39] | Age-matched control subjects, idiopathic PD patients | Controls: n = 6, PD patients: n = 5 | Midbrain | Control: 1 F, 5 M; PD: 1 F, 4 M | Controls: 65–95; PD patients: 65–85 | Post-mortem frozen unfixed | snRNAseq (10X Genomics Chromium Next GEM Single Cell 3′ Kit v3.1) | n = 3903 microglia nuclei | Disease-specific upregulation of microglia, into two branches: GPNMB-high resp. IL-1β-high microglia PD risk variants enrichment in neurons and microglia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badanjak, K.; Fixemer, S.; Smajić, S.; Skupin, A.; Grünewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094676
Badanjak K, Fixemer S, Smajić S, Skupin A, Grünewald A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(9):4676. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094676
Chicago/Turabian StyleBadanjak, Katja, Sonja Fixemer, Semra Smajić, Alexander Skupin, and Anne Grünewald. 2021. "The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 9: 4676. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094676