
Functional Roles of PARP2 in Assembling Protein–Protein Complexes Involved in Base Excision DNA Repair



Abstract
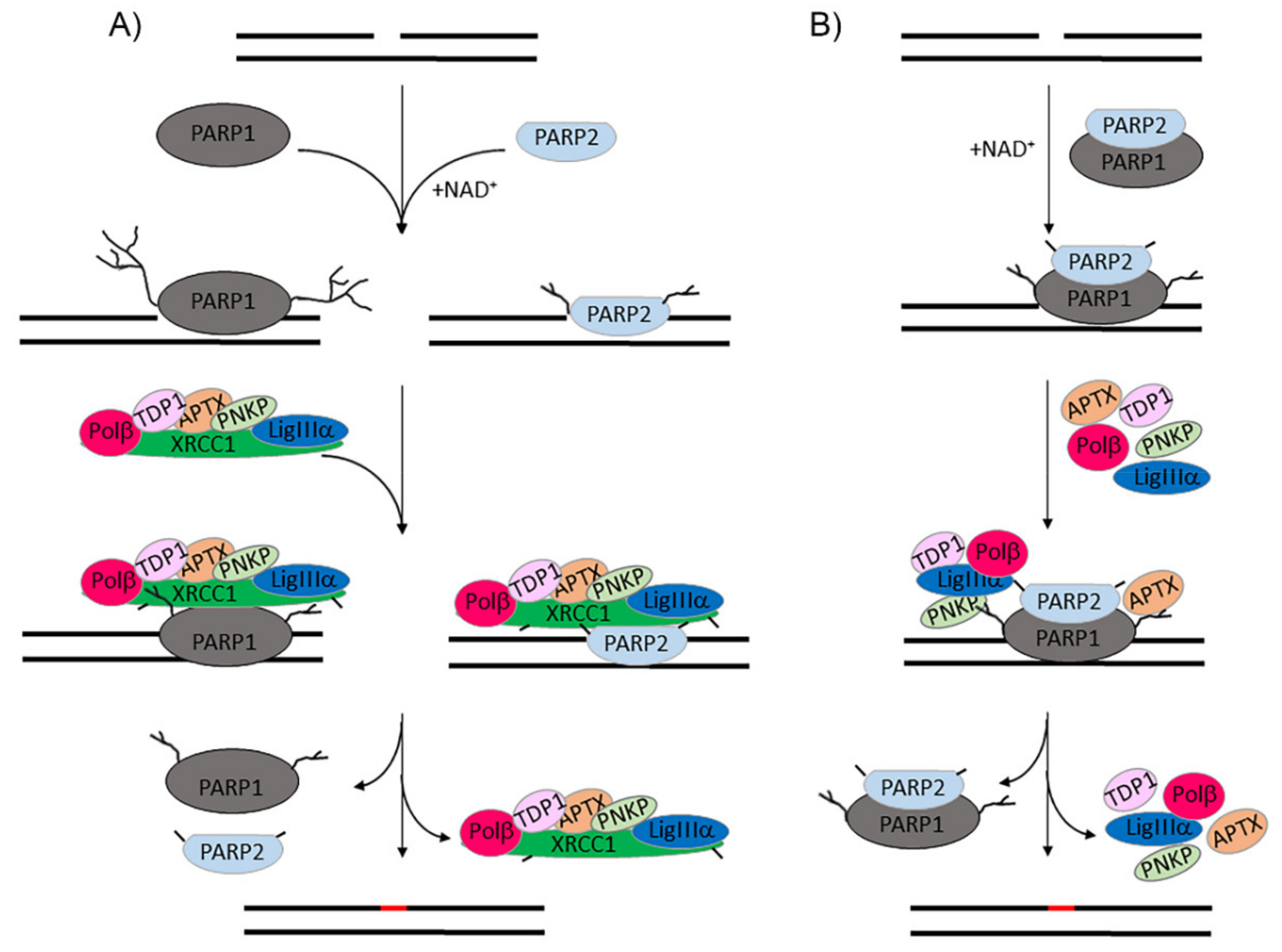
:1. Introduction
2. Results
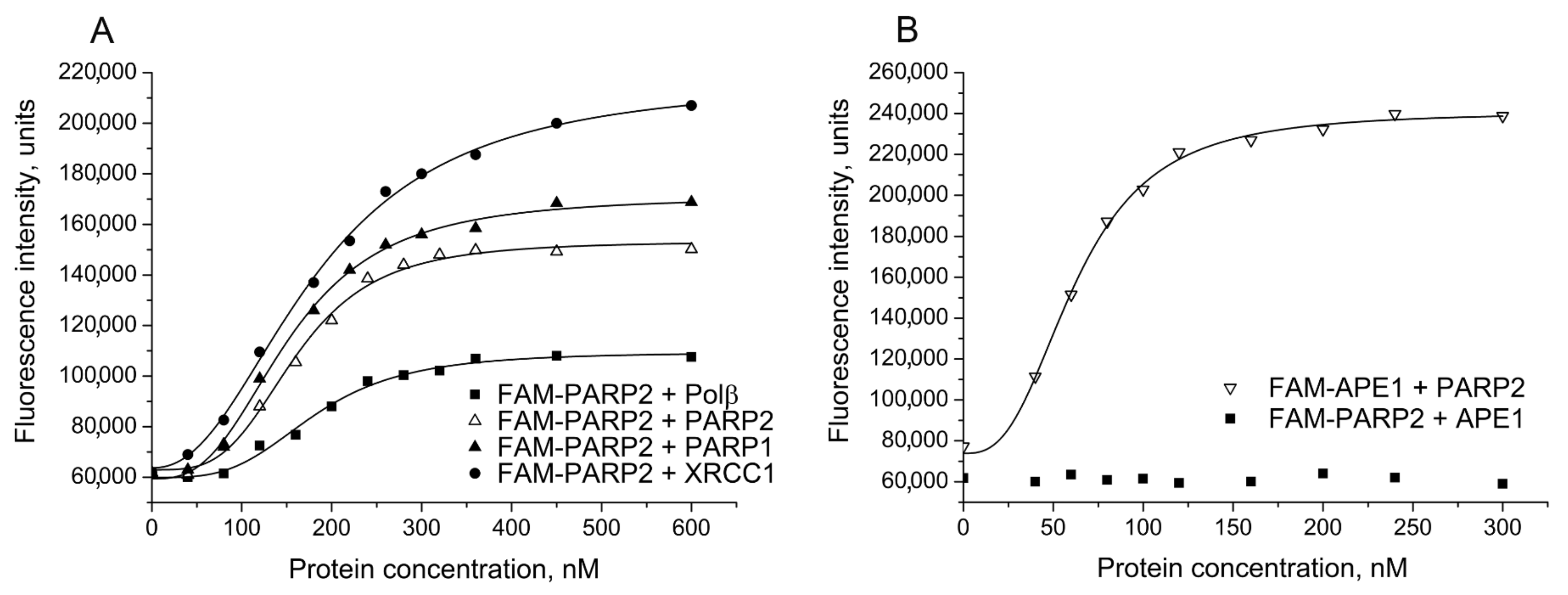
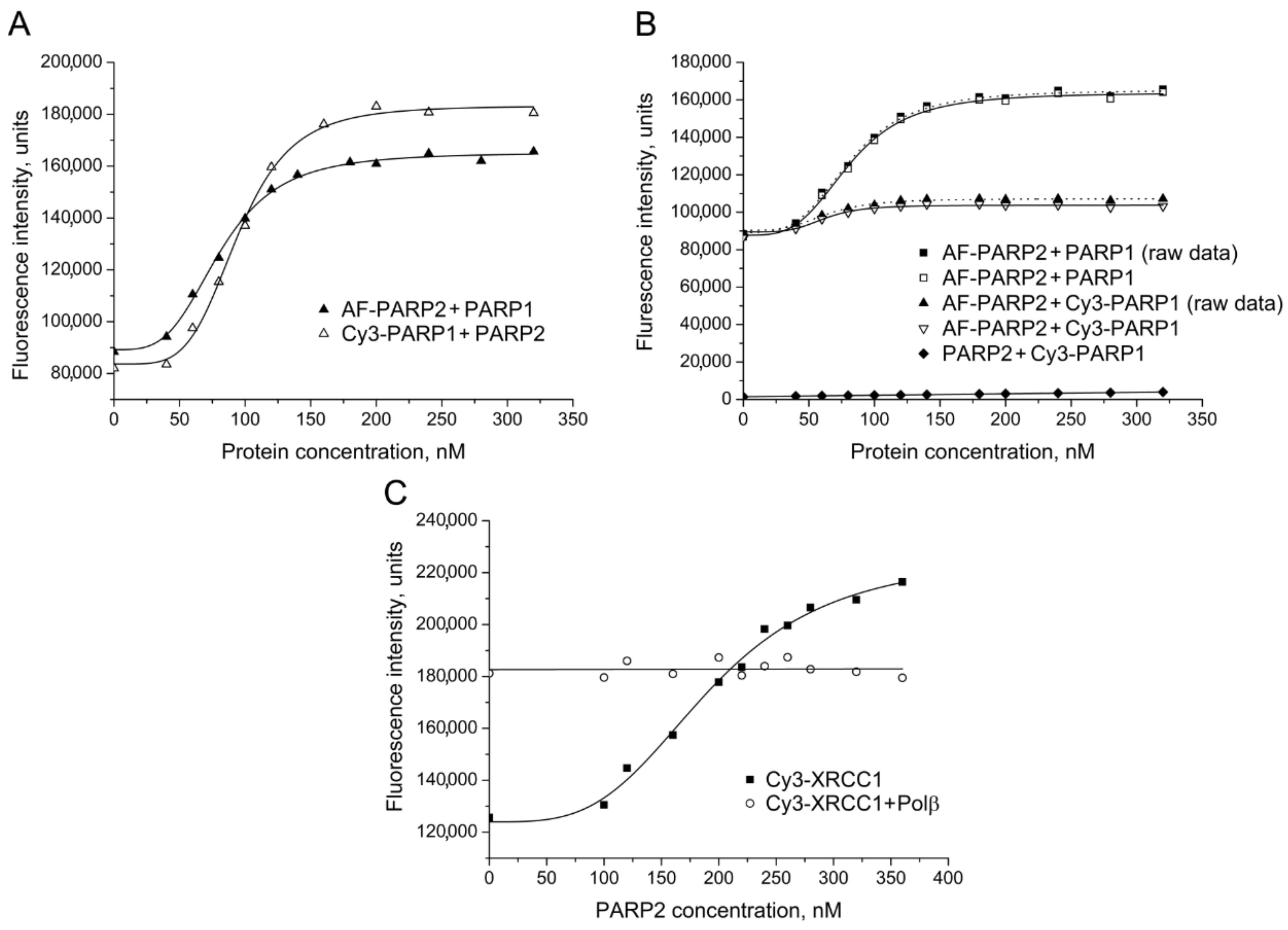
2.1. Detection and Quantification of PARP2–Protein Interactions by Fluorescence Titration and FRET Experiments
2.2. DLS Study of Oligomerization States of PARP2 and Its Hetero-Associated Complexes with BER Proteins
2.3. DLS Detection of Association of Poly(ADP-ribosyl)ated PARP2 in Solution
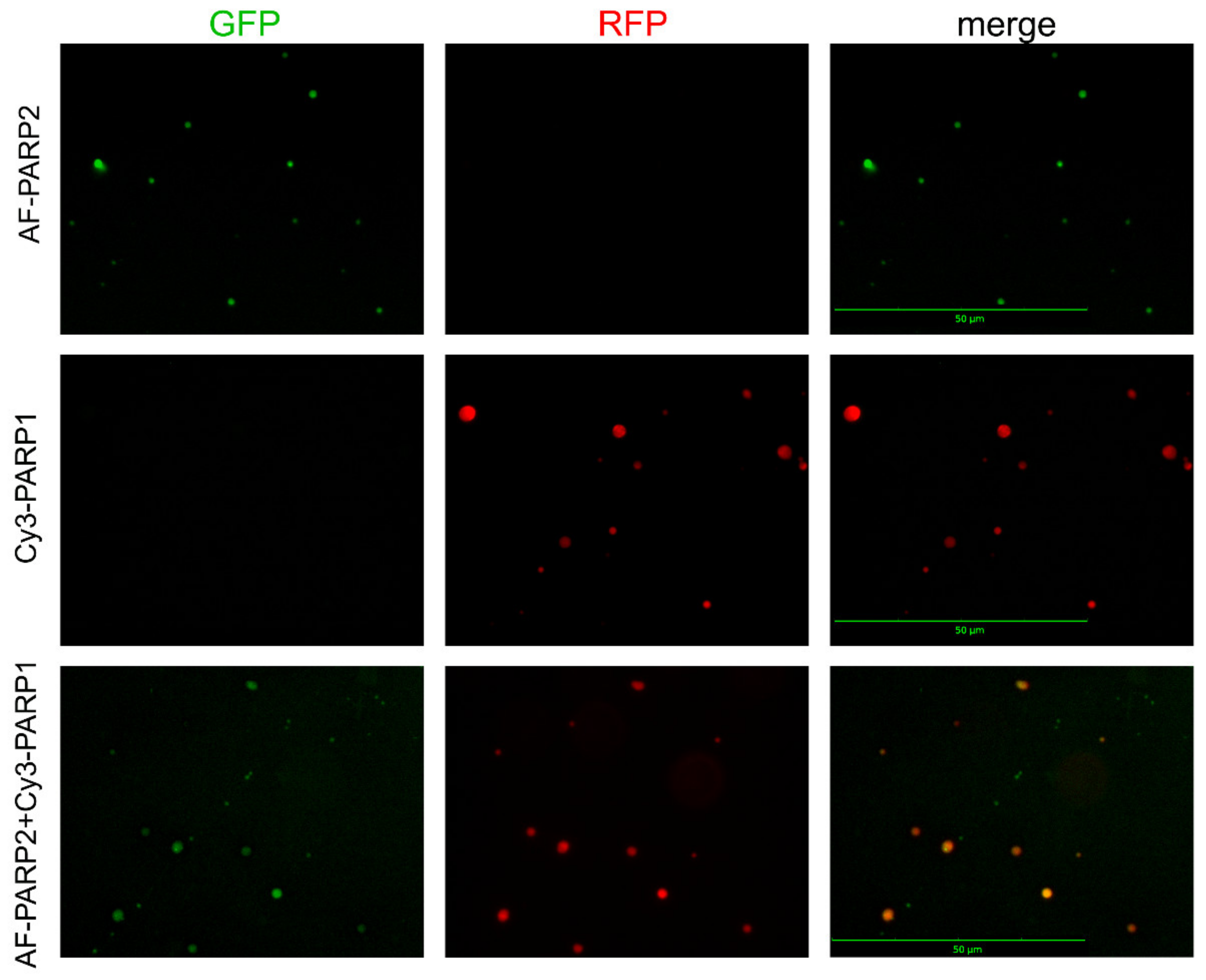
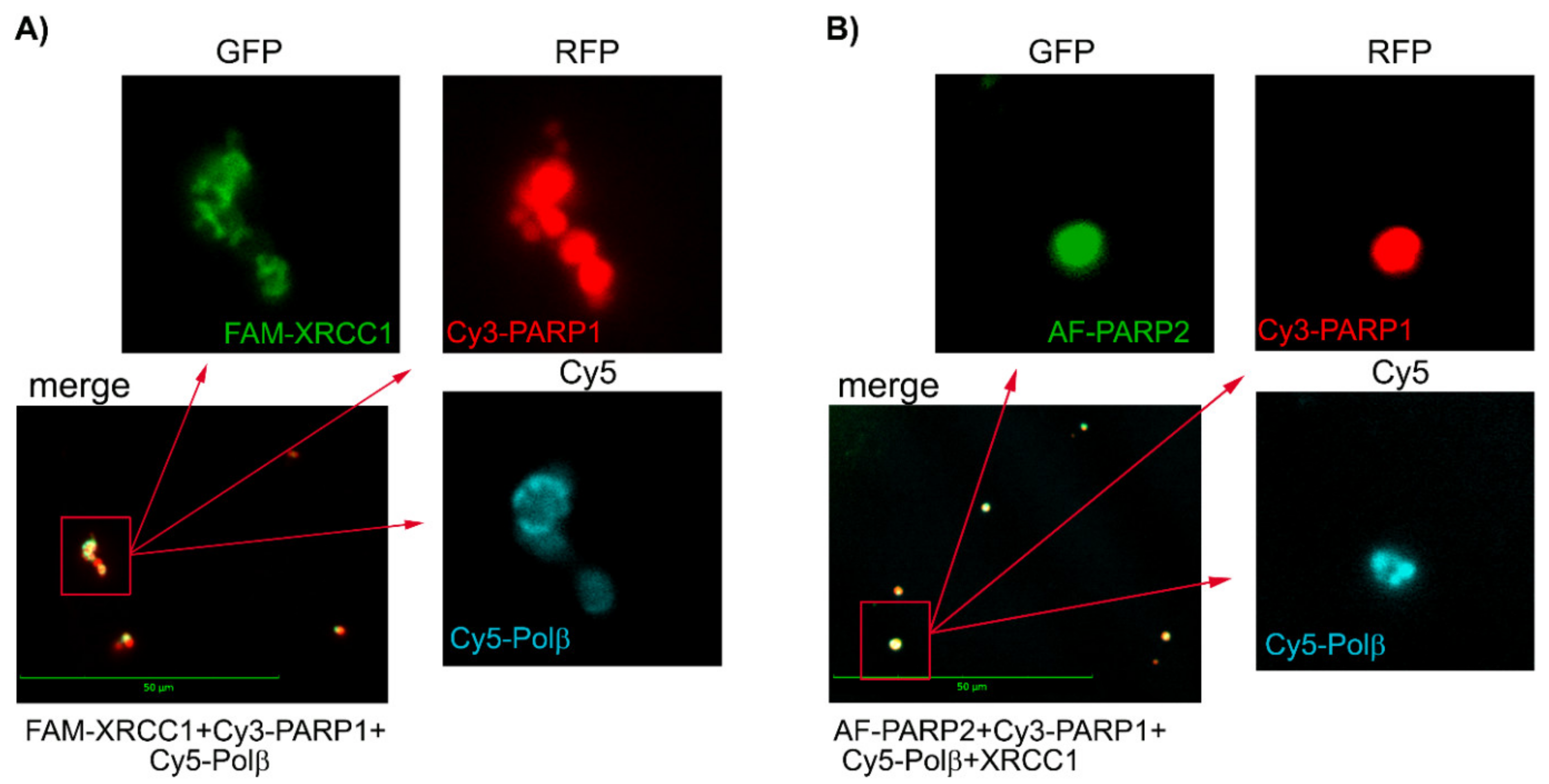
2.4. Visualization of PARylated Protein Associates Formed by PARP1 and PARP2
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Oligonucleotide Substrates
4.3. Dynamic Light-Scattering (DLS) Studies of PARP2 and Its Protein Complexes, and PARP2-Catalysed Poly(ADP-ribosyl)ation
4.4. Fluorescent Labelling of APE1, Polβ, PARP1, XRCC1, and PARP2
4.5. Fluorescence Studies of PARP2 Interaction with Proteins and DNAs
4.6. Visualization of PARP1 and PARP2 Associates Formed upon PARylation Reaction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PARP1 | poly(ADP)ribose polymerase 1 |
| PARP2 | poly(ADP)ribose polymerase 2 |
| Polβ | DNA polymerase β |
| XRCC1 | X-ray repair cross-complementing protein 1 |
| APE1 | apurinic/apyrimidinic endonuclease 1 |
| BER | base excision repair |
| SSB | single-strand break |
| FAM | 5(6)-carboxyfluorescein |
| AF488 | 5-Alexa Fluor 488 |
| Cy3 | sulfo-Cyanine 3 |
| Cy5 | sulfo-Cyanine 5 |
| SE | N-succinimidyl ester |
| FRET | fluorescence resonance energy transfer |
| DLS | dynamic light scattering |
| PAR | poly(ADP-ribose) |
References
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.A.; Lavrik, O.I. Protein-Protein Interactions in DNA Base Excision Repair. Biochemistry 2018, 83, 411–422. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic base excision repair: New approaches shine light on mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP-dependent protein ADP-ribosylation. Mol. Cell. 2015, 58, 935–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Hernandez, K.; Rodriguez-Vargas, J.-M.; Schreiber, V.; Dantzer, F. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin. Cell Dev. Biol. 2017, 63, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef]
- Hoch, N.C.; Polo, L.M. ADP-ribosylation: From molecular mechanisms to human disease. Genet. Mol. Biol. 2019, 43, e20190075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarm, K.; Smith, S. Nuclear PARPs and genome integrity. Genes Dev. 2020, 34, 285–301. [Google Scholar] [CrossRef]
- Lavrik, O.I. PARPs’ impact on base excision DNA repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Amé, J.-C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Ménissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Amé, J.-C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccio, A.A.; Cingolani, G.; Pascal, J.M. PARP-2 domain requirements for DNA damage-dependent activation and localization to sites of DNA damage. Nucleic Acids Res. 2016, 44, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amé, J.-C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [Green Version]
- Kutuzov, M.M.; Khodyreva, S.N.; Ame, J.-C.; Ilina, E.S.; Sukhanova, M.V.; Schreiber, V.; Lavrik, O.I. Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie 2013, 95, 1208–1215. [Google Scholar] [CrossRef]
- Kutuzov, M.M.; Khodyreva, S.N.; Ilina, E.S.; Sukhanova, M.V.; Ame, J.-C.; Lavrik, O.I. Interaction of PARP-2 with AP site containing DNA. Biochimie 2015, 112, 10–19. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Amé, J.-C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007, 35, 7665–7675. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef] [Green Version]
- Sukhanova, M.V.; Hamon, L.; Kutuzov, M.M.; Vandana, J.; Abrakhi, S.; Dobra, I.; Curmi, P.A.; Pastre, D.; Lavrik, O.I. A Single-Molecule Atomic Force Microscopy Study of PARP1 and PARP2 Recognition of Base Excision Repair DNA Intermediates. J. Mol. Biol. 2019, 431, 2655–2673. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.A.; Vasil’eva, I.A.; Anarbaev, R.O.; Antson, A.A.; Lavrik, O.I. Quantitative characterization of protein-protein complexes involved in base excision DNA repair. Nucleic Acids Res. 2015, 43, 6009–6022. [Google Scholar] [CrossRef] [Green Version]
- Vasil’eva, I.A.; Anarbaev, R.O.; Moor, N.A.; Lavrik, O.I. Dynamic light scattering study of base excision DNA repair proteins and their complexes. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Haugland, R.P. The Handbook—A Guide to Fluorescent Probes and Labeling Technologies, 10th ed.; Invitrogen Corp.: Carlsbad, CA, USA, 2005. [Google Scholar]
- Lumiprobe Life Science Solutions. Available online: https://www.lumiprobe.com; (accessed on 28 April 2021).
- Mani, R.S.; Karimi-Busheri, F.; Fanta, M.; Caldecott, K.W.; Cass, C.E.; Weinfeld, M. Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry 2004, 43, 16505–16514. [Google Scholar] [CrossRef] [PubMed]
- Fraústo da Silva, J.J.R.; Williams, R.J.P. The Biological Chemistry of the Elements: The Inorganic Chemistry of Life, 2nd ed.; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Lin, W.; Amé, J.-C.; Aboul-Ela, N.; Jacobson, E.L.; Jacobson, M.K. Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 1997, 272, 11895–11901. [Google Scholar] [CrossRef] [Green Version]
- Slade, D.; Dunstan, M.S.; Barkauskaite, E.; Weston, R.; Lafite, P.; Dixon, N.; Ahel, M.; Leys, D.; Ahel, I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 2011, 477, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. XRCC1 protein; Form and function. DNA Repair 2019, 81, 102664. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Roderick, A.; Stegeman, R.A.; Brosey, C.A.; Ellenberger, T. A quantitative assay reveals ligand specificity of the DNA scaffold repair protein XRCC1 and efficient disassembly of complexes of XRCC1 and the poly(ADP-ribose) polymerase 1 by poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 2015, 290, 3775–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutuzov, M.M.; Belousova, E.A.; Kurgina, T.A.; Ukraintsev, A.A.; Vasil’eva, I.A.; Khodyreva, S.N.; Lavrik, O.I. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci. Rep. 2021, 11, 4849. [Google Scholar] [CrossRef]
- Kouyama, K.; Mayanagi, K.; Nakae, S.; Nishi, Y.; Miwa, M.; Shirai, T. Single-particle analysis of full-length human poly(ADP-ribose) polymerase 1. Biophys. Physicobiol. 2019, 16, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.I.; Buki, K.G.; Hakam, A.; Kun, E. Macromolecular association of ADP-ribosyltransferase and its correlation with enzymic activity. Biochem. J. 1990, 270, 17–26. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.B.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singatulina, A.S.; Hamon, L.; Sukhanova, M.V.; Desforges, B.; Joshi, V.; Bouhss, A.; Lavrik, O.I.; Pastré, D. PARP-1 activation directs FUS to DNA damage sites to form PARG-reversible compartments enriched in damaged DNA. Cell Rep. 2019, 27, 1809–1821.e5. [Google Scholar] [CrossRef] [Green Version]
- Sukhanova, M.V.; Singatulina, A.S.; Pastré, D.; Lavrik, O.I. Fused in sarcoma (FUS) in DNA repair: Tango with poly(ADP-ribose) polymerase 1 and compartmentalisation of damaged DNA. Int. J. Mol. Sci. 2020, 21, 7020. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Alvarez, H.; Alvarez-Gonzalez, R. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J. Biol. Chem. 1993, 268, 22575–22580. [Google Scholar] [CrossRef]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998, 18, 3563–3571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moor, N.A.; Vasil’eva, I.A.; Kuznetsov, N.A.; Lavrik, O.I. Human apurinic/apyrimidinic endonuclease 1 is modified in vitro by poly(ADP-ribose) polymerase 1 under control of the structure of damaged DNA. Biochimie 2020, 168, 144–155. [Google Scholar] [CrossRef]
- Horton, J.K.; Gassman, N.R.; Dunigan, B.D.; Stefanick, D.F.; Wilson, S.H. DNA polymerase β-dependent cell survival independent of XRCC1 expression. DNA Repair 2015, 26, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Maltseva, E.A.; Krasikova, Y.S.; Sukhanova, M.V.; Rechkunova, N.I.; Lavrik, O.I. Replication protein A as a modulator of the poly(ADP-ribose)polymerase 1 activity. DNA Repair 2018, 72, 28–38. [Google Scholar] [CrossRef]
- Naumenko, K.N.; Sukhanova, M.V.; Hamon, L.; Kurgina, T.A.; Alemasova, E.E.; Kutuzov, M.M.; Pastré, D.; Lavrik, O.I. Regulation of poly(ADP-ribose) polymerase 1 activity by Y-Box-binding protein 1. Biomolecules 2020, 10, 1325. [Google Scholar] [CrossRef]
- Amé, J.-C.; Kalisch, T.; Dantzer, F.; Schreiber, V. Purification of recombinant poly(ADPribose) polymerases. Methods Mol. Biol. 2011, 780, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Belousova, E.A.; Vasil’eva, I.A.; Moor, N.A.; Zatsepin, T.S.; Oretskaya, T.S.; Lavrik, O.I. Clustered DNA lesions containing 5-formyluracil and AP site: Repair via the BER system. PLoS ONE 2013, 8, e68576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, P.R.; Beard, W.A.; Patterson, T.A.; Wilson, S.H. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 1997, 272, 1302–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Widen, S.G.; Williams, K.R.; Kedar, P.; Karpel, R.L.; Wilson, S.H. Studies of the domain structure of mammalian DNA polymerase β. Identification of a discrete template binding domain. J. Biol. Chem. 1990, 265, 2124–2131. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Labelled Protein 1 | Protein Partner | EC50 2, nM | E 3 |
|---|---|---|---|
| FAM-PARP2 | PARP2 | 152 ± 12 | |
| FAM-PARP2 | Polβ | 185 ± 18 * | |
| FAM-PARP2 | XRCC1 | 190 ± 18 * | |
| FAM-PARP2 | PARP1 | 146 ± 10 | |
| FAM-APE1 | PARP2 | 61 ± 8 *** | |
| AF-PARP2 | PARP1 | 79 ± 7 *** | |
| AF-PARP2 | Cy3-PARP1 | 76 ± 7 *** | 0.36 ± 0.04 |
| Cy3-PARP1 | PARP2 | 90 ± 6 *** | |
| Cy3-XRCC1 | PARP2 | 196 ± 10 ** |
| Labelled Protein 1 | DNA 2 | Protein Partner | EC50 3, nM | Effect on Binding Affinity 4 | Effect on FRET Efficiency 5 |
|---|---|---|---|---|---|
| FAM-PARP2 | gap-DNA | PARP2 | 101 ± 9 ** | 1.5 (+) | |
| FAM-PARP2 | gap-DNA | Polβ | 106 ± 10 ** | 1.7 (+) | |
| FAM-PARP2 | gap-DNA | XRCC1 | 164 ± 12 * | 1.2 (+) | |
| AF-PARP2 | nick-DNA | PARP1 | 44 ± 5 ** | 1.8 (+) | |
| AF-PARP2 | ds-DNA | PARP1 | 144 ± 16 ** | 1.8 (−) | |
| AF-PARP2 | nick-DNA | Cy3-PARP1 | −0.04 * | ||
| AF-PARP2 | ds-DNA | Cy3-PARP1 | +0.08 ** | ||
| FAM-PARP2 | gap-DNA | ≤18 | |||
| AF-PARP2 | nick-DNA | ≤15 | |||
| AF-PARP2 | ds-DNA | 35 ± 4 |
| Protein(s) 1 (Mw 2, kDa) | DLS RH 3, nm | Theoretical RH 4, nm | Oligomeric State 5 | |
|---|---|---|---|---|
| Protomer | Dimer (Tetramer) | |||
| Homo- and hetero-oligomerization of PARP2 | ||||
| PARP2 (62.0) | 5.0 ± 0.4 | 3.47 | 4.67 (6.28) | A2 |
| PARP2 + Polβ (100.3) | 5.6 ± 0.3 | 4.27 | 5.74 | (AB)2 |
| PARP2 + APE1 (97.5) | 4.9 ± 0.2 | 4.21 | 5.67 | AB+(AB)2 |
| PARP2 + XRCC1 (131.5) | 7.6 ± 0.3 | 4.79 | 6.44 (8.66) | (AB)2 |
| PARP2 + PARP1 (175.0) | 7.8 ± 0.6 | 5.41 | 7.28 | (AB)2 |
| Homo-oligomerization of BER proteins determined in our previous study [24] | ||||
| Polβ (38.3) | 3.5 ± 0.2 | 2.83 | 3.80 | A2 |
| APE1 (35.5) | 3.2 ± 0.2 | 2.74 | 3.68 | A2 |
| XRCC1 (69.5) | 5.7 ± 0.2 | 3.65 | 4.90 | A2 |
| PARP1 (113.0) | 8.0 ± 0.5 | 4.50 | 6.04(8.12) | A2 6 |
| Protein(s) 1 | RH 2, (nm) | |||
|---|---|---|---|---|
| Before Reaction Initiation | PARylated Protein Associate 3 | PARylated Protein after Addition of EDTA 3 | PARylated Protein after PARG Treatment 3 | |
| PARP2 | 5.0 ± 0.4 | 383 ± 45 | 5.6 ± 0.3 | 5.2 ± 0.3 |
| PARP2 + Polβ | 5.6 ± 0.3 | 370 ± 43 | 6.2 ± 0.5 | 5.8 ± 0.2 |
| PARP2 + APE1 | 4.9 ± 0.2 | 440 ± 51 | 5.2 ± 0.3 | n. d. 4 |
| PARP2 + XRCC1 | 7.6 ± 0.3 | 11.3 ± 1.6 | 6.7 ± 0.5 | 5.8 ± 0.4 |
| PARP2 + PARP1 | 7.8 ± 0.4 | 14.8 ± 1.4 | 9.9 ± 1.2 | n. d. 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasil’eva, I.; Moor, N.; Anarbaev, R.; Kutuzov, M.; Lavrik, O. Functional Roles of PARP2 in Assembling Protein–Protein Complexes Involved in Base Excision DNA Repair. Int. J. Mol. Sci. 2021, 22, 4679. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094679
Vasil’eva I, Moor N, Anarbaev R, Kutuzov M, Lavrik O. Functional Roles of PARP2 in Assembling Protein–Protein Complexes Involved in Base Excision DNA Repair. International Journal of Molecular Sciences. 2021; 22(9):4679. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094679
Chicago/Turabian StyleVasil’eva, Inna, Nina Moor, Rashid Anarbaev, Mikhail Kutuzov, and Olga Lavrik. 2021. "Functional Roles of PARP2 in Assembling Protein–Protein Complexes Involved in Base Excision DNA Repair" International Journal of Molecular Sciences 22, no. 9: 4679. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094679