
Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time


1
Laboratory of Cytogenomics and Animal Genomics (CAG), Department of Genetics and Biotechnology (DGB), University of Trás-os-Montes and Alto Douro (UTAD), 5000-801 Vila Real, Portugal
2
Biosystems and Integrative Sciences Institute (BioISI), Faculty of Sciences, University of Lisbon, 1749-016 Lisbon, Portugal
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(9), 4707; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094707
Submission received: 31 March 2021
/
Revised: 24 April 2021
/
Accepted: 27 April 2021
/
Published: 29 April 2021
(This article belongs to the Special Issue Repetitive DNA Sequences in Eukaryotic Genomes)
Abstract
:(Peri)centromeric repetitive sequences and, more specifically, satellite DNA (satDNA) sequences, constitute a major human genomic component. SatDNA sequences can vary on a large number of features, including nucleotide composition, complexity, and abundance. Several satDNA families have been identified and characterized in the human genome through time, albeit at different speeds. Human satDNA families present a high degree of sub-variability, leading to the definition of various subfamilies with different organization and clustered localization. Evolution of satDNA analysis has enabled the progressive characterization of satDNA features. Despite recent advances in the sequencing of centromeric arrays, comprehensive genomic studies to assess their variability are still required to provide accurate and proportional representation of satDNA (peri)centromeric/acrocentric short arm sequences. Approaches combining multiple techniques have been successfully applied and seem to be the path to follow for generating integrated knowledge in the promising field of human satDNA biology.
1. Introduction
Back in the 1960s, Britten and Kohne revealed the high abundance of repetitive sequences in eukaryotic genomes [1], opening up a new field of research. Notwithstanding, the biological importance of these repeated sequences was perpetually neglected for many years to come, as the repetitive portion of the genome was often dismissed as non-functional (simply ‘‘junk DNA’’) [2]. This non-functional term itself has long been troublesome, as it is argued that even in the early days some researchers stated the likelihood of a functionality for repetitive DNA [3].
Soon, the need to classify repetitive DNA sequences arose, first in a major classification related to repeat number and subsequently in group classifications according to their organization (arrays of tandem repeats or interspersed sequences) [4]. Tandem repeats are characterized by the adjacent alignment of sequence units in a hierarchically organized manner, while interspersed repeats have a scattered, multi-locus distribution across the genome [5,6].
The term “heterochromatin” was coined in 1928 [7], in parallel with the assessment of its distinctive state of constant compaction. As a consequence, heterochromatic genomic regions were portrayed as silent and inert, an assumption that became inevitably associated with repetitive sequences, being the major heterochromatic component [8]. In the wake of the disclosure of the repetitive fraction of the genome, a new class of tandemly repeated DNA sequences was first revealed in 1961 [9,10] and later identified in the 1970s as the constituent of satellite peaks in cesium chloride density gradients. Essentially, satellite bands were differentiated from the remaining genomic DNA by their A/T content [11]. The name satellite DNA (satDNA) was here to stay [12] and has been used ever since as a broader term for tandem repeats [13]. Fundamentally, satDNAs make up the eukaryotic centromeric and pericentromeric genomic regions [14], even though they can also be located at subtelomeric sites or even at interstitial regions [15,16,17]. SatDNA sequences can be distinguished by a multitude of dissimilar features, like nucleotide sequence composition, complexity, and abundance, although their major shared characteristics cannot be dismissed: the capacity to form heterochromatic regions and the intrinsic propensity to form long tandemly organized arrays [17]. The apparent unlikelihood of a discernible role ascribed to repetitive sequences (and therefore to satellite DNA) did not prevent the continuous emergence of a variety of studies, essentially focusing on investigating these sequences in terms of their associated functions [5,11,14,18,19,20,21,22,23,24,25]. Thus, the idea of a potential function was parsimoniously considered: the common presence of genomic repetitive sequences could have some underlying meaning [17].
We now know that a variety of vital cellular processes is influenced by satDNA arrays: cell cycle, gene expression, or even genome stability [26]. Being a constitutive element of key structures such as centromeres or telomeres, satDNA has been phenotypically associated with chromosome and cell function in multiple species, specifically in humans [27,28], in which several satDNA families have been progressively identified and characterized, albeit not at the same pace.
2. Human Satellite DNA Families
Going back to the 1960s, the discovery and classification of three clearly distinguishable human genomic DNA fractions in CsSO4 gradients established the identity of the corresponding classical satellite DNAs I, II, and III. More precisely, a set of repetitive sequences with analogous buoyant densities was found to compose each gradient fraction [29]. These DNA fractions presented a characteristic inter-sequence heterogeneity, which led to a new classification in 1987, as a prime family of simple repeats was identified for each fraction [30]. The three families were described as satellite DNA families I, II, and III [29] and were first reported to be present in all acrocentric chromosomes, as well as in chromosomes 3 and 4 [31]. Additionally, the centromeric alpha (α) satellite DNA family was also identified and described, soon becoming the most intensively studied human satDNA sequence. Later on, gamma (γ) and beta (β) satellites were likewise found among the diverse families of human satellite DNAs [32].
Through processes of amplification and homogenization, some satDNA monomers have the ability to form Higher-Order-Repeats (HORs) units [17]. In fact, it has been demonstrated that HOR structure can influence regulated functions (like gene expression [33,34] and replication efficiency [35,36,37]) and have a significant role in individual/populational diversity [38,39].
Today, the larger satellite regions of classical heterochromatin in the human genome (essentially present in chromosomes 1, 9, and 16 and acrocentric short arms) [40] remain poorly understood. The known complexity of these regions and associated lack of knowledge cause a significant void: the lack of an integrated portfolio describing different subfamilies, possibly facilitating variability studies within the same satellite family [41]. For instance, the heterochromatic band of chromosomes 1, 9, and 16 represents a source of human variation [41], given that assessable polymorphisms are found between individuals [42,43,44]. The Y satDNA repeats represent an additional example of how satDNA may constitute a valuable tool to study human variation, given that the frequency of satellite variants can fluctuate greatly between individuals [40]. Indeed, variations within satellite arrays have been shown to influence the overall size of the Y chromosome across human populations [41].
Twenty years ago, different satellite subfamilies were already recognized to have different features. In spite of the constant repeat unit size, sub-variability within the same satDNA family was reported [45], feasibly explained by a different organization and, therefore, different clustered localization [46]. The differential chromosomal location of satellite subfamilies has been mostly shown by clonal-based fluorescent in situ hybridization (FISH) studies [30,31,45,47,48,49,50,51]. In accordance with these early studies, it was demonstrated that different subfamilies do not correspond to different parts of the same array but instead coincide with genomically separated subgroups [41]. This type of genomic analysis is essential, since satellite-associated information gaps are responsible for uncertainty issues regarding the exact number of satDNA families/subfamilies (e.g., families reported as different might be related or different subfamilies may compose the same satDNA family) [52]. In the next sections, we introduce the present-day understanding about the most significantly described human satDNA (sub)families.
2.1. α Satellite DNA
Initially, α satellite DNA (αSAT) was isolated from a highly repetitive fraction present in the African green monkey genome [53]. Subsequently, α satellite repeats were shown to be present in all human centromeres and to be composed of tandem repeats of an AT-rich 171 bp-long monomer [54,55,56,57]. Alphoid monomers can form HORs composed of nmer repeats (being n the number of monomers) or be organized in a non-HOR manner as simple monomeric repeats [28]. HORs can be formed by 2 to 34 monomers [28,56,58,59]. Some monomers within α satellite HORs have a 17 bp sequence motif called the Centromere Protein B (CENP-B) box because of the ability of CENP-B to recognize and bind to these regions [60]. The CENP-B box location is structurally related to the chromosome-specific HOR array (varying accordingly) [5]. Moreover, the CENP-B box is present with high degree of conservation in other mammal genomes [61]. Studies show that an active CENP-B box is required for de novo centromere assembly in humans, acting in the recruitment of the Centromere Protein A (CENP-A) and stabilization of the Centromere Protein C (CENP-C) [62], both related to an active kinetochore and proper chromosome segregation, which might explain its retention in different genomes. Each human chromosome contains one or more exclusive α HOR array, except for chromosomes 13/21 and 14/22, which share the same HOR array [46,50,63]. Regarding acrocentric chromosomes, a variety of α satellite subfamilies can be found in the vicinity of the centromere: pTRA-1, pTRA-2, pTRA-4, and pTRA-7, all of them present in chromosomes 13, 14, and 21 [46,64]. These subfamilies are part of a catalog of 28 clone-isolated α subfamilies from all human chromosomes [64], although, presently, an accurate genomic analysis is still required to avoid redundant classifications.
α satellite soon became the model for the hierarchical HOR organization [29]. Alphoid sequences are deeply related to proper cell division (being the foundation for kinetochore formation); the occurrence of active centromeres; and, therefore, centromere identity. It is possible to distinguish human centromeres based on their α HOR specificity-conferring composition, namely, by the number and order of monomers (that share 50–70% of identity) [65]. By defining α monomer consensus sequences, it is possible to discern five suprachromosomal groups or subfamilies, based on the possible monomer combinations [65,66] (reviewed in [5,66]). The main suprachromosomal subfamilies (SF1-3) correspond to the kinetochore formation region and are associated with centromere functionality [67,68]. Hybridization studies performed at high stringency allow the mapping of individual HORs to specific chromosomes [56] because of sequence polymorphisms found between them [5]. At low stringency, subsets of HOR arrays co-hybridize, allowing one to study how suprachromosomal subfamilies relate to each other [58,69]. Beyond the occurrence of α HORs, α monomeric repeats are present in transitory, array-adjacent pericentromeric regions, feasibly evolving non-homogeneously from homogenous HORs [59,70]. The relative mutation rate of centromeric α satellite sequences (accelerated comparing to unique genomic portions) lines up with a layered and symmetric evolution in the following direction: active HOR repeats-ancient HOR repeats-monomeric repeats [71]. In fact, closeness to the functional core centromere is a determining factor for HOR homogenization, as distant monomers are considered older, more variable, and a trace of centromere primate evolution [72]. Therefore, HOR array chromosome specificity results from intrachromosomal homogenization [13].
2.2. Satellite DNA I
SatDNA I (SATI) is distinguished by the presence of 42 bp repeats, consecutively arranged in units of 2 types, A (17 bp) and B (25 bp) repeat units [29], which can tandemly organize in ABABA constructs [30,73]. SATI repeats can form HORs of 2.97 Kb [74]. The amplification of these sequence arrays arranged in a head-to-tail fashion resulted in the current complexity of the SATI DNA family [73]. SATI is the most AT-rich fraction of the human genome, being also the least abundant classical satellite [51]. This classical satellite was first described using a probe (pTRI-6) that hybridizes with all acrocentric chromosomes at low stringency and only with chromosomes 13 and 21 at high stringency [46,74]. Until this day, pTRI-6 remains the only sequence described as a SATI subfamily. In 1986, experiments with the restriction enzyme RsaI allowed for the detection of the ABABA construct [30]. In this study, the A-B 42 bp form was considered predominant, although a possible second form (B-B dimers) was also observed. Later, Meyne et al. determined that B-B repeats hybridize with chromosome 3 and acrocentric chromosomes. The predominant ABABA construct showed an analogous chromosomal location to that of the pTRI-6 subfamily (chromosomes 3 and 4 and acrocentric). Acrocentric hybridization signals could be found in two locations: proximal pericentromeric and more distal short arm regions. The authors highlighted the need for high-resolution molecular studies for variant analysis [73], a requirement that remains pressing a quarter a decade later.
2.3. Satellite DNA II/III
SatDNA II (SATII) associates with a poorly conserved repeat unit (ATTCC), and satDNA III (SATIII) was shown to be composed of pentameric repeats of the same motif (well-conserved and interspersed with a specific 10 bp sequence) [29,75]. The inconsistent arrangement of satellite II/III in complex repeats (as opposed to tandem repeats) has led to a poor characterization of these satellite families [41]. SATII and SATIII probably arose from the same pentameric repeat [30], yet today these sequences locate to different genomic regions [41,48].
SATII repeats were initially reported to predominantly locate at chromosome 1 [45,48] and chromosome 16 [76,77]. In particular, the chromosome 1 SATII array represents a chromosome-specific 1.77 kb unit [48]. To a smaller extent, SATII was also found in pericentromeric regions of chromosomes 2 and 10 [45]. In 2014, three different SATII subfamilies were analyzed, presenting different sequence composition and genomic location [41]. Today, the chromosomal location of SATII family is more broadly recognized, supported by increasing genomic and bioinformatic studies.
SATIII was localized to chromosomes 1, 9, and Y [76,77,78], as well as to acrocentric short arms [79]. SATIII repeats have likewise been progressively found in additional chromosomal locations (e.g., chromosomes 5, 10, 17, and 20) [29,45]. SATIII presence in the Y chromosome long arm is distinguished by a male-specific 3.6 Kb repeat unit [78,80,81]. With respect to SATIII acrocentric repeats, eleven different subfamilies have been identified and characterized: pTRS-47 [82], pTRS-63 [83], pTR9-s3 [31], pTRS-2 [46], pE-1, pE-2, pR-1, pR-2, pR-4, pK-1, and pW-1 [84]. The pTRS-47 and pTRS-63 subfamilies, located at chromosomes 14/22 and 14, respectively, seem to be particularly significant in a clinical context for their involvement in the breakpoint of human Robertsonian translocations [85]. Interestingly, computational clustering analysis of human sequences was able to identify a total of eleven SATIII subfamilies [41]. Although the number of identified subfamilies correlates with previous clone hybridization studies on acrocentric chromosomes, predicted chromosomal locations do not seem to match (not all identified subfamilies locate to acrocentric chromosomes). A comprehensive sequence analysis is essential since different sequence composition and physical locations (observed in both approaches) point to the existence of a higher number of SATIII subfamilies. Gaps in our understanding of SATII/SATIII repeats are strongly associated with limiting bioinformatic/sequencing approaches, due to their short irregular nature [41], as well as close sequence relation.
2.4. β Satellite DNA
β satDNA (βSAT) was initially named as Sau3A satDNA family [86] and effectively termed β satellite in 1989 [87]. β satellite repeats consist of tandem arrays of a 68 bp monomer organized in multimeric HORs, described to be present in all acrocentric chromosomes and chromosomes 1, 3, 9, 19, and Y [47,79,86,87,88,89], predominantly in pericentromeric regions [90]. Indeed, β satellite was distinguished in two different types of HORs (pB3 and pB4), composed of non-overlapping arrays with distinct genomic locations. pB3 is specifically localized in chromosome 9, and its representation is equivalent to 50–100 times per haploid genome. The second type of HOR, pB4, is 5 times more represented per haploid genome and is located in acrocentric chromosomes, where β satellite was found early on to map distally and proximally to rDNA [87]. Recently, β satellite was identified to be present in multiple eukaryotic taxa and to be the object of horizontal transfer (HT) events, contradicting previous claims of its exclusive presence in primates [91].
2.5. γ Satellite DNA
Originally, γ satDNA (γSAT) was isolated from a chromosome 8 specific clone [92]. Later on, another γ subfamily was described in chromosome X [93]. Known γ satellite subfamilies (GSAT, GSATX, and GSATII, with ~60% identity) are GC-rich tandem pericentromeric repeats of a vastly diverged 220 bp monomer and have been identified in all human chromosomes [40,94] usually forming clusters of 2–10 kb [92]. Kim et al. [94] proposed that γ satellite repeats may possibly work as barriers for heterochromatin expansion to chromosomal arms, being functionally similar to genomic insulators. This thesis emerged in accordance with previous statements regarding the existence of structural and functional constrains related to γ satellite [29].
Regardless of the common satDNA composition, centromeric and pericentromeric chromatin are structurally different, essentially because centromeres are epigenetically compatible with kinetochore assembly and chromosome segregation, while pericentromeric regions have a typical heterochromatic behavior [26]. Thus, the ubiquitous centromeric presence of α satellite sequences is contrasted by the nature of pericentromeric satellite families that clearly behave in a more non-homogenous manner [23,29,55], frequently leading to incongruences about their overall existence and location in the human genome [95]. Human centromeres are not only composed of satellite sequences, but also contain mobile elements, including LINEs and SINEs (Long/Small Interspersed Nuclear Elements), already described both in HOR arrays and monomeric repeats [13,95]. Hence, the centromeric region of human chromosomes is mostly composed of α HORs, eventually punctuated by transposable elements (TEs) [96,97], and progressively replaced by pericentromeric satellite families (classical satellites and β/γ satellites) [23]. Table 1 presents a summary of the available information about human satellite families.
3. Satellite DNA: Repetitively Challenging
The heterogeneity of known human satellite families constitutes evidence as to why the satDNA field has struggled with terms and definitions. If we take repeat unit size as an example, defining classical satellite sequences as microsatellites (≤10 bp) or minisatellites (between 10 and 100 bp) [6,100,101] is straightforward reasoning. However, these sequences tandemly organize in long arrays in heterochromatic regions, having a known satellite-like behavior. So, to categorize sequences with smaller monomer sizes as satDNAs (classically with longer repeat unit sizes) is also plausible [52,102]. The uncertainty found in human satDNA features (such as identification, existence, location, and others) is caused by the complex assembly of the (peri)centromeric regions [95,103]. The organization of αSAT (the most studied human satellite) in the human centromere clearly shows this intricacy: each HOR array is composed of the same monomer set; however, HOR composition is responsible for chromosome specificity, and HOR repeat number is distinguishable between individuals and even between homologous chromosomes of the same individual [66].
The vast structure of the human centromere has been paradoxically suppressed since the apparent completion of the human genome sequence map by the human genome project (HGP) consortium in 2003 [104], which in reality excluded 10% (or more) of genomic elements, specifically large portions of repetitive (peri)centromeric sequences [103] and acrocentric short arm sequences [105]. Today, the human genome reference GRCh38 still contains 161 Mbp of undetermined sequences (up to 5% of the genome) [106]. The latter fact is striking given that, using computational tools like the Tandem Repeats Finder [107] and Repeat-Masker, it was possible to generate evidence suggesting that ~1 million tandemly repeated sequences exist in the human genome (according to the UCSC annotation) [108,109].
Highly repetitive satellite DNA undoubtedly represents a major gap in human genome assemblies, significantly contributing to the lack of high-resolution sequencing studies in the field of centromere genomics, whose characterization has been substantially hindered by the repetitive nature of satDNA [26,110,111]. The availability of computer software algorithms for sequence analysis has been highly restricted to methods excluding repetitive sequences and disregarding their annotation [6,97,111]. Reads repetitive in nature (i.e., mapping to multiple locations) are generally overlooked. These problems cause misalignments and misassemblies [112] with a high number of contigs, assigning untraceable genomic positions to the analyzed repeats [113]. It is a known fact that satellite repeats are significantly represented in assembly pools, but the precise determination of their location in linear stretches within centromeric regions becomes unfeasible [5]. Theoretically, the correct placement of centromeric repeats in a linear assembly requires the availability of distinctive sequence information [114], which cannot be due to sequencing errors and is often not found in sequencing reads from homogenized satellite arrays [115,116,117], at least until recently.
An improved contiguity of the human reference genome is of crucial importance in the case of repetitive satellite sequences. Compared to the previous assembly, the GRCh38 human reference genome provides for the first time a representation of centromeric sequences, with the former gaps replaced by the insertion of millions of bases of chromosome-specific α satellite repeats identified from sequencing reads, modeling each centromere. Although this inclusion is expected to have a positive impact on the mapping and assembly of sequencing reads [118], the sole inclusion of αSAT sequences [95] undermines accurate and proportional representation of repetitive sequences, biasing data interpretation [119]. If we start from an incomplete and collapsed set of satellite data, we certainly ignore uncharted, possibly relevant information, especially information related to pericentromeric satDNA families. Acrocentric chromosomes, often lacking unique markers and sequence heterogeneity, are especially affected by this obstacle [114]. Within the same HOR array, HORs have little variability. So, centromere sequence assembly has to rely on monomer rearrangements [96,120,121] causing single nucleotide variants or large structural variants [122,123]. Hence, to the challenge of sequencing a single HOR array, acrocentric chromosomes add the difficulty of HOR array sharing and sequence similarities concerning pericentromeric subfamilies.
Recently, a group of scientists initiated the telomere-to-telomere (T2T) consortium with the aim of produce high-quality, end-to-end assemblies for every human chromosome, sharing all the generated data with the scientific community. This consortium is settled to fix the remaining gaps in the human genome (particularly the sections composed by highly repetitive DNA, typically difficult to include in cloning, sequencing, or assembling processes [124]) by combining different sequencing technologies and bioinformatic tools (progress detailed in the Section 4.3).
4. Satellite DNA Analysis: The Promise behind Long Reads and Technique Interdependency
4.1. Sequencing Methodologies over Time
DNA sequencing technologies were developed to solve the pressing need of efficiently identifying the order of monomers in the largest biological molecule. The evolution path of sequencing methods was forged by Sanger sequencing in the 1970s; it was firstly unmanageable in terms of reproducibility but continuously developed and was soon seen as a standard for many years to come [125]. The dideoxy Sanger method allowed the obtention of the first sequenced genome (bacteriophage Phi-X174) [126] and many years later, was behind the first draft of the human genome [127,128]. In a prior time when Sanger sequencing was in development, the use of polyacrylamide gels was slow, laborious, and simply not compatible with larger, complex genomes (the human genome appeared unachievable) [129]. So, and thanks to their genomic abundance, repetitive sequences were an early present obstacle when sequencing the human genome. The development of capillary electrophoresis and fluorescent labelling allowed the development of highly parallel sequencing in automated sequencing machines [130,131,132,133], turning the first sequence of the human genome a visible reality. In 2003, the first sequence of the human genome was achieved by the International Human Genome Sequencing Consortium (IHGSC) and the private biotechnology company Celera genomics, using the same method for DNA sequencing but following different approaches. As the Sanger methodology generates sequence information below one kb (kilobase) in length, the fast determination of long fragments required the development of approaches based on the ‘‘shotgun sequencing’’ of cloned segments derived from randomly fragmented large molecular weight DNA, followed by sequence assembly into in silico contigs [134,135]. With the first map of the human genome, the need to efficiently tackle human genetic diversity laid the foundations for the appearance of new sequencing methods capable of addressing such issues. Next-generation sequencing (NGS) emerged, supporting high-throughput and cost-effective analysis, and considerably improving the sequencing process [136,137,138]. Throughout the years, the introduction of high-performance platforms has allowed the easy and low-cost obtention of a very high number of short reads and a novel understanding of genome complexity [139]. Despite the benefits, the limitations of NGS (or third generation sequencing) spurred the development of alternative technologies in order to reach the technological highpoint of sequencing with reads great in length, accuracy, and the use of native DNA [138,140].
Long-read technologies, like PacBio (Pacific Biosciences) or Oxford Nanopore Sequencing, can surpass some limitations of short reads, such as the profiling of tandem repetitive sequences [141]. Despite enabling highly accurate genotyping in non-repetitive genomic regions, technologies like short-read Illumina sequencing are not able to provide contiguous de novo genome assemblies, limiting the reconstruction of long stretches of repetitive sequences [142]. By turn, sequencing methods that produce longer read lengths have been shown to support a more accurate assessment of the size of repeated monomers in satellite sequences [143]. With clear chemical and functional differences, the two predominant long-read technologies—henceforth termed PacBio and Nanopore sequencing—have great potential in the study of previously unreachable (peri)centromeric and acrocentric short arm regions s [144]. The different approaches used by each technology affect read length, accuracy, and throughput, establishing distinct limiting factors. For example, the sequencing-by-synthesis principle used by PacBio, often called single-molecule real-time (SMRT) sequencing, has a higher accuracy but read lengths that are limited by the lifespan of the polymerase [145], constantly incorporating fluorescently labelled deoxynucleoside triphosphates [144]. In contrast, for Nanopore sequencing, read length limits essentially depend on the ability to obtain unfragmented high-molecular weight (HMW) DNA, whereas accuracy is dependent on the ability of base-calling algorithms to deconvolute the ionic current fluctuations established by the passage of molecules through a pore into a precise identification of the order of nucleotides [142,145]. The current read length record using R10.3 nanopore flow cells stands at 4.3 Mb, while the most recent analysis algorithm Bonito CRF is consistently supporting single read accuracies of 98% and SNP accuracies of 99.92%, comparable to short read accuracy [146]. In turn, PacBio counts with a level of accuracy of 99.8% [147], thanks to the latest circular consensus sequencing (CCS) technology [148], which produces the so-called high-fidelity (Hi-Fi) reads (longer than 10 kb) [144]. Both PacBio and Nanopore sequencing offer the opportunity to address long tandem repeats by analyzing their length, nucleotide composition, and nucleotide modifications. Nanopore Sequencing is particularly promising given its polymerase-free chemistry and related ability to tackle extreme GC content [149], while PacBio offers the increased accuracy of HiFi reads, specifically interesting for assembling centromeric repeats [150,151].
Given that a wide variety of satellite repeats can be transcribed in noncoding RNAs (ncRNAs), RNA sequencing (RNA-seq) has also become a part of satellite DNA research. Satellite DNA expression may depend on time, tissue, developmental state, or stress conditions [18,20,26], clearly making RNA-seq studies noteworthy and subject to constant evolution. In this matter, it is important to emphasize on the different methodological constrains of analyzing small or long ncRNAs. Long ncRNAs (lncRNAs) represent transcripts longer than 200 nucleotides, and contrarily to small ncRNAs, the great majority of their functions remain poorly understood [152,153]. The challenging nature of lncRNAs is essentially related to their features (for example, low abundance, structure conservation rather than sequence conservation, or even inconsistent polyadenylation) [152]. These differences are pertinent, given that satellite DNA transcripts are variable in size (from small to long ncRNAs) [20,154]. RNA-seq with short-read technologies traditionally relies on cDNA synthesis, reduced read length, and high sequencing throughput. On the other hand, newer long-read technologies, through long-read RNA-seq or direct RNA sequencing (dRNA-seq), are allowing one to address several questions related to RNA characterization. SatDNA transcripts that have distinctive features like length, strand specificity, nucleotide modifications, and polyadenylation [26]. The possibility of directly sequencing RNA (brought by Nanopore sequencing) [155,156] has removed the need for cDNA conversion or PCR amplification, significantly removing associated bias [157,158] and supporting the analysis of epigenetic modifications and 3′ poly(A) tails [159]. However, Nanopore sequencing of RNA molecules associates with higher error rates than DNA sequencing [158,160]. In this regard, PacBio (despite not allowing dRNA-seq) can face the problem of high error rates by increasing read coverage with consensus circular sequencing (CCS) [157].
4.2. Technique Interdependency
While assembling a genome, specific satellite-associated gaps arise due to the organization in HOR tandem arrays, causing read “pile-ups” during the overlap mapping stage that cannot be resolved [161]. With long-read sequencing, analysis of whole sections of repetitive DNA becomes more accessible, including HOR structure, TE interruption [162], or the accurate determination of monomer size [143]. The overall centromeric structure is finally within reach in a variety of circumstances.
Nevertheless, the picture did not immediately present itself as all rosy for long-read technologies, as improved read length was often accompanied by increased error rate. Presently, the attempt to overcome this association is based on high-quality consensus and improved read coverage [114,163], which can still be improved by additional strategies for de novo genome assembly, especially in repetitive regions. This is where error correction enters, using non-hybrid or hybrid methods, in a process generally termed ‘‘polishing’’ (Figure 1). Non-hybrid correction is solely based on long reads; hybrid methods rely on the accuracy of supplementary short-read information [145]. The latter is usually applied in the context of a long-read-generated high-level genome assembly with repeat complexity, which is subsequently polished with short-read data for high-quality resolution (though the opposite is also applicable) [163,164,165,166].
Mapping strategies and cytogenetic approaches can also be applied to aid in the assembly process (Figure 1) [147], experimentally validating array structure [114]. Cytogenetic methodologies include optical mapping by Bionano, which uses restriction enzymes and fluorescent labels to obtain linearized genomic optical maps [147,167]; andfibre-FISH, where fluorescently labelled probes hybridize to stretched DNA [110,147,168]. Optical mapping, for instance, has been used in improving contig size or in scaffolding [169,170]. Additionally, PFGE Southern-blotting, making use of specific satellite array probes [122], and combined with other methods like quantitative digital droplet PCR [120], can help discover HOR array copy number and structure by comparing the resulting data with long-read assembly information [114,120]. Polishing methods and mapping-assisted assembly are co-dependent and intrinsically related. The obtainment of higher error rates with long-read sequencing greatly hampers precise sequence annotation [171], limiting the purpose of scaffolding methods (like optical mapping), which rely on base accuracy [172] (possibly guaranteed by polishing strategies) [173].
To sum up, genome sequencing projects can be greatly facilitated and provide integrated knowledge if combined with other fields of genome biology such as cytogenetics and cellular biology [174]. In addition to the already mentioned Bionano and fibre-FISH, several cytogenetic approaches have been used in interdependency with “orthodox” genomic studies (a changing concept, since genomic studies are progressively integrating research from all fields). These cytogenetic methodologies include chromosome microdissection [175,176,177], chromosome flow sorting [178], and assessment of chromatin interactions (e.g., Hi-C sequencing) [179,180]. Technologies like chromosome flow sorting, chromosome microdissection, and even magnetic bead capture offer an inventive, technical tackling of complex repetitive genomic regions, through the ability to isolate specific chromosomes/chromosomal regions [181,182]. Hence, this targeted capture can be applied to the task of sequencing a chromosome for which there is limited sequence information [181], which can be particularly interesting in the case of (peri)centromeric/acrocentric short arm sequences, given the extensive degree of sequence sharing between different chromosomes.
Cytogenetically-assisted chromosome-level assemblies undoubtedly offer more insightfulness than a disarray of fragmented contigs/scaffolds. To fully understand a genome, it is also necessary to recognize genome architecture and chromosome function. In order to improve genome assembly, all the above-mentioned technologies deeply depend on the development of adequate bioinformatic pipelines and software tools, which progressively need to adapt to new data forms. The concept of technique interdependency intricates algorithm development since it demands large data compacting structures, flexibility, and consequent speed-increased analysis [183]. Recently, a set of long-read tools has been put together to evaluate state-of-art software possibilities and identify missing useful pipelines with development potential (long-read-tools.org) [145]. Thus, the evolution of genome sequencing and mapping technologies must be supported by innovative bioinformatic tools with ability to overcome the computational challenge of combined data.
4.3. The Achieved Vs. the Achievable
In the last fifty years, since the first efforts to characterize human satellite DNA families, a variety of research landmarks have spearheaded the development of the field of satDNA biology (Figure 2). The appearance of restriction enzyme treatment for satDNA analysis [184] or the development of in situ hybridization [185,186] supported the attainment of the first descriptions of the main human satellite families. With the progressive growth of sequencing technologies, several satDNAs have been sequenced and analyzed in specific and independent studies. However, they have somewhat been left aside from genome assemblies, essentially due to the previously discussed technological constrains of short-read sequencing. Nonetheless, in more recent years, the emergence of terms like ‘‘repeatome’’ [187] or ‘‘satellitome’’ [188] highlights the advantages of gathering total information about satDNA diversity using sequencing data. In turn, long-read technologies have produced the possibility of finally assembling satellite sequences. Human X [120] and Y [96] chromosomes were the first targeted for a linear centromeric assembly with characterization of centromeric array data (α HOR units DXZ1 and DYZ3, respectively). Sequencing of the DYZ3 centromeric array relied on the use of a previously developed satDNA BAC library spanning the entire region, which is considered a laborious and possibly bias-prone process. On the other hand, sequencing and polishing of DXZ1 relied on the presence of unique markers [114]. Both historical landmarks set a high-quality precedent for the potential of centromere genomic studies. Twenty years after the first draft of the human genome, the T2T (responsible for fully sequencing the X chromosome) just released a new human genome assembly for all chromosomes using the CHM13 haploid cell line, excitingly bringing the first wide-ranging glance of (peri)centromeric and acrocentric short arm regions. This project provides a near-completed assembly of the human genome, as only 5 rDNA-related gaps remain. Thus, high-resolution analysis excitingly became a reality for highly repetitive genomic regions [106,124].
Additionally, HiCanu software development [150], aimed at quality improvement for pre-existing reads, has resulted in linear assembly predictions spanning centromeric regions of chromosomes 2, 3, 7, 8, 10, 12, 16, 19, and 20, an achievement that clearly demonstrates the importance of upgrading assembler software. Beyond that, the linear assembly of chromosome 7 allowed one to confirm the presence of other repetitive elements like pericentromeric satellite families [114]. Bioinformatic tools can also be applied to determine HOR structure and variability, allowing one to distinguish apparently shared satDNA repeats, especially between acrocentric chromosomes [28]. Unraveling (peri)centromeric structural diversity will eventually allow us to expand our knowledge about the evolution of these type of sequences [71].
In the previous section, we emphasized the advances of technique interdependency, for example in the form of cytogenetically-assisted assembly. Along this line, future genomic innovation will probably rely on the combination of the maximum number of informative techniques. Ideally, high-quality array characterization should extend from αSAT repeats to other satellite families, primarily by exploring a broad inventory covering the variability of satDNA (sub)families. Despite clear efforts in representing centromere sequences in the human reference genome, all available human assemblies (previously to T2T consortium) drastically underrepresent SATII/SATIII repeats [39], a gap that has not even been acknowledged for the remaining families. Nevertheless, this type of underestimations caused by the collapse of identical repeats can be avoided by using raw read information for each satellite family [41,67]. Henceforth, it is essential to genomically update/confirm the available information about satDNA (sub)families, as several questions are unavoidable: number of (sub)families, genomic representativity, biological significance, and overall existence. Through the analysis of the CHM13 genome assembly from T2T, the meticulous analysis of satDNA (sub)families is finally a near reality.
Since human satDNA families present such a complex variability between individuals, and even between homologous chromosomes, the next genomic challenges will probably be centered on exploiting these differences: upscaling linear assembly to diploid chromosomes by optimizing phased assembly [114]; and extending the concept of ‘‘reference’’ genome to a multidimensional pan-genome approach, fully representing human populational haplogroup variation [71]. This last goal is being currently addressed by the Human Pangenome Reference Consortium, which recently launched 64 assembled human haplotypes from 32 genomes uncovering previously uncharted human variation [189]. Essentially, it is clear that satDNA analysis can benefit from the most representative survey of human satellite variants and their possible functional significance [39].
Figure 2.
Landmarks in human satellite DNA research. The depicted timeline represents the knowledge evolution of satellite DNA biology in terms of classification, representativity, and significance, as well as the potential future and ongoing character of substantial breakthroughs in the area [7,9,10,29,30,31,45,46,51,52,53,58,73,74,75,79,87,92,96,107,117,120,127,128,150,162,173,184,185,186,187,188,190,191,192,193,194,195,196,197,198,199].
Figure 2.
Landmarks in human satellite DNA research. The depicted timeline represents the knowledge evolution of satellite DNA biology in terms of classification, representativity, and significance, as well as the potential future and ongoing character of substantial breakthroughs in the area [7,9,10,29,30,31,45,46,51,52,53,58,73,74,75,79,87,92,96,107,117,120,127,128,150,162,173,184,185,186,187,188,190,191,192,193,194,195,196,197,198,199].

5. Concluding Remarks
There is no doubt that satDNA has established a past, present, and future in breaking barriers. Some of the largest have been recently overcome with the breakthrough of telomere-to-telomere assembly. Still, the detailed characterization of human satDNA families must walk a long path. To achieve better centromere contiguity and overall knowledge of (peri)centromeric/acrocentric short arm regions, it is vital to bet on the thorough tactic of technique interdependency. Better than a flawless, versatile, and stand-alone technique, a combined approach, where one technique’s disadvantages are counteracted by another, should be used for attaining accuracy.
In this review, we tried to trail the route of obstacles in satDNA analysis and to draw attention to the disparities in the amount of available information for different families. Previous attitudes towards repetitive sequences, combined with the subsequent awareness of their biological importance, establish a clear dichotomy, alerting one to the yet uncharted world of human satDNA diversity. Thus, by broadening our satDNA knowledge through time, it is highly likely that we will yet encounter new forms of functionality.
Author Contributions
M.L., S.L., and R.C. conceptualization; M.L. and S.L. data curation and investigation; M.L. wrote the original draft; R.C. funding acquisition; M.L., S.L., M.G.-C., and R.C. manuscript revision. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ph.D. grant (SFRH/BD/147488/2019), by a Scientific Employment Stimulus 2017 junior research contract in the biological sciences field, from the Science and Technology Foundation (FCT) from Portugal.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Britten, R.J.; Kohne, D.E. Repeated sequences in DNA. Science 1968, 161, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. So much’junk’DNA in our genome. In Proceedings of Evolution of Genetic Systems. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar] [PubMed]
- Palazzo, A.F.; Gregory, T.R. The case for junk DNA. PLoS Genet. 2014, 10, e1004351. [Google Scholar] [CrossRef] [Green Version]
- López-Flores, I.; Garrido-Ramos, M. The repetitive DNA content of eukaryotic genomes. In Repetitive DNA; Karger Publishers: Basel, Switzerland, 2012; Volume 7, pp. 1–28. [Google Scholar]
- McNulty, S.M.; Sullivan, B.A. Alpha satellite DNA biology: Finding function in the recesses of the genome. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2018, 26, 115–138. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef]
- Heitz, E. Das Heterochromatin der Moose; Bornträger: Berlin, Germany, 1928. [Google Scholar]
- Podgornaya, O.I.; Ostromyshenskii, D.I.; Enukashvily, N.I. Who Needs This Junk, or Genomic Dark Matter. Biochem. Biokhimiia 2018, 83, 450–466. [Google Scholar] [CrossRef] [PubMed]
- Kit, S. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J. Mol. Biol. 1961, 3, 711–IN712. [Google Scholar] [CrossRef]
- Sueoka, N. Compositional correlation between deoxyribonucleic acid and protein. Cold Spring Harb. Symp. Quant. Biol. 1961, 26, 35–43. [Google Scholar] [CrossRef]
- Plohl, M.; Luchetti, A.; Mestrovic, N.; Mantovani, B. Satellite DNAs between selfishness and functionality: Structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 2008, 409, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Yasmineh, W.; Yunis, J. Localization of repeated DNA sequences in CsCl gradients by hybridization with complementary RNA. Exp. Cell Res. 1974, 88, 340–344. [Google Scholar] [CrossRef]
- Hartley, G.; O’Neill, R.J. Centromere repeats: Hidden gems of the genome. Genes 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, M.; Yamashita, Y.M. Function of Junk: Pericentromeric Satellite DNA in Chromosome Maintenance. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, R.; Ferreira, D.; Mendes-da-Silva, A.; Meles, S.; Adega, F. FA-SAT Is an Old Satellite DNA Frozen in Several Bilateria Genomes. Genome Biol. Evol. 2017, 9, 3073–3087. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Dalal, Y. Centromeric chromatin: What makes it unique? Curr. Opin. Genet. Dev. 2005, 15, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Meštrović, N.; Mravinac, B. Satellite DNA evolution. In Repetitive DNA; Karger Publishers: Basel, Switzerland, 2012; Volume 7, pp. 126–152. [Google Scholar]
- Biscotti, M.A.; Canapa, A.; Forconi, M.; Olmo, E.; Barucca, M. Transcription of tandemly repetitive DNA: Functional roles. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Escudeiro, A.; Adega, F.; Anjo, S.I.; Manadas, B.; Chaves, R. FA-SAT ncRNA interacts with PKM2 protein: Depletion of this complex induces a switch from cell proliferation to apoptosis. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Meles, S.; Escudeiro, A.; Mendes-da-Silva, A.; Adega, F.; Chaves, R. Satellite non-coding RNAs: The emerging players in cells, cellular pathways and cancer. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Lee, J.; Han, K. Transposable elements: No more ‘Junk DNA’. Genom. Inform. 2012, 10, 226. [Google Scholar] [CrossRef]
- Makałowski, W. Genomic scrap yard: How genomes utilize all that junk. Gene 2000, 259, 61–67. [Google Scholar] [CrossRef]
- Puppo, I.; Saifitdinova, A.; Tonyan, Z. The Role of Satellite DNA in Causing Structural Rearrangements in Human Karyotype. Russ. J. Genet. 2020, 56, 41–47. [Google Scholar] [CrossRef]
- Veiko, N.N.; Ershova, E.S.; Malinovskaya, E.M.; Konkova, M.S.; Veiko, R.V.; Umriukhin, P.E.; Martynov, A.V.; Kutsev, S.I.; Kostyuk, S.V. Copy number variation of human satellite III (1q12) with Aging. Front. Genet. 2019, 10, 704. [Google Scholar]
- Yandım, C.; Karakülah, G. Expression dynamics of repetitive DNA in early human embryonic development. BMC Genom. 2019, 20, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatskikh, A.S.; Kotov, A.A.; Adashev, V.E.; Bazylev, S.S.; Olenina, L.V. Functional Significance of Satellite DNAs: Insights from Drosophila. Front. Cell Dev. Biol. 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Aldrup-MacDonald, M.; Sullivan, B. The Past, Present, and Future of Human Centromere Genomics. Genes 2014, 5, 33–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glunčić, M.; Vlahović, I.; Paar, V. Discovery of 33mer in chromosome 21–the largest alpha satellite higher order repeat unit among all human somatic chromosomes. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Wevrick, R.; Fisher, R.B.; Ferguson-Smith, M.A.; Lin, C.C. Human centromeric DNAs. Hum. Genet. 1997, 100, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.; Frommer, M.; Paul, C.; Vincent, P.C. Sequence relationships of three human satellite DNAs. J. Mol. Biol. 1986, 187, 145–155. [Google Scholar] [CrossRef]
- Vissel, B.; Nagy, A.; Choo, K. A satellite III sequence shared by human chromosomes 13, 14, and 21 that is contiguous with α satellite DNA. Cytogenet. Genome Res. 1992, 61, 81–86. [Google Scholar] [CrossRef]
- Plohl, M.; Mestrovic, N.; Mravinac, B. Centromere identity from the DNA point of view. Chromosoma 2014, 123, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, L.L.; Byron, M.; Carone, D.M.; Whitfield, T.W.; Pouliot, G.P.; Fischer, A.; Jones, P.; Lawrence, J.B. Demethylated HSATII DNA and HSATII RNA foci sequester PRC1 and MeCP2 into cancer-specific nuclear bodies. Cell Rep. 2017, 18, 2943–2956. [Google Scholar] [CrossRef]
- McNulty, S.M.; Sullivan, L.L.; Sullivan, B.A. Human Centromeres Produce Chromosome-Specific and Array-Specific Alpha Satellite Transcripts that Are Complexed with CENP-A and CENP-C. Dev. Cell, 2017; 42, 226–240.e6. [Google Scholar] [CrossRef]
- Bersani, F.; Lee, E.; Kharchenko, P.V.; Xu, A.W.; Liu, M.; Xega, K.; MacKenzie, O.C.; Brannigan, B.W.; Wittner, B.S.; Jung, H. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15148–15153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delpu, Y.; McNamara, T.; Griffin, P.; Kaleem, S.; Narayan, S.; Schildkraut, C.; Miga, K.; Tahiliani, M. Chromosomal rearrangements at hypomethylated Satellite 2 sequences are associated with impaired replication efficiency and increased fork stalling. bioRxiv 2019, 554410. [Google Scholar] [CrossRef]
- Erliandri, I.; Fu, H.; Nakano, M.; Kim, J.-H.; Miga, K.H.; Liskovykh, M.; Earnshaw, W.C.; Masumoto, H.; Kouprina, N.; Aladjem, M.I. Replication of alpha-satellite DNA arrays in endogenous human centromeric regions and in human artificial chromosome. Nucleic Acids Res. 2014, 42, 11502–11516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahović, I.; Glunčić, M.; Rosandić, M.; Ugarković, Đ.; Paar, V. Regular higher order repeat structures in beetle Tribolium castaneum genome. Genome Biol. Evol. 2017, 9, 2668–2680. [Google Scholar] [PubMed] [Green Version]
- Miga, K.H. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburton, P.E.; Hasson, D.; Guillem, F.; Lescale, C.; Jin, X.; Abrusan, G. Analysis of the largest tandemly repeated DNA families in the human genome. BMC Genom. 2008, 9, 533. [Google Scholar] [CrossRef] [Green Version]
- Altemose, N.; Miga, K.H.; Maggioni, M.; Willard, H.F. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput. Biol. 2014, 10, e1003628. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.; Drinkwater, R.; Burgoyne, L.; Skinner, J. Hypervariable lengths of human DNA associated with a human satellite III sequence found in the 3.4 kb Y-specific fragment. Nucleic Acids Res. 1987, 15, 3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.Y.; Benn, P.A.; Tannenbaum, H.L.; Perlis, T.E.; Carlson, A.D.; Opitz, J.M.; Reynolds, J.F. Chromosomal polymorphisms of 1, 9, 16, and Y in 4 major ethnic groups: A large prenatal study. Am. J. Med Genet. 1987, 26, 95–101. [Google Scholar] [CrossRef]
- Podugolnikova, O.; Korostelev, A. The quantitative analysis of polymorphism on human chromosomes 1, 9, 16, and Y. IV. Heterogeneity of a normal population. Hum. Genet. 1980, 54, 163. [Google Scholar] [CrossRef] [PubMed]
- Tagarro, I.; Fernández-Peralta, A.M.; González-Aguilera, J.J. Chromosomal localization of human satellites 2 and 3 by a FISH method using oligonucleotides as probes. Hum. Genet. 1994, 93, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Trowell, H.E.; Nagy, A.; Vissel, B.; Choo, K.A. Long-range analyses of the centromeric regions of human chromosomes 13, 14 and 21: Identification of a narrow domain containing two key centromeric DNA elements. Hum. Mol. Genet. 1993, 2, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Agresti, A.; Rainaldi, G.; Lobbiani, A.; Magnani, I.; Di Lernia, R.; Meneveri, R.; Siccardi, A.; Ginelli, E. Chromosomal location by in situ hybridization of the human Sau3A family of DNA repeats. Hum. Genet. 1987, 75, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Cooke, H.J.; Hindley, J. Cloning of human satellite III DNA: Different components are on different chromosomes. Nucleic Acids Res. 1979, 6, 3177–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosden, J.; Mitchell, A.; Buckland, R.; Clayton, R.; Evans, H. The location of four human satellite DNAs on human chromosomes. Exp. Cell Res. 1975, 92, 148–158. [Google Scholar] [CrossRef]
- Jørgensen, A.L.; Kølvraa, S.; Jones, C.; Bak, A.L. A subfamily of alphoid repetitive DNA shared by the NOR-bearing human chromosomes 14 and 22. Genomics 1988, 3, 100–109. [Google Scholar] [CrossRef]
- Tagarro, I.; Wiegant, J.; Raap, A.K.; González-Aguilera, J.J.; Fernández-Peralta, A.M. Assignment of human satellite 1 DNA as revealed by fluorescent in situ hybridization with oligonucleotides. Hum. Genet. 1994, 93, 125–128. [Google Scholar] [CrossRef]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Maio, J.J. DNA strand reassociation and polyribonucleotide binding in the African green monkey, Cercopithecus aethiops. J. Mol. Biol. 1971, 56, 579–595. [Google Scholar] [CrossRef]
- Manuelidis, L. Chromosomal localization of complex and simple repeated human DNAs. Chromosoma 1978, 66, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.; Schueler, M.; Willard, H. Sequence organization and functional annotation of human centromeres. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 141–149. [Google Scholar] [CrossRef]
- Willard, H.F. Chromosome-specific organization of human alpha satellite DNA. Am. J. Hum. Genet. 1985, 37, 524. [Google Scholar] [PubMed]
- Willard, H.F.; Waye, J.S. Chromosome-specific subsets of human alpha satellite DNA: Analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat. J. Mol. Evol. 1987, 25, 207–214. [Google Scholar] [CrossRef] [PubMed]
- AIexandrov, I.; Medvedev, L.; Mashkova, T.; Kisselev, L.; Romanova, L.; Yurov, Y. Definition of a new alpha satellite suprachromosomal family characterized by monomeric organization. Nucleic Acids Res. 1993, 21, 2209–2215. [Google Scholar] [CrossRef]
- Shepelev, V.; Uralsky, L.; Alexandrov, A.; Yurov, Y.; Rogaev, E.I.; Alexandrov, I. Annotation of suprachromosomal families reveals uncommon types of alpha satellite organization in pericentromeric regions of hg38 human genome assembly. Genom. Data 2015, 5, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muro, Y.; Masumoto, H.; Yoda, K.; Nozaki, N.; Ohashi, M.; Okazaki, T. Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box. J. Cell Biol. 1992, 116, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, K.F.; Glass, C.A. CENP-B is a highly conserved mammalian centromere protein with homology to the helix-loop-helix family of proteins. Chromosoma 1991, 100, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devilee, P.; Cremer, T.; Slagboom, P.; Bakker, E.; Scholl, H.P.; Hager, H.; Stevenson, A.; Cornelisse, C.; Pearson, P. Two subsets of human alphoid repetitive DNA show distinct preferential localization in the pericentric regions of chromosomes 13, 18, and 21. Cytogenet. Genome Res. 1986, 41, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.; Vissel, B.; Nagy, A.; Earle, E.; Kalitsis, P. A survey of the genomic distribution of alpha satellite DNA on all the human chromosomes, and derivation of a new consensus sequence. Nucleic Acids Res. 1991, 19, 1179. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.L.; Chew, K.; Sullivan, B.A. α satellite DNA variation and function of the human centromere. Nucleus 2017, 8, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.L.; Sullivan, B.A. Genomic and functional variation of human centromeres. Exp. Cell Res. 2020, 111896. [Google Scholar] [CrossRef] [PubMed]
- Hayden, K.E.; Strome, E.D.; Merrett, S.L.; Lee, H.-R.; Rudd, M.K.; Willard, H.F. Sequences associated with centromere competency in the human genome. Mol. Cell. Biol. 2013, 33, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, J.G.; Thakur, J.; Kasinathan, S.; Henikoff, S. A unique chromatin complex occupies young α-satellite arrays of human centromeres. Sci. Adv. 2015, 1, e1400234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waye, J.S.; Willard, H.F. Nucleotide sequence heterogeneity of alpha satellite repetitive DNA: A survey of alphoid sequences from different human chromosomes. Nucleic Acids Res. 1987, 15, 7549–7569. [Google Scholar] [CrossRef] [Green Version]
- Schueler, M.G.; Dunn, J.M.; Bird, C.P.; Ross, M.T.; Viggiano, L.; Rocchi, M.; Willard, H.F.; Green, E.D.; Program, N.C.S. Progressive proximal expansion of the primate X chromosome centromere. Proc. Natl. Acad. Sci. USA 2005, 102, 10563–10568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, G.A.; Vollger, M.R.; Hsieh, P.; Mao, Y.; Liskovykh, M.A.; Koren, S.; Nurk, S.; Mercuri, L.; Dishuck, P.C.; Rhie, A. The structure, function and evolution of a complete human chromosome 8. Nature 2021, 1–7. [Google Scholar] [CrossRef]
- Shepelev, V.A.; Alexandrov, A.A.; Yurov, Y.B.; Alexandrov, I.A. The evolutionary origin of man can be traced in the layers of defunct ancestral alpha satellites flanking the active centromeres of human chromosomes. PLoS Genet 2009, 5, e1000641. [Google Scholar] [CrossRef]
- Meyne, J.; Goodwin, E.H.; Moyzis, R.K. Chromosome localization and orientation of the simple sequence repeat of human satellite I DNA. Chromosoma 1994, 103, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kalitsis, P.; Earle, E.; Vissel, B.; Shaffer, L.G.; Choo, K.A. A chromosome 13-specific human satellite I DNA subfamily with minor presence on chromosome 21: Further studies on Robertsonian translocations. Genomics 1993, 16, 104–112. [Google Scholar] [CrossRef]
- Jeanpierre, M. Human satellites 2 and 3. Ann. De Genet. 1994, 37, 163–171. [Google Scholar]
- Moyzis, R.K.; Torney, D.C.; Meyne, J.; Buckingham, J.M.; Wu, J.-R.; Burks, C.; Sirotkin, K.M.; Goad, W.B. The distribution of interspersed repetitive DNA sequences in the human genome. Genomics 1989, 4, 273–289. [Google Scholar] [CrossRef]
- Schwarzacher-Robinson, T.; Cram, L.; Meyne, J.; Moyzis, R. Characterization of human heterochromatin by in situ hybridization with satellite DNA clones. Cytogenet. Cell Genet. 1988, 47, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Nakahori, Y.; Mitani, K.; Yamada, M.; Nakagome, Y. A human Y-chromosome specific repeated DNA family (DYZ1) consists of a tandem array of pentanucleotides. Nucleic Acids Res. 1986, 14, 7569–7580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.A. The Centromere; Oxford University Press: Oxford, UK, 1997; Volume 320. [Google Scholar]
- Cooke, H. Repeated sequence specific to human males. Nature 1976, 262, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, L.M.; Smith, K.D.; Boyer, S.H. Human Y-chromosome-specific reiterated DNA. Science 1976, 191, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.; Earle, E.; McQuillan, C. A homologous subfamily of satellite III DNA on human chromosomes 14 and 22. Nucleic Acids Res. 1990, 18, 5641–5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.; Earle, E.; Vissel, B.; Kalitsis, P. A chromosome 14-specific human satellite III DNA subfamily that shows variable presence on different chromosomes 14. Am. J. Hum. Genet. 1992, 50, 706. [Google Scholar] [PubMed]
- Bandyopadhyay, R.; McQuillan, C.; Page, S.; Choo, K.; Shaffer, L. Identification and characterization of satellite III subfamilies to the acrocentric chromosomes. Chromosome Res. 2001, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Earle, E.; Shaffer, L.; Kalitsis, P.; McQuillan, C.; Dale, S.; Choo, K. Identification of DNA sequences flanking the breakpoint of human t (14q21q) Robertsonian translocations. Am. J. Hum. Genet. 1992, 50, 717. [Google Scholar] [PubMed]
- Meneveri, R.; Agresti, A.; Della Valle, G.; Talarico, D.; Siccardi, A.; Ginelli, E. Identification of a human clustered G+ C-rich DNA family of repeats (Sau3A family). J. Mol. Biol. 1985, 186, 483–489. [Google Scholar] [CrossRef]
- Waye, J.S.; Willard, H.F. Human beta satellite DNA: Genomic organization and sequence definition of a class of highly repetitive tandem DNA. Proc. Natl. Acad. Sci. USA 1989, 86, 6250–6254. [Google Scholar] [CrossRef] [Green Version]
- Meneveri, R.; Agresti, A.; Marozzi, A.; Saccone, S.; Rocchi, M.; Archidiacono, N.; Corneo, G.; Valle, G.D.; Ginelli, E. Molecular organization and chromosomal location of human GC-rich heterochromatic blocks. Gene 1993, 123, 227–234. [Google Scholar] [CrossRef]
- Eichler, E.E.; Hoffman, S.M.; Adamson, A.A.; Gordon, L.A.; McCready, P.; Lamerdin, J.E.; Mohrenweiser, H.W. Complex β-satellite repeat structures and the expansion of the zinc finger gene cluster in 19p12. Genome Res. 1998, 8, 791–808. [Google Scholar] [CrossRef] [Green Version]
- Cardone, M.; Ballarati, L.; Ventura, M.; Rocchi, M.; Marozzi, A.; Ginelli, E.; Meneveri, R. Evolution of beta satellite DNA sequences: Evidence for duplication-mediated repeat amplification and spreading. Mol. Biol. Evol. 2004, 21, 1792–1799. [Google Scholar] [CrossRef]
- Yang, J.; Yuan, B.; Wu, Y.; Li, M.; Li, J.; Xu, D.; Gao, Z.-h.; Ma, G.; Zhou, Y.; Zuo, Y. The wide distribution and horizontal transfers of beta satellite DNA in eukaryotes. Genomics 2020. [Google Scholar] [CrossRef]
- Lin, C.; Sasi, R.; Fan, Y.-S. Isolation and identification of a novel tandemly repeated DNA sequence in the centromeric region of human chromosome 8. Chromosoma 1993, 102, 333–339. [Google Scholar] [CrossRef]
- Lee, C.; Li, X.; Jabs, E.; Lin, C. Human gamma X satellite DNA: An X chromosome specific centromeric DNA sequence. Chromosoma 1995, 104, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Ebersole, T.; Kouprina, N.; Noskov, V.N.; Ohzeki, J.-I.; Masumoto, H.; Mravinac, B.; Sullivan, B.A.; Pavlicek, A.; Dovat, S. Human gamma-satellite DNA maintains open chromatin structure and protects a transgene from epigenetic silencing. Genome Res. 2009, 19, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H. The Promises and Challenges of Genomic Studies of Human Centromeres. Prog. Mol. Subcell. Biol. 2017, 56, 285–304. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Turner, D.J.; Stoddart, D.; Bulazel, K.V.; Paten, B.; Haussler, D.; Willard, H.F.; Akeson, M.; Miga, K.H. Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol. 2018, 36, 321–323. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H. Completing the human genome: The progress and challenge of satellite DNA assembly. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Gosden, J.; Lawrie, S.; Gosden, C. Satellite DNA sequences in the human acrocentric chromosomes: Information from translocations and heteromorphisms. Am. J. Hum. Genet. 1981, 33, 243. [Google Scholar]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [Green Version]
- Jeffreys, A.J.; Wilson, V.; Thein, S.L. Hypervariable ‘minisatellite’regions in human DNA. Nature 1985, 314, 67–73. [Google Scholar] [CrossRef]
- Litt, M.; Luty, J.A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 1989, 44, 397. [Google Scholar]
- Garrido-Ramos, M.A. Satellite DNA in Plants: More than Just Rubbish. Cytogenet Genome Res 2015, 146, 153–170. [Google Scholar] [CrossRef]
- Black, E.M.; Giunta, S. Repetitive Fragile Sites: Centromere Satellite DNA As a Source of Genome Instability in Human Diseases. Genes 2018, 9, 615. [Google Scholar] [CrossRef] [Green Version]
- Collins, F.S.; Green, E.D.; Guttmacher, A.E.; Guyer, M.S. A vision for the future of genomics research. Nature 2003, 422, 835. [Google Scholar] [CrossRef]
- Eichler, E.E.; Clark, R.A.; She, X. An assessment of the sequence gaps: Unfinished business in a finished human genome. Nat. Rev. Genet. 2004, 5, 345. [Google Scholar] [CrossRef]
- Phillippy, A. The (Near) Complete Sequence of a Human Genome. Available online: https://genomeinformatics.github.io/CHM13v1/ (accessed on 20 April 2021).
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, S.; Frith, M.C.; Mizuguchi, T.; Miyatake, S.; Toyota, T.; Adachi, H.; Oma, Y.; Kino, Y.; Mitsuhashi, H.; Matsumoto, N. Tandem-genotypes: Robust detection of tandem repeat expansions from long DNA reads. Genome Biol. 2019, 20, 58. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef]
- Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-Carvalho, M.; Chaves, R. Decoding the Role of Satellite DNA in Genome Architecture and Plasticity—An Evolutionary and Clinical Affair. Genes 2020, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [Green Version]
- Ameur, A.; Kloosterman, W.P.; Hestand, M.S. Single-molecule sequencing: Towards clinical applications. Trends Biotechnol. 2018, 37, 72–85. [Google Scholar] [CrossRef]
- Cao, M.D.; Nguyen, S.H.; Ganesamoorthy, D.; Elliott, A.G.; Cooper, M.A.; Coin, L.J. Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nat. Commun. 2017, 8, 14515. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H. Centromere studies in the era of ‘telomere-to-telomere’genomics. Exp. Cell Res. 2020, 112127. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B. Comparison of the two major classes of assembly algorithms: Overlap–layout–consensus and de-bruijn-graph. Brief. Funct. Genom. 2012, 11, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Luce, A.C.; Sharma, A.; Mollere, O.S.; Wolfgruber, T.K.; Nagaki, K.; Jiang, J.; Presting, G.G.; Dawe, R.K. Precise centromere mapping using a combination of repeat junction markers and chromatin immunoprecipitation–polymerase chain reaction. Genetics 2006, 174, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Dai, Y.; Yu, H.; Zhao, S.; Samuels, D.C.; Shyr, Y. Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 2017, 109, 83–90. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Myers, G.; Morishita, S. Long-read Data Revealed Structural Diversity in Human Centromere Sequences. BioRxiv 2019, 784785. [Google Scholar] [CrossRef] [Green Version]
- Mahtani, M.M.; Willard, H.F. Pulsed-field gel analysis of α-satellite DNA at the human X chromosome centromere: High-frequency polymorphisms and array size estimate. Genomics 1990, 7, 607–613. [Google Scholar] [CrossRef]
- Schindelhauer, D.; Schwarz, T. Evidence for a fast, intrachromosomal conversion mechanism from mapping of nucleotide variants within a homogeneous α-satellite DNA array. Genome Res. 2002, 12, 1815–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miga, K.H. Breaking through the Unknowns of the Human Reference Genome; Nature Publishing Group: London, UK, 2021. [Google Scholar]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.; Slocombe, P.M.; Smith, M. Nucleotide sequence of bacteriophage φX174 DNA. Nature 1977, 265, 687. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janitz, M. Next-Generation Genome Sequencing: Towards Personalized Medicine; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Ansorge, W.; Sproat, B.; Stegemann, J.; Schwager, C.; Zenke, M. Automated DNA sequencing: Ultrasensitive detection of fluorescent bands during electrophoresis. Nucleic Acids Res. 1987, 15, 4593–4602. [Google Scholar] [CrossRef] [Green Version]
- Luckey, J.A.; Drossman, H.; Kostichka, A.J.; Mead, D.A.; D’Cunha, J.; Norris, T.B.; Smith, L.M. High speed DNA sequencing by capillary electrophoresis. Nucleic Acids Res. 1990, 18, 4417–4421. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.M.; Fung, S.; Hunkapiller, M.W.; Hunkapiller, T.J.; Hood, L.E. The synthesis of oligonucleotides containing an aliphatic amino group at the 5′ terminus: Synthesis of fluorescent DNA primers for use in DNA sequence analysis. Nucleic Acids Res. 1985, 13, 2399–2412. [Google Scholar] [CrossRef]
- Swerdlow, H.; Gesteland, R. Capillary gel electrophoresis for rapid, high resolution DNA sequencing. Nucleic Acids Res. 1990, 18, 1415–1419. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staden, R. A strategy of DNA sequencing employing computer programs. Nucleic Acids Res. 1979, 6, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- de Lannoy, C.; de Ridder, D.; Risse, J. The long reads ahead: De novo genome assembly using the MinION. F1000Research 2017, 6, 1023. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Q.; Wang, Z. The evolution of nanopore sequencing. Front. Genet. 2015, 5, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S.; Cechova, M.; Makova, K.D. Noise-Cancelling Repeat Finder: Uncovering tandem repeats in error-prone long-read sequencing data. Bioinformatics 2019, 35, 4809–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacheux, L.; Ponger, L.; Gerbault-Seureau, M.; Richard, F.A.; Escudé, C. Diversity and distribution of alpha satellite DNA in the genome of an Old World monkey: Cercopithecus solatus. BMC Genom. 2016, 17, 916. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- ONT. At NCM, Announcements Include Single-Read Accuracy of 99.1% on New Chemistry and Sequencing a Record 10 Tb in a Single PromethION Run. Available online: https://nanoporetech.com/about-us/news/ncm-announcements-include-single-read-accuracy-991-new-chemistry-and-sequencing(accessed on 21 March 2021).
- Kraft, F.; Kurth, I. Long-read sequencing to understand genome biology and cell function. Int. J. Biochem. Cell Biol. 2020, 105799. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- De Roeck, A.; De Coster, W.; Bossaerts, L.; Cacace, R.; De Pooter, T.; Van Dongen, J.; D’Hert, S.; De Rijk, P.; Strazisar, M.; Van Broeckhoven, C. NanoSatellite: Accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol. 2019, 20, 239. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. BioRxiv 2020. [Google Scholar] [CrossRef]
- Vollger, M.R.; Logsdon, G.A.; Audano, P.A.; Sulovari, A.; Porubsky, D.; Peluso, P.; Wenger, A.M.; Concepcion, G.T.; Kronenberg, Z.N.; Munson, K.M. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann. Hum. Genet. 2020, 84, 125–140. [Google Scholar] [CrossRef]
- Cao, H.; Wahlestedt, C.; Kapranov, P. Strategies to annotate and characterize long noncoding RNAs: Advantages and pitfalls. Trends Genet. 2018, 34, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Interactions between short and long noncoding RNAs. FEBS Lett. 2018, 592, 2874–2883. [Google Scholar] [CrossRef] [Green Version]
- Saksouk, N.; Simboeck, E.; Déjardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 2015, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J. Nanopore native RNA sequencing of a human poly (A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Wilson, R.K.; Eichler, E.E. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015, 16, 627. [Google Scholar] [CrossRef] [PubMed]
- Sevim, V.; Bashir, A.; Chin, C.-S.; Miga, K.H. Alpha-CENTAURI: Assessing novel centromeric repeat sequence variation with long read sequencing. Bioinformatics 2016, 32, 1921–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-Generation Sequencing Technologies. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-generation sequencing and emerging technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Zhou, P.; Mudge, J.; Gurtowski, J.; Lee, H.; Ramaraj, T.; Walenz, B.P.; Liu, J.; Stupar, R.M.; Denny, R. Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genom. 2017, 18, 541. [Google Scholar] [CrossRef] [PubMed]
- Minei, R.; Hoshina, R.; Ogura, A. De novo assembly of middle-sized genome using MinION and Illumina sequencers. BMC Genom. 2018, 19, 700. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, M.H.; Pang, A.W.; Bunikis, I.; Höijer, I.; Vinnere-Petterson, O.; Suh, A.; Wolf, J.B. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017, 27, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Louzada, S.; Komatsu, J.; Yang, F. Fluorescence in situ hybridization onto DNA fibres generated using molecular combing. In Fluorescence In Situ Hybridization (FISH); Springer: Berlin/Heidelberg, Germany, 2017; pp. 275–293. [Google Scholar]
- Deschamps, S.; Zhang, Y.; Llaca, V.; Ye, L.; Sanyal, A.; King, M.; May, G.; Lin, H. A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Etherington, G.J.; Heavens, D.; Baker, D.; Lister, A.; McNelly, R.; Garcia, G.; Clavijo, B.; Macaulay, I.; Haerty, W.; Di Palma, F. Sequencing smart: De novo sequencing and assembly approaches for a non-model mammal. GigaScience 2020, 9, giaa045. [Google Scholar] [CrossRef]
- Watson, M.; Warr, A. Errors in long-read assemblies can critically affect protein prediction. Nat. Biotechnol. 2019, 37, 124–126. [Google Scholar] [CrossRef] [Green Version]
- Yeo, S.; Coombe, L.; Warren, R.L.; Chu, J.; Birol, I. ARCS: Scaffolding genome drafts with linked reads. Bioinformatics 2018, 34, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long read assembly. Bioinformatics 2020. [CrossRef]
- Deakin, J.E.; Potter, S.; O’Neill, R.; Ruiz-Herrera, A.; Cioffi, M.B.; Eldridge, M.D.; Fukui, K.; Marshall Graves, J.A.; Griffin, D.; Grutzner, F. Chromosomics: Bridging the gap between genomes and chromosomes. Genes 2019, 10, 627. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Li, W.; Song, Q. Chromosome-range whole-genome high-throughput experimental haplotyping by single-chromosome microdissection. In Haplotyping; Springer: Berlin/Heidelberg, Germany, 2017; pp. 161–169. [Google Scholar]
- Seifertova, E.; Zimmerman, L.B.; Gilchrist, M.J.; Macha, J.; Kubickova, S.; Cernohorska, H.; Zarsky, V.; Owens, N.D.; Sesay, A.K.; Tlapakova, T. Efficient high-throughput sequencing of a laser microdissected chromosome arm. BMC Genom. 2013, 14, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traut, W.; Vogel, H.; Glöckner, G.; Hartmann, E.; Heckel, D.G. High-throughput sequencing of a single chromosome: A moth W chromosome. Chromosome Res. 2013, 21, 491–505. [Google Scholar] [CrossRef]
- Makunin, A.I.; Kichigin, I.G.; Larkin, D.M.; O’Brien, P.C.; Ferguson-Smith, M.A.; Yang, F.; Proskuryakova, A.A.; Vorobieva, N.V.; Chernyaeva, E.N.; O’Brien, S.J. Contrasting origin of B chromosomes in two cervids (Siberian roe deer and grey brocket deer) unravelled by chromosome-specific DNA sequencing. BMC Genom. 2016, 17, 618. [Google Scholar] [CrossRef] [Green Version]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Cubero, M.J.; Santiago, O.; Martinez-Labarga, C.; Martinez-Garcia, B.; Marrero-Diaz, R.; Rubio-Roldan, A.; Perez-Gutierrez, A.M.; Carmona-Saez, P.; Lorente, J.A.; Martinez-Gonzalez, L.J. Methodology for Y Chromosome Capture: A complete genome sequence of Y chromosome using flow cytometry, laser microdissection and magnetic streptavidin-beads. Sci. Rep. 2018, 8, 9436. [Google Scholar] [CrossRef]
- Kuderna, L.F.; Lizano, E.; Julià, E.; Gomez-Garrido, J.; Serres-Armero, A.; Kuhlwilm, M.; Alandes, R.A.; Alvarez-Estape, M.; Juan, D.; Simon, H. Selective single molecule sequencing and assembly of a human Y chromosome of African origin. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018, 19, 329. [Google Scholar] [CrossRef]
- Singer, M.F. Highly repeated sequences in mammalian genomes. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 1982; Volume 76, pp. 67–112. [Google Scholar]
- Gall, J.G. The origin of in situ hybridization–a personal history. Methods 2016, 98, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzacher, T.; Heslop-Harrison, P. Practical In Situ Hybridization; BIOS Scientific Publishers Ltd: Milton Park, UK, 2000. [Google Scholar]
- Kim, Y.B.; Oh, J.H.; McIver, L.J.; Rashkovetsky, E.; Michalak, K.; Garner, H.R.; Kang, L.; Nevo, E.; Korol, A.B.; Michalak, P. Divergence of Drosophila melanogaster repeatomes in response to a sharp microclimate contrast in Evolution Canyon, Israel. Proc. Natl. Acad. Sci. USA 2014, 111, 10630–10635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ruano, F.J.; López-León, M.D.; Cabrero, J.; Camacho, J.P.M. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci. Rep. 2016, 6, 28333. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Mari, R.S. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. A satellite DNA isolated from human tissues. J. Mol. Biol. 1967, 23, 619. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. Isolation of the complementary strands of a human satellite DNA. J. Mol. Biol. 1968, 33, 331–335. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. Repeated sequences in human DNA. J. Mol. Biol. 1970, 48, 319–327. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. Renaturation properties and localization in heterochromatin of human satellite DNA’s. Biochim. Et Biophys. Acta (Bba)-Nucleic Acids Protein Synth. 1971, 247, 528–534. [Google Scholar] [CrossRef]
- Mikheenko, A.; Bzikadze, A.V.; Gurevich, A.; Miga, K.H.; Pevzner, P.A. TandemMapper and TandemQUAST: Mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. BioRxiv 2019. [Google Scholar] [CrossRef]
- Mikheenko, A.; Bzikadze, A.V.; Gurevich, A.; Miga, K.H.; Pevzner, P.A. TandemTools: Mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 2020, 36, i75–i83. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Ávila Robledillo, L.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [Green Version]
- Novak, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [Green Version]
- Pech, M.; Igo-Kemenes, T.; Zachau, H.G. Nucleotide sequence of a highly repetitive component of rat DNA. Nucleic Acids Res. 1979, 7, 417–432. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Telomere-to-telomere (T2T) assembly of human chromosomes relied on several different techniques. This combined approach was indispensable for closing the remaining gaps found in early assemblies (mostly related with telomeric, centromeric, and other interstitial regions like segmental duplications). The use of both PacBio and Nanopore sequencing, together with “polishing” methods, was applied as a sequencing strategy. The methodologies presented in the left allowed improve sequence mapping and assembly.
Figure 1.
Telomere-to-telomere (T2T) assembly of human chromosomes relied on several different techniques. This combined approach was indispensable for closing the remaining gaps found in early assemblies (mostly related with telomeric, centromeric, and other interstitial regions like segmental duplications). The use of both PacBio and Nanopore sequencing, together with “polishing” methods, was applied as a sequencing strategy. The methodologies presented in the left allowed improve sequence mapping and assembly.


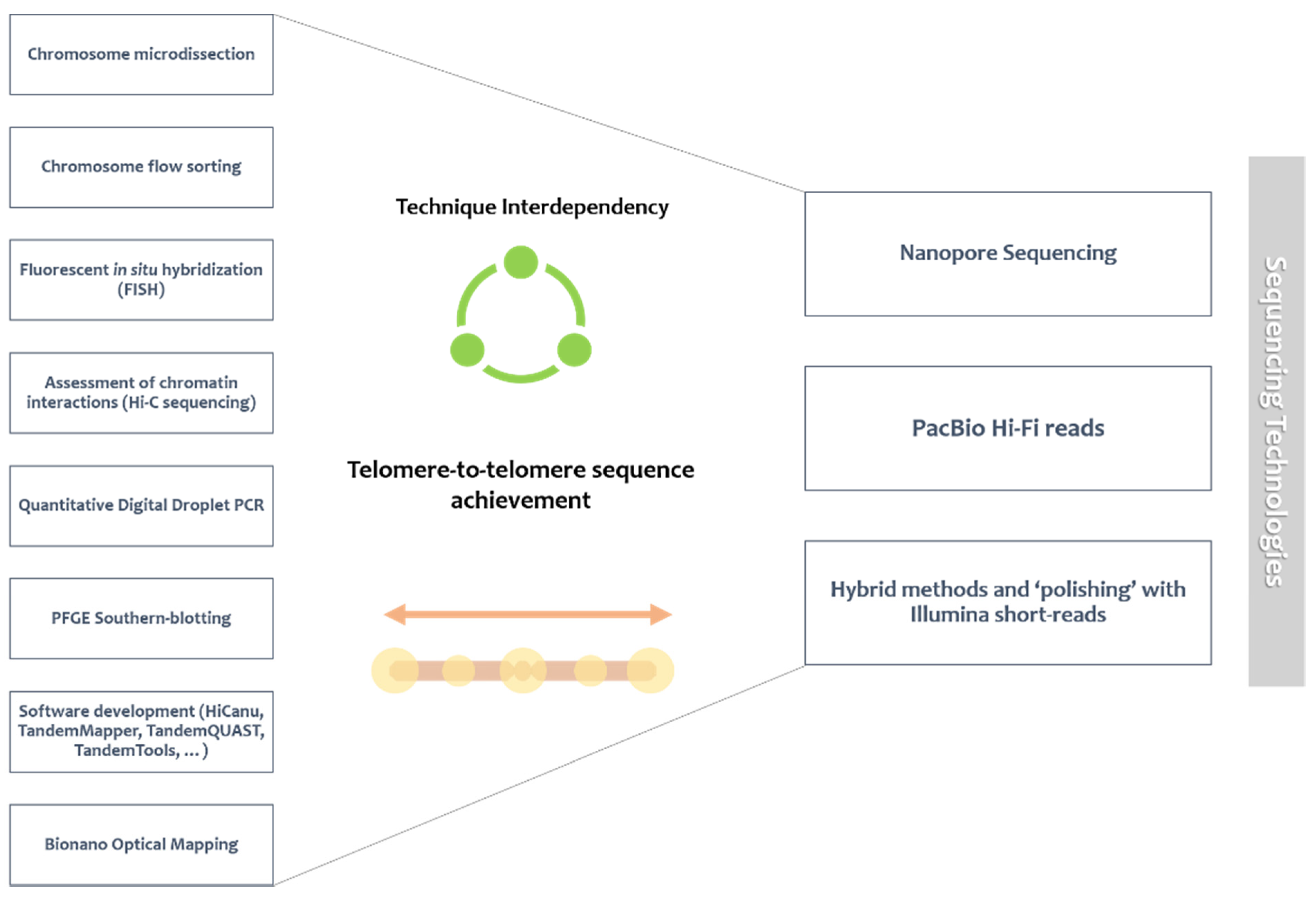{kind=link}
{kind=link}
Table 1.
Summary of currently recognized human satDNA families features. Different satDNA families present distinct traits and can be divided in AT-rich or GC-rich satellites [29,33,41,47,74,87,88,89,94,95,98,99]. 1-SATII presents large blocks on chromosomes 1 and 16. 2-SATIII is widely represented on chromosome 9 [45].
Table 1.
Summary of currently recognized human satDNA families features. Different satDNA families present distinct traits and can be divided in AT-rich or GC-rich satellites [29,33,41,47,74,87,88,89,94,95,98,99]. 1-SATII presents large blocks on chromosomes 1 and 16. 2-SATIII is widely represented on chromosome 9 [45].
| Repeat Unit Size | Identified Subfamilies | HOR Formation | Chromosomal Presence | Genome Representativity | ||
|---|---|---|---|---|---|---|
| αSAT | 171 bp | SFs; 28 identified (e.g., pTRA-1/2/4/7) | ✓ | All | 3–5% | AT-rich |
| SATI | 42 bp | pTRI-6 | ✓ | 3; 4; All acrocentric | 0.12% | |
| SATII | 5 bp | 3 mentioned, no name identified | ✓ | 11; 2; 5; 7; 10; 13–17; 21; 22 | 1.5% (together w/ SATIII) | GC-rich |
| SATIII | 5 bp | pTRS-47; pTRS-63; | ✓ | Y; 1; 3–5; 7; 92; 10; 13–18 ;20–22 | 1.5% (together w/ SATII) | |
| pTR9-s3; pTRS-2; | ||||||
| pE-1/2; pR-1/2/4; | ||||||
| pK-1; pW-1 | ||||||
| βSAT | 68 bp | pB3/4 | ✓ | Y; 1; 3; 9; 19; All acrocentric | 0.02% | |
| γSAT | 220 bp | GSAT; GSATX; GSATII | - | All | 0.13% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lopes, M.; Louzada, S.; Gama-Carvalho, M.; Chaves, R. Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time. Int. J. Mol. Sci. 2021, 22, 4707. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094707
AMA Style
Lopes M, Louzada S, Gama-Carvalho M, Chaves R. Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time. International Journal of Molecular Sciences. 2021; 22(9):4707. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094707
Chicago/Turabian StyleLopes, Mariana, Sandra Louzada, Margarida Gama-Carvalho, and Raquel Chaves. 2021. "Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time" International Journal of Molecular Sciences 22, no. 9: 4707. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094707
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.