
MitoQ Is Able to Modulate Apoptosis and Inflammation

 , ,
, ,  ,
,
Abstract
: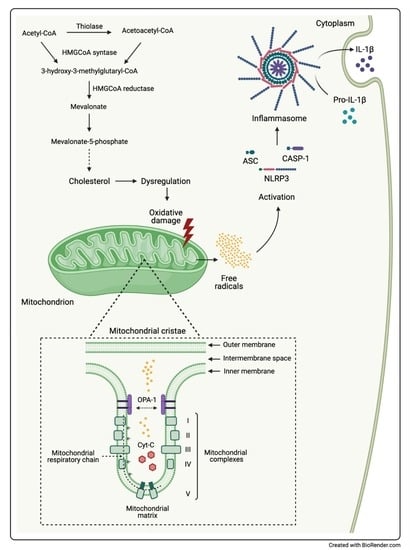
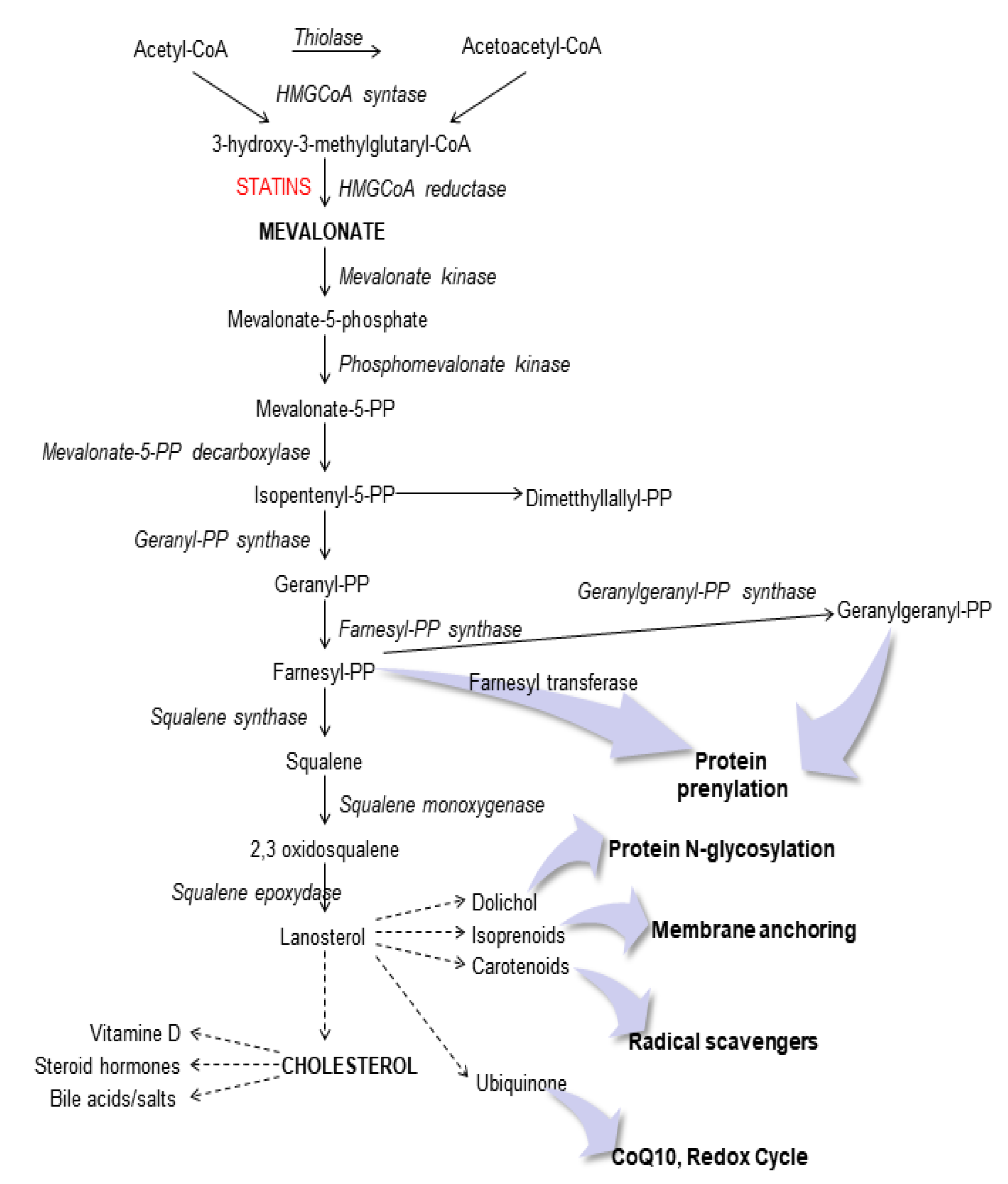
1. Introduction
2. Results
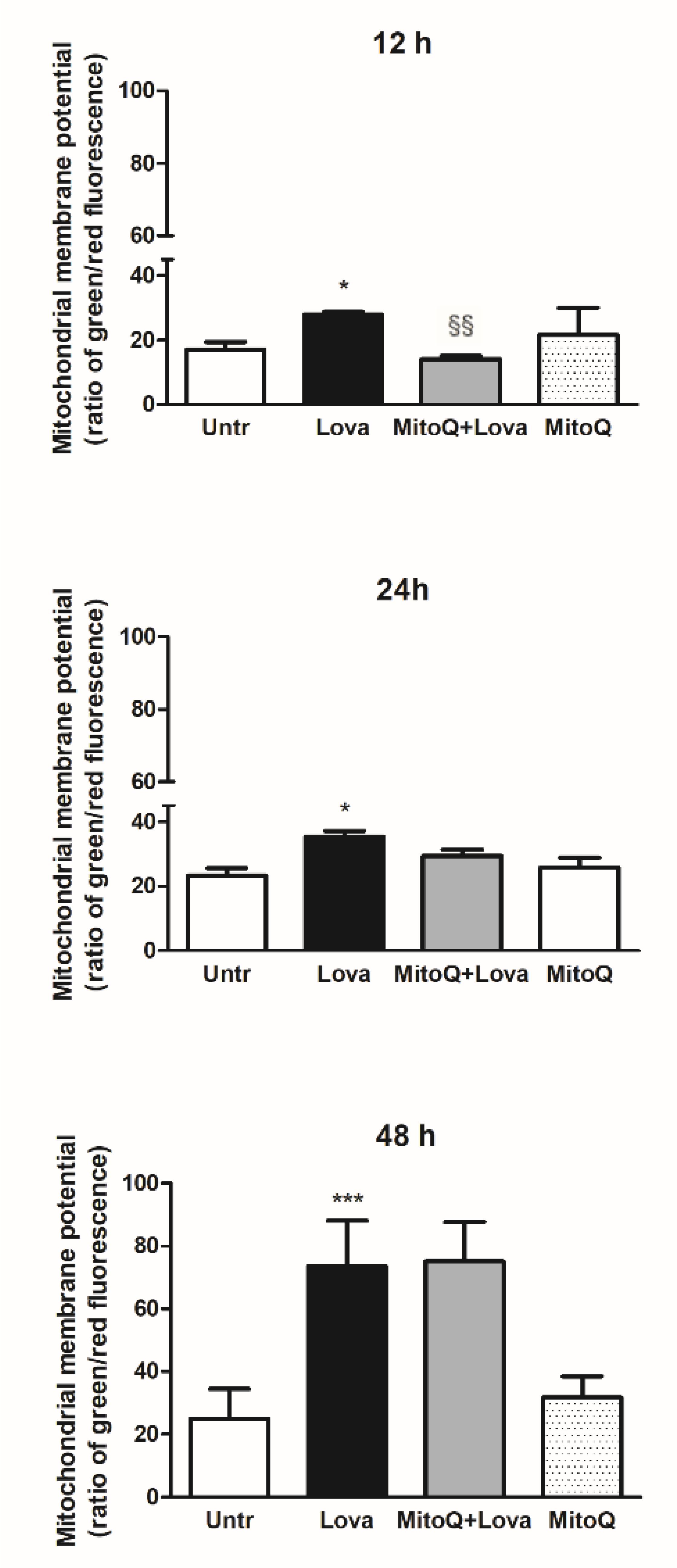
2.1. Effect of MitoQ on Mitochondrial Electrochemical Potential Gradient
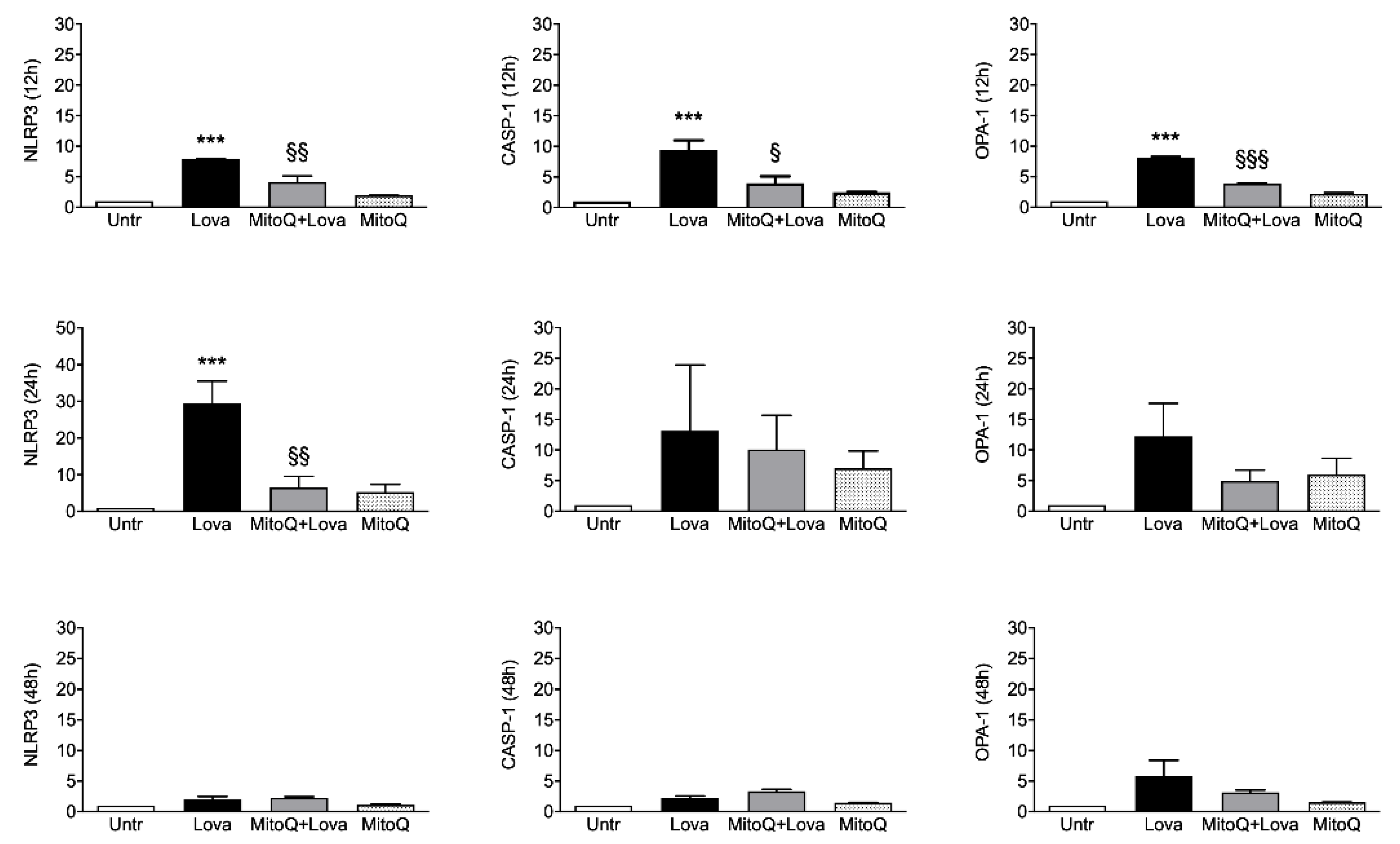
2.2. Inflammatory Genes Expression Related to MitoQ Activity
2.3. Pro-Inflammatory Cytokines Secretion
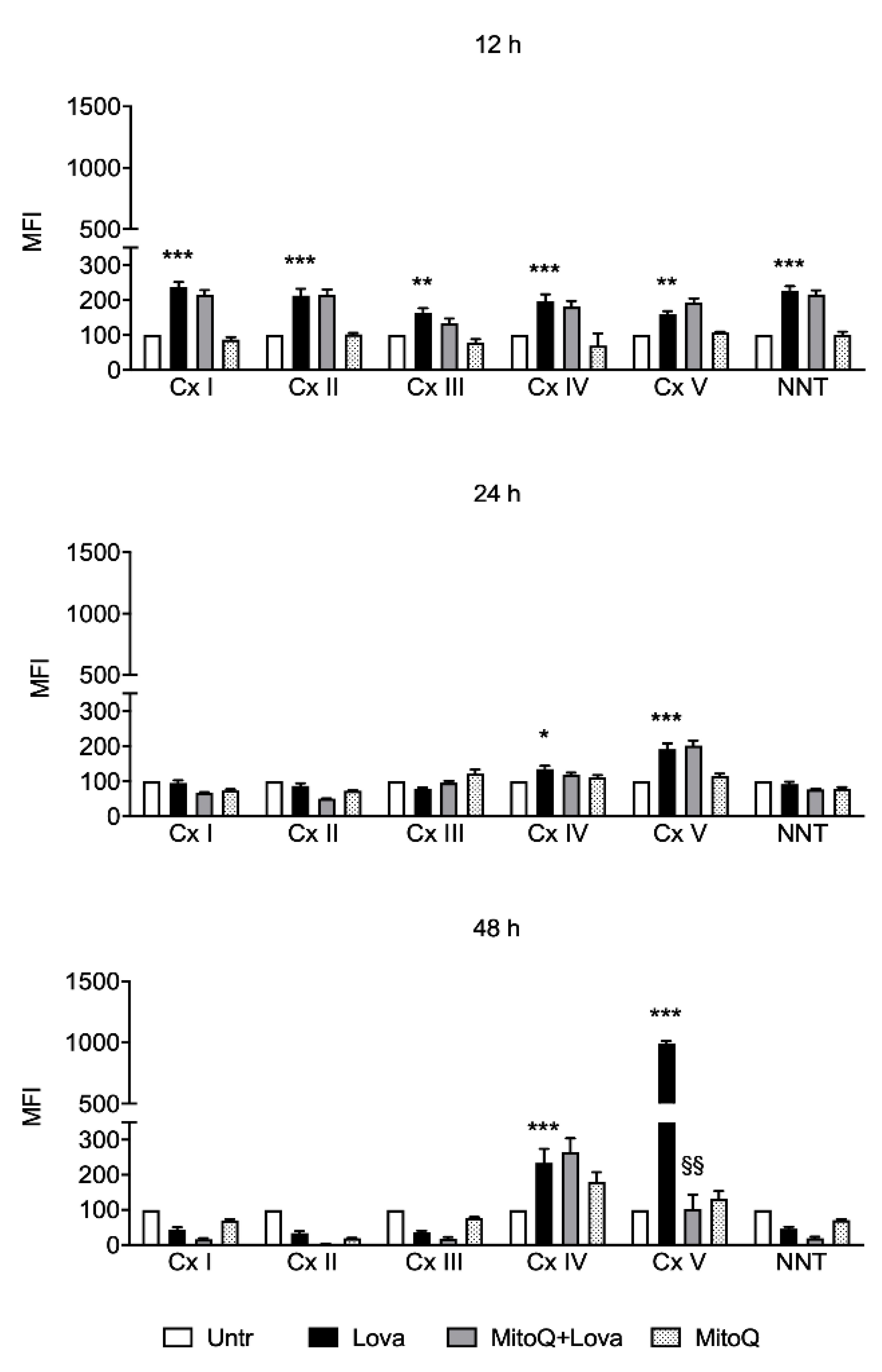
2.4. Influence of MitoQ on Mitochondrial Complexes in DAOY Cell Line Treated with Lovastatin
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Culture
4.2. JC-1 Assay
4.3. RNA Isolation, Reverse Transcription and Real Time-PCR (qPCR)
4.4. Determination of Cytokines Release
4.5. Cellular Metabolism Analysis
4.6. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef]
- Martín, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannsen, D.L.; Ravussin, E. The role of mitochondria in health and disease. Curr. Opin. Pharmacol. 2009, 9, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Williamson, J.; Davison, G. Targeted Antioxidants in Exercise-Induced Mitochondrial Oxidative Stress: Emphasis on DNA Damage. Antioxidants 2020, 9, 1142. [Google Scholar] [CrossRef]
- Battogtokh, G.; Choi, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Huh, K.M.; Cho, Y.-Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880. [Google Scholar] [CrossRef]
- Clinical Trials. Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=&term=mitoq&cntry=&state=&city=&dist= (accessed on 25 April 2021).
- Gliozzi, M.; Musolino, V.; Bosco, F.; Scicchitano, M.; Scarano, F.; Nucera, S.; Zito, M.C.; Ruga, S.; Carresi, C.; Macrì, R.; et al. Cholesterol homeostasis: Researching a dialogue between the brain and peripheral tissues. Pharmacol. Res. 2021, 163, 105215. [Google Scholar] [CrossRef]
- Martin, L.A.; Kennedy, B.E.; Karten, B. Mitochondrial cholesterol: Mechanisms of import and effects on mitochondrial function. J. Bioenerg. Biomembr. 2014, 48, 137–151. [Google Scholar] [CrossRef]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, T.L. Mitochondrial Trafficking in Neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.-H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Lewis, T.L., Jr.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.-K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [Green Version]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; De La Cruz-Ojeda, P.; De La Mata, M.; Cotán, D.; Oropesa-Ávila, M.; De Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, A.; Loganes, C.; Valencic, E.; Piscianz, E.; Monasta, L.; Bilel, S.; Bortul, R.; Celeghini, C.; Zweyer, M.; Tommasini, A. Neuronal Dysfunction Associated with Cholesterol Deregulation. Int. J. Mol. Sci. 2018, 19, 1523. [Google Scholar] [CrossRef] [Green Version]
- Macaulay, R.J.; Wang, W.; Dimitroulakos, J.; Becker, L.E.; Yeger, H. Lovastatin-induced apoptosis of human medulloblastoma cell lines in vitro. J. Neurooncol. 1999, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Macaulay, R.J. Mevalonate Prevents Lovastatin-Induced Apoptosis in Medulloblastoma Cell Lines. Can. J. Neurol. Sci. 1999, 26, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, A.; Tricarico, P.M.; Piscianz, E.; Kleiner, G.; Brumatti, L.V.; Crovella, S. Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis. 2013, 4, e585. [Google Scholar] [CrossRef]
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef] [PubMed]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Jo, E.-K. Crosstalk between Autophagy and Inflammasomes. Mol. Cells 2013, 36, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol 2019, 9, e3128. [Google Scholar] [CrossRef]
- Perelman, A.; Wachtel, C.; Cohen, M.E.; Haupt, S.; Shapiro, H.M.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Björkhem, I.; Leoni, V.; Meaney, S. Genetic connections between neurological disorders and cholesterol metabolism. J. Lipid Res. 2010, 51, 2489–2503. [Google Scholar] [CrossRef] [Green Version]
- Marzban, H.; Kong, J.; Mehr, S.; Vriend, J.; Li, J.; Guan, T.; Chung, S.; Mirzaei, N.; Marzban, A.; Shojaei, S.; et al. Mevalonate Cascade and Neurodevelopmental and Neurodegenerative Diseases: Future Targets for Therapeutic Application. Curr. Mol. Pharmacol. 2017, 10, 115–140. [Google Scholar] [CrossRef]
- Platt, F.M.; Wassif, C.; Colaco, A.; Dardis, A.; Lloyd-Evans, E.; Bembi, B.; Porter, F.D. Disorders of cholesterol metabolism and their unanticipated convergent isms omechanf disease. Annu. Rev. Genom. Hum. Genet. 2014, 15, 173–194. [Google Scholar] [CrossRef]
- Colina-Tenorio, L.; Horten, P.; Pfanner, N.; Rampelt, H. Shaping the mitochondrial inner membrane in health and disease. J. Intern. Med. 2020, 287, 645–664. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-B.; Nagar, H.; Choi, S.; Jung, S.-B.; Kim, H.-W.; Kang, S.K.; Lee, J.W.; Lee, J.H.; Park, J.-W.; Irani, K.; et al. IDH2 deficiency impairs mitochondrial function in endothelial cells and endothelium-dependent vasomotor function. Free Radic. Biol. Med. 2016, 94, 36–46. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Wu, J.; Sun, H. CoenzymeQ10-Induced Activation of AMPK-YAP-OPA1 Pathway Alleviates Atherosclerosis by Improving Mitochondrial Function, Inhibiting Oxidative Stress and Promoting Energy Metabolism. Front. Pharmacol. 2020, 11, 1034. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Innis-Whitehouse, W.; Lopez, A.; Keniry, M.; Gilkerson, R. Oxidative insults disrupt OPA1-mediated mitochondrial dynamics in cultured mammalian cells. Redox Rep. 2018, 23, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landes, T.; Leroy, I.; Bertholet, A.; Diot, A.; Khosrobakhsh, F.; Daloyau, M.; Davezac, N.; Miquel, M.-C.; Courilleau, D.; Guillou, E.; et al. OPA1 (dys)functions. Semin. Cell Dev. Biol. 2010, 21, 593–598. [Google Scholar] [CrossRef]
- Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules 2020, 10, 1437. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Reinecke, F.; Smeitink, J.A.; van der Westhuizen, F.H. XPHOS gene expression and control in mitochondrial disorders. Biochim Biophys. Acta 2009, 1792, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.X.; Thompson, K.; Taylor, R.W.; Oláhová, M. Mitochondrial OXPHOS Biogenesis: Co-Regulation of Protein Synthesis, Import, and Assembly Pathways. Int. J. Mol. Sci. 2020, 21, 3820. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.-C.; Chen, L.-C.; Tsang, N.-M.; Chuang, W.-Y.; Liao, T.-C.; Yuan, S.-N.; OuYang, C.-N.; Ojcius, D.M.; Wu, C.-C.; Chang, Y.-S. Mitochondrial Oxidative Phosphorylation Complex Regulates NLRP3 Inflammasome Activation and Predicts Patient Survival in Nasopharyngeal Carcinoma. Mol. Cell. Proteom. 2020, 19, 142–154. [Google Scholar] [CrossRef]
- He, Z.-F.; Jin, X.-R.; Lin, J.-J.; Zhang, X.; Liu, Y.; Xu, H.-L.; Xie, D.-Y. NALP3 orchestrates cellular bioenergetics to facilitate non-small cell lung cancer cell growth. Life Sci. 2020, 241, 117165. [Google Scholar] [CrossRef]
- Bernardi, S.; Marcuzzi, A.; Piscianz, E.; Tommasini, A.; Fabris, B. The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 4058. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Cacquevel, M.; Lebeurrier, N.; Chéenne, S.; Vivien, D. Cytokines in neuroinflammation and Alzheimer’s disease. Curr. Drug Targets 2004, 5, 529–534. [Google Scholar] [CrossRef]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Bartfai, T.; Schultzberg, M. Cytokines in neuronal cell types. Neurochem. Int. 1993, 22, 435–444. [Google Scholar] [CrossRef]
- Benveniste, E.N. Cytokine circuits in brain. Implications for AIDS dementia complex. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1994, 72, 71–88. [Google Scholar] [PubMed]
- Porrini, A.M.; Reder, A.T. IFN-gamma, IFN-beta, and PGE1 affect monokine secretion: Relevance to monocyte activation in multiple sclerosis. Cell Immunol. 1994, 157, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic, J.; Arsenijevic, A.; Stojanovic, B.; Kanjevac, T.; Arsenijevic, D.; Radosavljevic, G.; Milovanovic, M.; Arsenijevic, N. Interleukin-17 in Chronic Inflammatory Neurological Diseases. Front. Immunol. 2020, 11, 947. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, J.; Chen, L.; Liu, T.; Xu, G.; Li, C.; Yuan, W.; Xu, H.; Su, Z. IL-17 contributed to the neuropathic pain following peripheral nerve injury by promoting astrocyte proliferation and secretion of proinflammatory cytokines. Mol. Med. Rep. 2016, 15, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Yin, C.; Fang, J.; Liu, B. The NLRP3 inflammasome: An emerging therapeutic target for chronic pain. J. Neuroinflammation 2021, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine (pg/mL) | Untr | Lova | MitoQ + Lova | MitoQ | p-Value | |
|---|---|---|---|---|---|---|
| IL-1β | 10.56 ± 1.46 | 14.58 ± 0.88 | 7.75 ± 1.31 | 12.94 ± 1.73 | * | §§§ |
| IL-2 | 41.73 ± 7.07 | 61.84 ± 1.79 | 36.72 ± 1.81 | 48.53 ± 5.03 | * | §§ |
| IL-4 | 15.22 ± 1.48 | 24.02 ± 0.79 | 13.48 ± 2.48 | 22.55 ± 4.28 | * | §§ |
| IL-17 | 31.70 ± 4.44 | 50.96 ± 8.42 | 28.16 ± 7.81 | 40.25 ± 8.94 | * | § |
| IL-6 | 15.65 × 103 ± 24.56 × 102 | 59.17 × 103 ± 73.19 × 102 | 23.34 × 103 ± 15.67 × 103 | 18.00 × 103 ± 86.42 × 102 | ** | § |
| IL-8 | 96.85 × 102 ± 21.37 × 102 | 22.87 × 103 ± 19.54 × 102 | 69.60 × 102 ± 23.29 × 102 | 17.97 × 103 ± 84.44 × 102 | * | § |
| IFN-γ | 72.24 × 101 ± 20.30 × 101 | 11.51 × 102 ± 70.48 | 54.51 × 101 ± 84.15 | 98.12 × 101 ± 16.65 × 101 | * | §§ |
| TNF-α | 25.75 × 101 ± 39.14 | 44.59 × 101 ± 29.76 | 25.33 × 101 ± 60.59 | 36.44 ×101 ± 69.89 | ** | §§ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piscianz, E.; Tesser, A.; Rimondi, E.; Melloni, E.; Celeghini, C.; Marcuzzi, A. MitoQ Is Able to Modulate Apoptosis and Inflammation. Int. J. Mol. Sci. 2021, 22, 4753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094753
Piscianz E, Tesser A, Rimondi E, Melloni E, Celeghini C, Marcuzzi A. MitoQ Is Able to Modulate Apoptosis and Inflammation. International Journal of Molecular Sciences. 2021; 22(9):4753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094753
Chicago/Turabian StylePiscianz, Elisa, Alessandra Tesser, Erika Rimondi, Elisabetta Melloni, Claudio Celeghini, and Annalisa Marcuzzi. 2021. "MitoQ Is Able to Modulate Apoptosis and Inflammation" International Journal of Molecular Sciences 22, no. 9: 4753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094753