
Epigenetic-Based Therapy—A Prospective Chance for Medulloblastoma Patients’ Recovery

 , , , , , and
, , , , , and
Abstract
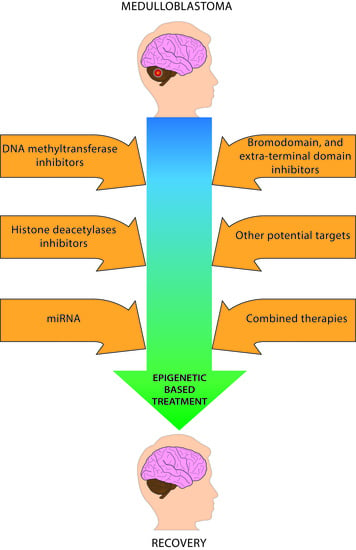
:
1. Introduction
2. Epigenetic Changes in Medulloblastoma
2.1. Histone Modifications
2.2. MicroRNAs (miRNAs, miRs)
2.3. Long Noncoding RNAs (lncRNAs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lncRNA | Expression [Up- (↑) or Down- (↓) Regulation] | Target | Results [Increases (↑) or Decreases (↓)] | MB Subgroup | Ref. | |
|---|---|---|---|---|---|---|
| Direct (Sponging Activity) | Indirect | |||||
| LOXL1-AS1 | ↑ | - | PIK3–AKT pathway | ↑ cell proliferation and metastasis | - | [68] |
| TP73-AS1 | ↑ | miR-494-3p | EIF5A2 | ↑ cell proliferation, migration, invasion, and colony formation; ↓ apoptosis | - | [69,70] |
| HOTAIR | ↑ | miR-1 and miR-206 | YY1 | ↑ tumor growth, migration, invasion, and epithelial-to- mesenchymal transmission; ↓ apoptosis | - | [71] |
| CCAT1 | ↑ | - | MAPK, ERK, MEK | ↑ cell proliferation and metastasis | - | [71] |
| CRNDE | ↑ | miR-29c-3p | - | ↑ tumor growth; ↓ chemosensitivity | - | [72,73] |
| UCA1 | ↑ | - | - | ↑ cell proliferation, migration, invasion, metastasis, and angiogenesis | - | [74,75] |
| SPRY4-IT1 | ↑ | - | MMP-2 | ↑ cell proliferation, migration, and invasion | - | [76] |
| ANRIL | ↑ | miR-323 | p38, MAPK, ERK, AKT, BRI3, CDK6 | ↑ cell proliferation and migration; ↓ apoptosis | - | [77] |
| Linc-NeD125 | ↑ | miR-19a-3p, miR-19b-3p, miR-106a-5p | CDK6, MYCN, SNCAIP, KDM6A | ↑ cell proliferation, migration, and invasion | Group 4 | [78] |
| lnc-HLX-2-7 | ↑ | - | HLX | ↑ cell proliferation, viability, and colony formation; ↓ apoptosis | Group 3 | [79] |
| Nkx2-2as | ↓ | miR-103/107, miR-548m | SHH pathway | tumor development | - | [80] |
2.4. Methylation
3. Epigenetic Therapeutics in Medulloblastoma
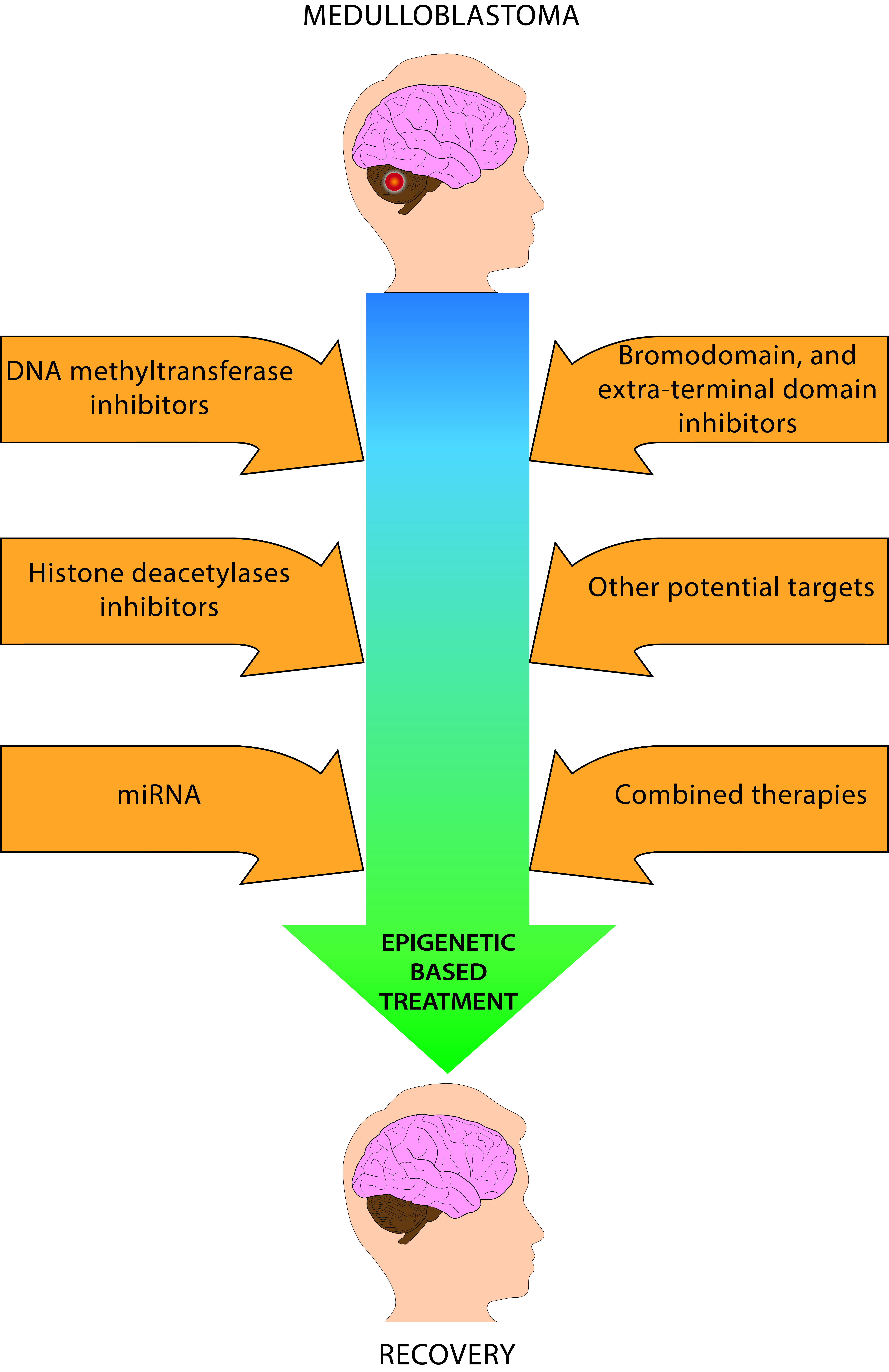
3.1. DNA Methyltransferase Inhibitors (DNMTis)
3.2. Inhibition of miRNA
3.3. Histone Deacetylase Inhibitors (HDACis)
3.3.1. Suberoylanilide Hydroxamic Acid
3.3.2. Panobinostat
3.3.3. Trichostatin A
3.3.4. Valproic Acid
3.3.5. Sodium Butyrate
3.3.6. Quisinostat
3.3.7. Entinostat
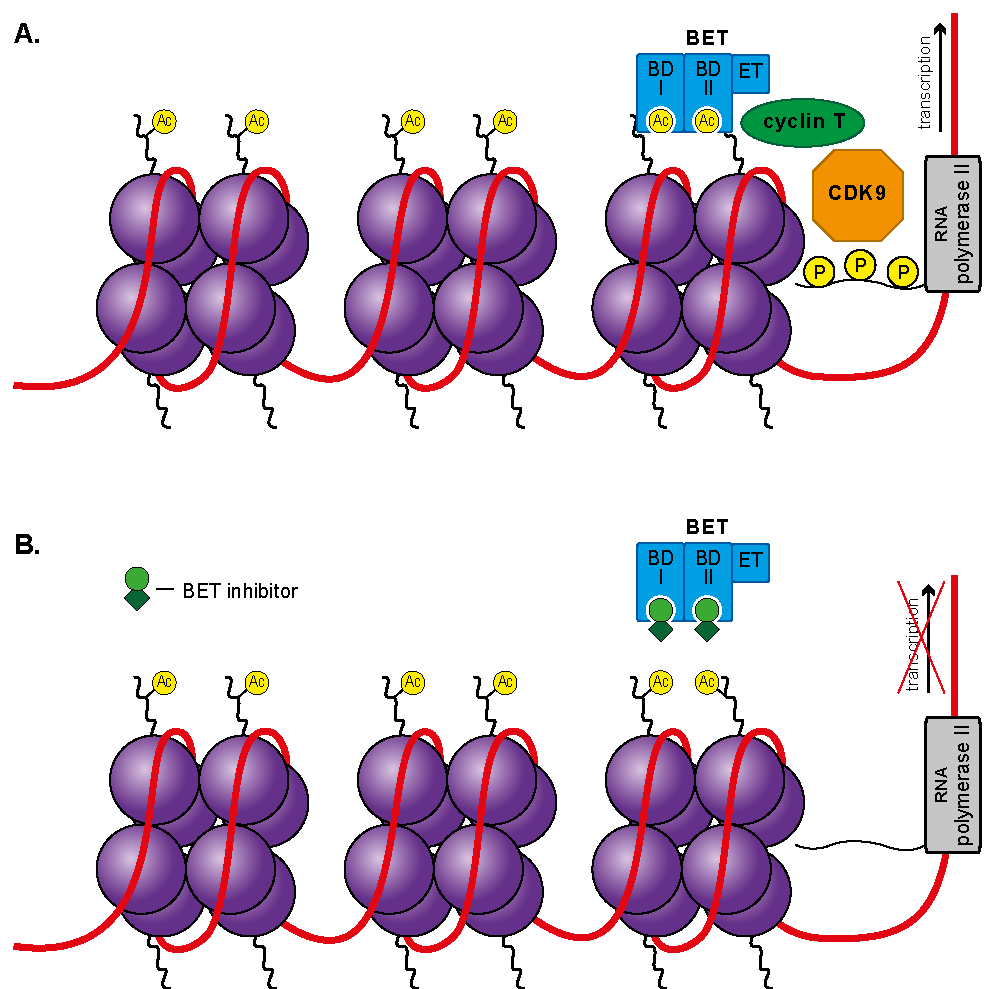
3.4. Bromodomain and Extra-Terminal Domain Inhibitors (BETis)
3.5. lncRNAs
3.6. LSD1 and EZH2 Inhibitors
3.7. Inhibition of MBD2
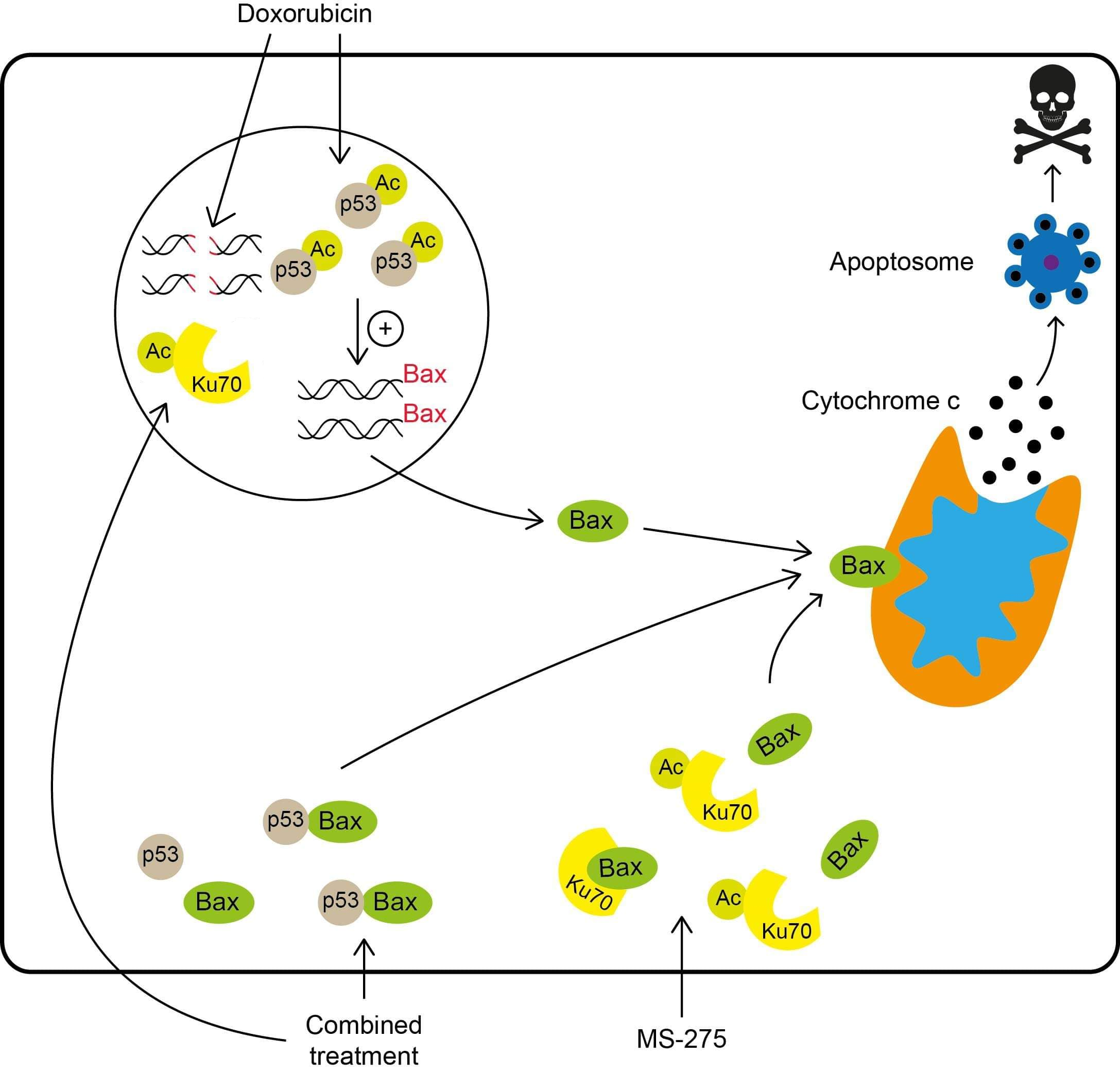
3.8. Combined Therapies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rossi, A.; Caracciolo, V.; Russo, G.; Reiss, K.; Giordano, A. Medulloblastoma: From Molecular Pathology to Therapy. Clin. Cancer Res. 2008, 14, 971–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, F.; Landeiro, J.A.; De Castro, I. Adult hemispheric cerebellar medulloblastoma. Surg. Neurol. Int. 2018, 9, 34. [Google Scholar] [CrossRef]
- McNeil, D.E.; Coté, T.R.; Clegg, L.; Rorke, L.B. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: A SEER update. Med Pediatr. Oncol. 2002, 39, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Smoll, N.R.; Drummond, K.J. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J. Clin. Neurosci. 2012, 19, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Housepian, E.M.; Herbert, C. An Operative Staging System and a Megavoltage Radiotherapeutic Technic for Cerebellar Medulloblastomas. Radiology 1969, 93, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Millard, N.E.; De Braganca, K.C. Medulloblastoma. J. Child Neurol. 2016, 31, 1341–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhang, Y.; Wang, J.; Du, J.; Qiu, X.; Wang, Y.; Li, C. Raynald A Retrospective Study of Progression-Free and Overall Survival in Pediatric Medulloblastoma Based on Molecular Subgroup Classification: A Single-Institution Experience. Front. Neurol. 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Korah, M.P.; Esiashvili, N.; Mazewski, C.M.; Hudgins, R.J.; Tighiouart, M.; Janss, A.J.; Schwaibold, F.P.; Crocker, I.R.; Curran, W.J.; Marcus, R.B. Incidence, Risks, and Sequelae of Posterior Fossa Syndrome in Pediatric Medulloblastoma. Int. J. Radiat. Oncol. 2010, 77, 106–112. [Google Scholar] [CrossRef]
- Gurney, J.G.; Kadan-Lottick, N.S.; Packer, R.J.; Neglia, J.P.; Sklar, C.A.; Punyko, J.A.; Stovall, M.; Yasui, Y.; Nicholson, H.S.; Wolden, S.L.; et al. Endocrine and cardiovascular late effects among adult survivors of childhood brain tumors. Cancer 2003, 97, 663–673. [Google Scholar] [CrossRef]
- Crawford, J.R.; Macdonald, T.J.; Packer, R.J. Medulloblastoma in childhood: New biological advances. Lancet Neurol. 2007, 6, 1073–1085. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Borowska, A.; Jóźwiak, J. Medulloblastoma: Molecular pathways and histopathological classification. Arch. Med Sci. 2016, 12, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, T.; Haberler, C. Update on the integrated histopathological and genetic classification of medulloblastoma—A practical diagnostic guideline. Clin. Neuropathol. 2016, 35, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreault, S.; Ramaswamy, V.; Achrol, A.; Chao, K.; Liu, T.; Shih, D.; Remke, M.; Schubert, S.; Bouffet, E.; Fisher, P.; et al. MRI Surrogates for Molecular Subgroups of Medulloblastoma. Am. J. Neuroradiol. 2014, 35, 1263–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Kumar, V.; McGuire, T.; Coulter, D.W.; Sharp, J.G.; Mahato, R.I. Challenges and Recent Advances in Medulloblastoma Therapy. Trends Pharmacol. Sci. 2017, 38, 1061–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless Pathway Activation and Chromosome 6 Loss Characterise a Distinct Molecular Sub-Group of Medulloblastomas Associated with a Favourable Prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.W.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Meeteren, A.S.-V.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Roussel, M.F.; Stripay, J.L. Epigenetic Drivers in Pediatric Medulloblastoma. Cerebellum 2018, 17, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Dhall, G. Medulloblastoma. J. Child Neurol. 2009, 24, 1418–1430. [Google Scholar] [CrossRef]
- Moosavi, A.; Ardekani, A.M. Role of Epigenetics in Biology and Human Diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.-W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nat. Cell Biol. 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekharan, M.B.; Huang, F.; Sun, Z.-W. Histone H2B ubiquitination and beyond. Epigenetics 2010, 5, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajtler, K.W.; Weingarten, C.; Thor, T.; Künkele, A.; Heukamp, L.C.; Büttner, R.; Suzuki, T.; Miyata, N.; Grotzer, M.; Rieb, A.; et al. The KDM1A histone demethylase is a promising new target for the epigenetic therapy of medulloblastoma. Acta Neuropathol. Commun. 2013, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.-J.; Huang, C.; Meng, X.-M.; Li, J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 2015, 116, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.N. Epigenetic and Chromatin Modifiers As Targeted Therapy of Hematologic Malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef]
- Nebbioso, A.; Carafa, V.; Benedetti, R.; Altucci, L. Trials with ‘epigenetic’ drugs: An update. Mol. Oncol. 2012, 6, 657–682. [Google Scholar] [CrossRef] [Green Version]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone Acetyltransferase PCAF Is Required for Hedgehog–Gli-Dependent Transcription and Cancer Cell Proliferation. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef] [Green Version]
- Vibhakar, R.; Foltz, G.; Yoon, J.-G.; Field, L.; Lee, H.; Ryu, G.-Y.; Pierson, J.; Davidson, B.; Madan, A. Dickkopf-1 is an epigenetically silenced candidate tumor suppressor gene in medulloblastoma1. Neuro-Oncology 2007, 9, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [Green Version]
- Saini, H.K.; Griffiths-Jones, S.; Enright, A.J. Genomic analysis of human microRNA transcripts. Proc. Natl. Acad. Sci. USA 2007, 104, 17719–17724. [Google Scholar] [CrossRef] [Green Version]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [Green Version]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nat. Cell Biol. 2008, 455, 1124–1128. [Google Scholar] [CrossRef]
- Ferretti, E.; De Smaele, E.; Po, A.; Di Marcotullio, L.; Tosi, E.; Espinola, M.S.B.; Di Rocco, C.; Riccardi, R.; Giangaspero, F.; Farcomeni, A.; et al. MicroRNA profiling in human medulloblastoma. Int. J. Cancer 2008, 124, 568–577. [Google Scholar] [CrossRef]
- Jiang, Z.; Cushing, L.; Ai, X.; Lü, J. miR-326 Is Downstream of Sonic Hedgehog Signaling and Regulates the Expression of Gli2 and Smoothened. Am. J. Respir. Cell Mol. Biol. 2014, 51, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Murphy, B.L.; Obad, S.; Bihannic, L.; Ayrault, O.; Zindy, F.; Kauppinen, S.; Roussel, M.F. Silencing of the miR-17∼92 Cluster Family Inhibits Medulloblastoma Progression. Cancer Res. 2013, 73, 7068–7078. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Fernandez-L, A.; Hagan, J.P.; Ellison, D.W.; Grajkowska, W.; Gillespie, Y.; Grundy, R.; Van Meter, T.; Rutka, J.T.; Croce, C.M.; et al. The miR-17/92 Polycistron Is Up-regulated in Sonic Hedgehog–Driven Medulloblastomas and Induced by N-myc in Sonic Hedgehog–Treated Cerebellar Neural Precursors. Cancer Res. 2009, 69, 3249–3255. [Google Scholar] [CrossRef] [Green Version]
- Mollashahi, B.; Aghamaleki, F.S.; Movafagh, A. The Roles of miRNAs in Medulloblastoma: A Systematic Review. J. Cancer Prev. 2019, 24, 79–90. [Google Scholar] [CrossRef]
- Olive, V.; Jiang, I.; He, L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int. J. Biochem. Cell Biol. 2010, 42, 1348–1354. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, G.; Hayashi, K.; Xi, Y.; Kudo, K.; Uchida, K.; Takasaki, K.; Yamamoto, M.; Ju, J. Non-coding MicroRNAs hsa-let-7g and hsa-miR-181b are Associated with Chemoresponse to S-1 in Colon Cancer. Cancer Genom. Proteom. 2006, 3, 317–324. [Google Scholar]
- Garzia, L.; Andolfo, I.; Cusanelli, E.; Marino, N.; Petrosino, G.; De Martino, D.; Esposito, V.; Galeone, A.; Navas, L.; Esposito, S.; et al. MicroRNA-199b-5p Impairs Cancer Stem Cells through Negative Regulation of HES1 in Medulloblastoma. PLoS ONE 2009, 4, e4998. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, A.; Kunder, R.; Goel, A.; Sarin, R.; Moiyadi, A.; Shenoy, A.; Mamidipally, C.; Noronha, S.; Kannan, S.; Shirsat, N.V. Distinctive microRNA signature of medulloblastomas associated with the WNT signaling pathway. J. Cancer Res. Ther. 2010, 6, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gong, Y.H.; Chao, T.F.; Peng, X.Z.; Yuan, J.G.; Ma, Z.Y.; Jia, G.; Zhao, J.Z. Identification of differentially expressed microRNAs by micro-array: A possible role for microRNAs gene in medulloblastomas. Chin. Med. J. 2009, 122, 2405–2411. [Google Scholar] [PubMed]
- Cho, W.C. MicroRNAs: Potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int. J. Biochem. Cell Biol. 2010, 42, 1273–1281. [Google Scholar] [CrossRef]
- Pierson, J.; Hostager, B.; Fan, R.; Vibhakar, R. Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J. Neurooncol. 2008, 90, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Salm, F.; Dimitrova, V.; Von Bueren, A.O.; Ćwiek, P.; Rehrauer, H.; Djonov, V.; Anderle, P.; Arcaro, A. The Phosphoinositide 3-Kinase p110α Isoform Regulates Leukemia Inhibitory Factor Receptor Expression via c-Myc and miR-125b to Promote Cell Proliferation in Medulloblastoma. PLoS ONE 2015, 10, e0123958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.-Q.; Kim, Y.-H.; Giulio, F.; Shalaby, T.; Nobusawa, S.; Yang, H.; Zhou, Z.; Grotzer, M.; Ohgaki, H. Genetic Alterations in MicroRNAs in Medulloblastomas. Brain Pathol. 2011, 22, 230–239. [Google Scholar] [CrossRef]
- Venkataraman, S.; Birks, D.K.; Balakrishnan, I.; Alimova, I.; Harris, P.S.; Patel, P.R.; Handler, M.H.; Dubuc, A.; Taylor, M.D.; Foreman, N.K.; et al. MicroRNA 218 Acts as a Tumor Suppressor by Targeting Multiple Cancer Phenotype-associated Genes in Medulloblastoma. J. Biol. Chem. 2013, 288, 1918–1928. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah, N.; Rajamanickam, S.; Panneerdoss, S.; Timilsina, S.; Yadav, P.; Onyeagucha, B.C.; Garcia, M.; Vadlamudi, R.; Chen, Y.; Brenner, A.; et al. MiR-584-5p potentiates vincristine and radiation response by inducing spindle defects and DNA damage in medulloblastoma. Nat. Commun. 2018, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Weeraratne, S.D.; Amani, V.; Neiss, A.; Teider, N.; Scott, D.K.; Pomeroy, S.L.; Cho, Y.-J. miR-34a confers chemosensitivity through modulation of MAGE-A and p53 in medulloblastoma. Neuro-Oncology 2010, 13, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, S.; Alimova, I.; Fan, R.; Harris, P.; Foreman, N.; Vibhakar, R. MicroRNA 128a Increases Intracellular ROS Level by Targeting Bmi-1 and Inhibits Medulloblastoma Cancer Cell Growth by Promoting Senescence. PLoS ONE 2010, 5, e10748. [Google Scholar] [CrossRef]
- Kondo, Y.; Shinjo, K.; Katsushima, K. Long non-coding RNA s as an epigenetic regulator in human cancers. Cancer Sci. 2017, 108, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Faculty of 1000 evaluation for Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Joshi, P.; Katsushima, K.; Zhou, R.; Meoded, A.; Stapleton, S.; Jallo, G.; Raabe, E.; Eberhart, C.G.; Perera, R.J. The therapeutic and diagnostic potential of regulatory noncoding RNAs in medulloblastoma. Neuro-Oncol. Adv. 2019, 1, vdz023. [Google Scholar] [CrossRef]
- Joshi, P.; Jallo, G.; Perera, R.J. In silico analysis of long non-coding RNAs in medulloblastoma and its subgroups. Neurobiol. Dis. 2020, 141, 104873. [Google Scholar] [CrossRef]
- Song, H.; Han, L.-M.; Gao, Q.; Sun, Y. Long non-coding RNA CRNDE promotes tumor growth in medulloblastoma. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 2588–2597. [Google Scholar]
- Sun, X.-H.; Fan, W.-J.; An, Z.-J.; Sun, Y. Inhibition of Long Noncoding RNA CRNDE Increases Chemosensitivity of Medulloblastoma Cells by Targeting miR-29c-3p. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2020, 28, 95–102. [Google Scholar] [CrossRef]
- Varon, M.; Levy, T.; Mazor, G.; Ben David, H.; Marciano, R.; Krelin, Y.; Prasad, M.; Elkabets, M.; Pauck, D.; Ahmadov, U.; et al. The long noncoding RNA TP73-AS1 promotes tumorigenicity of medulloblastoma cells. Int. J. Cancer 2019, 145, 3402–3413. [Google Scholar] [CrossRef]
- Li, B.; Shen, M.; Yao, H.; Chen, X.; Xiao, Z. Long Noncoding RNA TP73-AS1 Modulates Medulloblastoma Progression In Vitro And In Vivo By Sponging miR-494-3p And Targeting EIF5A2. Onco Targets Ther. 2019, 12, 9873–9885. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, N.; Fu, J.; Zhou, W. Long noncoding RNA HOTAIR promotes medulloblastoma growth, migration and invasion by sponging miR-1/miR-206 and targeting YY1. Biomed. Pharmacother. 2020, 124, 109887. [Google Scholar] [CrossRef]
- Gao, R.; Zhang, R.; Zhang, C.; Zhao, L.; Zhang, Y. Long noncoding RNA CCAT1 promotes cell proliferation and metastasis in human medulloblastoma via MAPK pathway. Tumori J. 2018, 104, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Zhang, R.; Zhang, C.; Liang, Y.; Tang, W. LncRNA LOXL1-AS1 Promotes the Proliferation and Metastasis of Medulloblastoma by Activating the PI3K/AKT Pathway. Anal. Cell. Pathol. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhengyuan, X.; Hu, X.; Qiang, W.; Nanxiang, L.; Junbin, C.; Wangming, Z. Silencing of Urothelial Carcinoma Associated 1 Inhibits the Proliferation and Migration of Medulloblastoma Cells. Med Sci. Monit. 2017, 23, 4454–4461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, N.F.; Manoochehri, H.; Khoei, S.G.; Sheykhhasan, M. The Functional Role of Long Non-coding RNA UCA1 in Human Multiple Cancers: A Review Study. Curr. Mol. Med. 2021, 21, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.-F.; Ji, H.-L.; Luo, Y.-K.; Mao, T.-M.; Chen, X.; Zhou, K.-Y. [Effect of long noncoding RNA SPRY4-IT1 on proliferation and metastasis of medulloblastoma]. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2017, 33, 78–82. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Chen, X. Potential Role of Long Non-Coding RNA ANRIL in Pediatric Medulloblastoma Through Promotion on Proliferation and Migration by Targeting miR-323. J. Cell. Biochem. 2017, 118, 4735–4744. [Google Scholar] [CrossRef]
- Laneve, P.; Po, A.; Favia, A.; Legnini, I.; Alfano, V.; Rea, J.; Di Carlo, V.; Bevilacqua, V.; Miele, E.; Mastronuzzi, A.; et al. The long noncoding RNA linc-NeD125 controls the expression of medulloblastoma driver genes by microRNA sponge activity. Oncotarget 2017, 8, 31003–31015. [Google Scholar] [CrossRef] [Green Version]
- Katsushima, K.; Lee, B.; Kunhiraman, H.; Zhong, C.; Murad, R.; Yin, J.; Liu, B.; Garancher, A.; Gonzalez-Gomez, I.; Monforte, H.L.; et al. The long noncoding RNA lnc-HLX-2-7 is oncogenic in Group 3 medulloblastomas. Neuro-Oncology 2021, 23, 572–585. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, T.; Wang, S.; Xiong, Y.; Zhang, R.; Zhang, X.; Zhao, J.; Yang, A.-G.; Wang, L.; Jia, L. Nkx2-2as Suppression Contributes to the Pathogenesis of Sonic Hedgehog Medulloblastoma. Cancer Res. 2017, 78, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Li, X.; Liang, D.; Li, T.; Zhu, P.; Guo, H.; Wu, X.; Wen, L.; Gu, T.-P.; Hu, B.; et al. Active and Passive Demethylation of Male and Female Pronuclear DNA in the Mammalian Zygote. Cell Stem Cell 2014, 15, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M.; Lacey, M. DNA methylation and differentiation: Silencing, upregulation and modulation of gene expression. Epigenomics 2013, 5, 553–568. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Scourzic, L.; Mouly, E.; Bernard, O.A. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Märtens, K.; Mägi, R.; Kolde, R.; Koltšina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [Green Version]
- Day, K.; Waite, L.L.; Thalacker-Mercer, A.; West, A.; Bamman, M.M.; Brooks, J.D.; Myers, R.M.; Absher, D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013, 14, R102. [Google Scholar] [CrossRef] [Green Version]
- Pócza, T.; Krenács, T.; Turányi, E.; Csáthy, J.; Jakab, Z.; Hauser, P. High expression of DNA methyltransferases in primary human medulloblastoma. Folia Neuropathol. 2016, 2, 105–113. [Google Scholar] [CrossRef] [Green Version]
- De Bont, J.M.; Packer, R.J.; Michiels, E.M.; den Boer, M.L.; Pieters, R. Biological background of pediatric medulloblastoma and ependymoma: A review from a translational research perspective. Neuro-Oncology 2008, 10, 1040–1060. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, J.C.; Lusher, M.E.; Anderton, J.A.; Bailey, S.; Gilbertson, R.J.; Pearson, A.D.; Ellison, D.W.; Clifford, S.C. Identification of tumour-specific epigenetic events in medulloblastoma development by hypermethylation profiling. Carcinogenesis 2003, 25, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2011, 81, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Yang, X.; Lay, F.; Han, H.; Jones, P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 2010, 31, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Seelan, R.S.; Mukhopadhyay, P.; Pisano, M.M.; Greene, R.M. Effects of 5-Aza-2′-deoxycytidine (decitabine) on gene expression. Drug Metab. Rev. 2018, 50, 193–207. [Google Scholar] [CrossRef]
- Mehdipour, P.; Murphy, T.; De Carvalho, D.D. The role of DNA-demethylating agents in cancer therapy. Pharmacol. Ther. 2020, 205, 107416. [Google Scholar] [CrossRef]
- Patties, I.; Kortmann, R.-D.; Glasow, A. Inhibitory effects of epigenetic modulators and differentiation inducers on human medulloblastoma cell lines. J. Exp. Clin. Cancer Res. 2013, 32, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemura, Y.; Satoh, M.; Hatanaka, K.; Kubota, S. Zebularine exerts its antiproliferative activity through S phase delay and cell death in human malignant mesothelioma cells. Biosci. Biotechnol. Biochem. 2018, 82, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Ben-Kasus, T.; Ben-Zvi, Z.; Marquez, V.E.; Kelley, J.A.; Agbaria, R. Metabolic activation of zebularine, a novel DNA methylation inhibitor, in human bladder carcinoma cells. Biochem. Pharmacol. 2005, 70, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Sass, P.; Sosnowski, P.; Podolak-Popinigis, J.; Górnikiewicz, B.; Kamińska, J.; Deptuła, M.; Nowicka, E.; Wardowska, A.; Ruczyński, J.; Rekowski, P.; et al. Epigenetic inhibitor zebularine activates ear pinna wound closure in the mouse. EBioMedicine 2019, 46, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.F.; Borges, K.S.; Suazo, V.K.; Geron, L.; Corrêa, C.A.P.; Castro-Gamero, A.M.; De Vasconcelos, E.J.R.; De Oliveira, R.S.; Neder, L.; Yunes, J.A.; et al. The DNA methyltransferase inhibitor zebularine exerts antitumor effects and reveals BATF2 as a poor prognostic marker for childhood medulloblastoma. Investig. New Drugs 2016, 35, 26–36. [Google Scholar] [CrossRef]
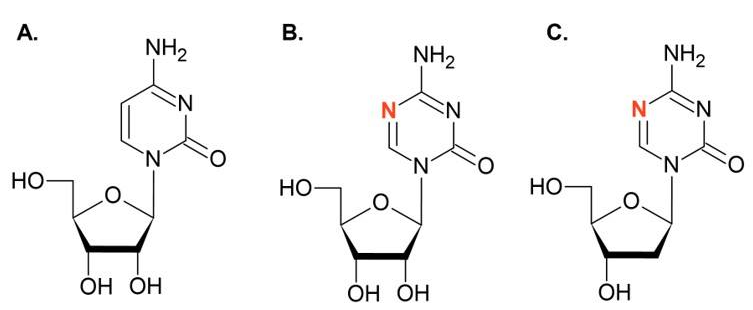
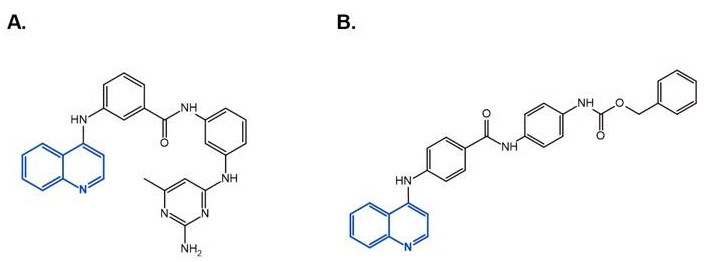
- Valente, S.; Liu, Y.; Schnekenburger, M.; Zwergel, C.; Cosconati, S.; Gros, C.; Tardugno, M.; Labella, D.; Florean, C.; Minden, S.; et al. Selective Non-nucleoside Inhibitors of Human DNA Methyltransferases Active in Cancer Including in Cancer Stem Cells. J. Med. Chem. 2014, 57, 701–713. [Google Scholar] [CrossRef]
- Zwergel, C.; Schnekenburger, M.; Sarno, F.; Battistelli, C.; Manara, M.C.; Stazi, G.; Mazzone, R.; Fioravanti, R.; Gros, C.; Ausseil, F.; et al. Identification of a novel quinoline-based DNA demethylating compound highly potent in cancer cells. Clin. Epigenetics 2019, 11, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Kumar, V.; Chaudhary, A.K.; Coulter, D.W.; McGuire, T.; Mahato, R.I. Impact of miRNA-mRNA Profiling and Their Correlation on Medulloblastoma Tumorigenesis. Mol. Ther. Nucleic Acids 2018, 12, 490–503. [Google Scholar] [CrossRef]
- De Antonellis, P.; Medaglia, C.; Cusanelli, E.; Andolfo, I.; Liguori, L.; De Vita, G.; Carotenuto, M.; Bello, A.; Formiggini, F.; Galeone, A.; et al. MiR-34a Targeting of Notch Ligand Delta-Like 1 Impairs CD15+/CD133+ Tumor-Propagating Cells and Supports Neural Differentiation in Medulloblastoma. PLoS ONE 2011, 6, e24584. [Google Scholar] [CrossRef] [Green Version]
- Silber, J.; Hashizume, R.; Felix, T.; Hariono, S.; Yu, M.; Berger, M.S.; Huse, J.T.; Vandenberg, S.R.; James, C.D.; Hodgson, J.G.; et al. Expression of miR-124 inhibits growth of medulloblastoma cells. Neuro-Oncology 2012, 15, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Burgess, A.; Ruefli, A.; Beamish, H.; Warrener, R.; Saunders, N.; Johnstone, R.; Gabrielli, B. Histone deacetylase inhibitors specifically kill nonproliferating tumour cells. Oncogene 2004, 23, 6693–6701. [Google Scholar] [CrossRef] [Green Version]
- Zwergel, C.; Valente, S.; Jacob, C.; Mai, A. Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin. Drug Discov. 2015, 10, 599–613. [Google Scholar] [CrossRef]
- Marks, P.A. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA). Cell Cycle 2004, 3, 534–535. [Google Scholar] [CrossRef]
- Yuan, J.; Luceño, N.L.; Sander, B.; Golas, M.M. Synergistic anti-cancer effects of epigenetic drugs on medulloblastoma cells. Cell. Oncol. 2017, 40, 263–279. [Google Scholar] [CrossRef]
- Taylor, P.; Fangusaro, J.; Rajaram, V.; Goldman, S.; Helenowski, I.B.; Macdonald, T.; Hasselblatt, M.; Riedemann, L.; Laureano, A.; Cooper, L.; et al. REST Is a Novel Prognostic Factor and Therapeutic Target for Medulloblastoma. Mol. Cancer Ther. 2012, 11, 1713–1723. [Google Scholar] [CrossRef] [Green Version]
- Häcker, S.; Karl, S.; Mader, I.; Cristofanon, S.; Schweitzer, T.; Krauss, J.; Rutkowski, S.; Debatin, K.-M.; Fulda, S. Histone deacetylase inhibitors prime medulloblastoma cells for chemotherapy-induced apoptosis by enhancing p53-dependent Bax activation. Oncogene 2011, 30, 2275–2281. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Liu, K.-W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC- Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewska, M.; Zakrzewski, K.; Grešner, S.M.; Piaskowski, S.; Zalewska-Szewczyk, B.; Liberski, P.P. Polycomb genes expression as a predictor of poor clinical outcome in children with medulloblastoma. Child’s Nerv. Syst. 2010, 27, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, M.D.C.; Ghisleni, E.C.; Cardoso, P.S.; Siniglaglia, M.; Falcon, T.; Brunetto, A.T.; Brunetto, A.L.; De Farias, C.B.; Taylor, M.D.; Nör, C.; et al. HDAC and MAPK/ERK Inhibitors Cooperate To Reduce Viability and Stemness in Medulloblastoma. J. Mol. Neurosci. 2020, 70, 981–992. [Google Scholar] [CrossRef]
- Arts, J.; King, P.; Mariën, A.; Floren, W.; Beliën, A.; Janssen, L.; Pilatte, I.; Roux, B.; DeCrane, L.; Gilissen, R.; et al. JNJ-26481585, a Novel “Second-Generation” Oral Histone Deacetylase Inhibitor, Shows Broad-Spectrum Preclinical Antitumoral Activity. Clin. Cancer Res. 2009, 15, 6841–6851. [Google Scholar] [CrossRef] [Green Version]
- Pak, E.; MacKenzie, E.L.; Zhao, X.; Pazyra-Murphy, M.F.; Park, P.M.C.; Wu, L.; Shaw, D.L.; Addleson, E.C.; Cayer, S.S.; Lopez, B.G.-C.; et al. A large-scale drug screen identifies selective inhibitors of class I HDACs as a potential therapeutic option for SHH medulloblastoma. Neuro-Oncology 2019, 21, 1150–1163. [Google Scholar] [CrossRef]
- Zhang, S.; Gong, Z.; Oladimeji, P.O.; Currier, D.G.; Deng, Q.; Liu, M.; Chen, T.; Li, Y. A high-throughput screening identifies histone deacetylase inhibitors as therapeutic agents against medulloblastoma. Exp. Hematol. Oncol. 2019, 8, 30. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [Green Version]
- Phi, J.H.; Choi, S.A.; Kwak, P.A.; Lee, J.Y.; Wang, K.-C.; Hwang, D.W.; Kim, S.-K. Panobinostat, a histone deacetylase inhibitor, suppresses leptomeningeal seeding in a medulloblastoma animal model. Oncotarget 2017, 8, 56747–56757. [Google Scholar] [CrossRef] [Green Version]
- Hellwig, M.; Merk, D.J.; Lutz, B.; Schüller, U. Preferential sensitivity to HDAC inhibitors in tumors with CREBBP mutation. Cancer Gene Ther. 2020, 27, 294–300. [Google Scholar] [CrossRef]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Coco, F.L.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3–RENKCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Di Marcotullio, L.; Ferretti, E.; De Smaele, E.; Argenti, B.; Mincione, C.; Zazzeroni, F.; Gallo, R.; Masuelli, L.; Napolitano, M.; Maroder, M.; et al. RENKCTD11 is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc. Natl. Acad. Sci. USA 2004, 101, 10833–10838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-N.; Shu, Q.; Su, J.M.-F.; Perlaky, L.; Blaney, S.M.; Lau, C.C. Valproic acid induces growth arrest, apoptosis, and senescence in medulloblastomas by increasing histone hyperacetylation and regulating expression of p21Cip1, CDK4, and CMYC. Mol. Cancer Ther. 2005, 4, 1912–1922. [Google Scholar] [CrossRef] [Green Version]
- Mascaro-Cordeiro, B.; Oliveira, I.D.; Tesser-Gamba, F.; Pavon, L.F.; Saba-Silva, N.; Cavalheiro, S.; Dastoli, P.; Toledo, S.R.C. Valproic acid treatment response in vitro is determined by TP53 status in medulloblastoma. Child’s Nerv. Syst. 2018, 34, 1497–1509. [Google Scholar] [CrossRef]
- Ecke, I.; Petry, F.; Rosenberger, A.; Tauber, S.; Mönkemeyer, S.; Hess, I.; Dullin, C.; Kimmina, S.; Pirngruber, J.; Johnsen, S.A.; et al. Antitumor Effects of a Combined 5-Aza-2′Deoxycytidine and Valproic Acid Treatment on Rhabdomyosarcoma and Medulloblastoma in Ptch Mutant Mice. Cancer Res. 2009, 69, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Jones, D.T.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, D.R.; Seo, K.-W.; Jung, J.-W.; Kim, H.-S.; Yang, S.-R.; Kang, K.-S. The regulatory role of c-MYC on HDAC2 and PcG expression in human multipotent stem cells. J. Cell. Mol. Med. 2011, 15, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Ecker, J.; Oehme, I.; Mazitschek, R.; Korshunov, A.; Kool, M.; Hielscher, T.; Kiss, J.; Selt, F.; Konrad, C.; Lodrini, M.; et al. Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol. Commun. 2015, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, E.; Nicolaides, T. Bromodomain Inhibitor Review: Bromodomain and Extra-terminal Family Protein Inhibitors as a Potential New Therapy in Central Nervous System Tumors. Cureus 2016, 8, e620. [Google Scholar] [CrossRef] [Green Version]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.G.; Thor, T.; Odersky, A.; Heukamp, L.; El-Hindy, N.; Beckers, A.; Slpeleman, F.; Althoff, K.; Schäfers, S.; Schramm, A.; et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013, 4, 2080–2095. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, M.C.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zeng, W.; Gai, X.; Xu, Q.; Li, C.; Liang, Z.; Tuo, H.; Liu, Q. Role of the Hedgehog pathway in hepatocellular carcinoma (Review). Oncol. Rep. 2013, 30, 2020–2026. [Google Scholar] [CrossRef] [Green Version]
- Milla, L.A.; González-Ramírez, C.N.; Palma, V. Sonic Hedgehog in cancer stem cells: A novel link with autophagy. Biol. Res. 2012, 45, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Oliver, T.G.; Grasfeder, L.L.; Carroll, A.L.; Kaiser, C.; Gillingham, C.L.; Lin, S.M.; Wickramasinghe, R.; Scott, M.P.; Wechsler-Reya, R.J. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 7331–7336. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Sewastianik, T.; Prochorec-Sobieszek, M.; Chapuy, B.; Juszczyński, P. MYC deregulation in lymphoid tumors: Molecular mechanisms, clinical consequences and therapeutic implications. Biochim. et Biophys. Acta (BBA) Bioenerg. 2014, 1846, 457–467. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stuetz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nat. Cell Biol. 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Venkataraman, S.; Alimova, I.; Balakrishnan, I.; Harris, P.; Birks, D.K.; Griesinger, A.; Amani, V.; Cristiano, B.; Remke, M.; Taylor, M.D.; et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget 2014, 5, 2355–2371. [Google Scholar] [CrossRef] [Green Version]
- Swartling, F.J. Myc proteins in brain tumor development and maintenance. Upsala J. Med Sci. 2012, 117, 122–131. [Google Scholar] [CrossRef]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Callegari, K.; Maegawa, S.; Bravo-Alegria, J.; Gopalakrishnan, V. Pharmacological inhibition of LSD1 activity blocks REST-dependent medulloblastoma cell migration. Cell Commun. Signal. 2018, 16, 60. [Google Scholar] [CrossRef] [Green Version]
- Inui, K.; Zhao, Z.; Yuan, J.; Jayaprakash, S.; Le, L.T.M.; Drakulic, S.; Sander, B.; Golas, M.M. Stepwise assembly of functional C-terminal REST/NRSF transcriptional repressor complexes as a drug target. Protein Sci. 2017, 26, 997–1011. [Google Scholar] [CrossRef]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stütz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.H.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nat. Cell Biol. 2014, 511, 428–434. [Google Scholar] [CrossRef]
- Lee, C.; Rudneva, V.A.; Erkek, S.; Zapatka, M.; Chau, L.Q.; Tacheva-Grigorova, S.K.; Garancher, A.; Rusert, J.M.; Aksoy, O.; Lea, R.; et al. Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Velinder, M.; Singer, J.; Bareyan, D.; Meznarich, J.; Tracy, C.M.; Fulcher, J.M.; McClellan, D.; Lucente, H.; Franklin, S.; Sharma, S.; et al. GFI1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit LSD1. Biochem. J. 2016, 473, 3355–3369. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Gamo, K.; Yabuki, M.; Takagi, S.; Toyoshima, K.; Nakayama, K.; Nakayama, A.; Morimoto, M.; Miyashita, H.; Dairiki, R.; et al. A Novel LSD1 Inhibitor T-3775440 Disrupts GFI1B-Containing Complex Leading to Transdifferentiation and Impaired Growth of AML Cells. Mol. Cancer Ther. 2016, 16, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-H.; Holder, M.; Grau, D.; Saldaña-Meyer, R.; Yu, J.-R.; Ganai, R.A.; Zhang, J.; Wang, M.; LeRoy, G.; Dobenecker, M.-W.; et al. Distinct Stimulatory Mechanisms Regulate the Catalytic Activity of Polycomb Repressive Complex 2. Mol. Cell 2018, 70, 435–448.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimova, I.; Birks, D.K.; Harris, P.S.; Knipstein, J.A.; Venkataraman, S.; Marquez, V.E.; Foreman, N.K.; Vibhakar, R. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro-Oncology 2012, 15, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026575. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, K.H.K.C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Zhu, D.; Osuka, S.; Zhang, Z.; Reichert, Z.R.; Yang, L.; Kanemura, Y.; Jiang, Y.; You, S.; Zhang, H.; Devi, N.S.; et al. BAI1 Suppresses Medulloblastoma Formation by Protecting p53 from Mdm2-Mediated Degradation. Cancer Cell 2018, 33, 1004–1016.e5. [Google Scholar] [CrossRef] [Green Version]
- Malyutina, A.; Majumder, M.M.; Wang, W.; Pessia, A.; Heckman, C.A.; Tang, J. Drug combination sensitivity scoring facilitates the discovery of synergistic and efficacious drug combinations in cancer. PLoS Comput. Biol. 2019, 15, e1006752. [Google Scholar] [CrossRef] [Green Version]
- Patties, I.; Kortmann, R.-D.; Menzel, F.; Glasow, A. Enhanced inhibition of clonogenic survival of human medulloblastoma cells by multimodal treatment with ionizing irradiation, epigenetic modifiers, and differentiation-inducing drugs. J. Exp. Clin. Cancer Res. 2016, 35, 94. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-J.; Chiang, C.-H.; Song, W.-S.; Tsai, S.-K.; Woung, L.-C.; Chang, C.-H.; Jeng, S.-Y.; Tsai, C.-Y.; Hsu, C.-C.; Lee, H.-F.; et al. Inhibition of phosphorylated STAT3 by cucurbitacin I enhances chemoradiosensitivity in medulloblastoma-derived cancer stem cells. Child’s Nerv. Syst. 2012, 28, 363–373. [Google Scholar] [CrossRef]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.-J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Jiang, W.; Sui, Y.; Meng, W.; Hou, L.; Li, T.; Li, M.; Zhang, L.; Mo, J.; Wang, J.; et al. CDK7 inhibition suppresses aberrant hedgehog pathway and overcomes resistance to smoothened antagonists. Proc. Natl. Acad. Sci. USA 2019, 116, 12986–12995. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Lindner, S.; Bei, Y.; Garcia, H.D.; Timme, N.; Althoff, K.; Odersky, A.; Schramm, A.; Lissat, A.; Künkele, A.; et al. Synergistic activity of BET inhibitor MK-8628 and PLK inhibitor Volasertib in preclinical models of medulloblastoma. Cancer Lett. 2019, 445, 24–33. [Google Scholar] [CrossRef]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.J.; Wee, Z.N.; et al. PDK1 Signaling Toward PLK1–MYC Activation Confers Oncogenic Transformation, Tumor-Initiating Cell Activation, and Resistance to mTOR-Targeted Therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [Green Version]
- Timme, N.; Han, Y.; Liu, S.; Yosief, H.O.; García, H.D.; Bei, Y.; Klironomos, F.; MacArthur, I.C.; Szymansky, A.; von Stebut, J.; et al. Small-Molecule Dual PLK1 and BRD4 Inhibitors are Active Against Preclinical Models of Pediatric Solid Tumors. Transl. Oncol. 2020, 13, 221–232. [Google Scholar] [CrossRef]
- Zwergel, C.; Romanelli, A.; Stazi, G.; Besharat, Z.M.; Catanzaro, G.; Tafani, M.; Valente, S.; Mai, A. Application of Small Epigenetic Modulators in Pediatric Medulloblastoma. Front. Pediatr. 2018, 6, 370. [Google Scholar] [CrossRef] [Green Version]





| Feature | WNT | SHH | Group 3 | Group 4 |
|---|---|---|---|---|
| Occurrence | 10% | 30% | 25% | 35% |
| Age group | Older children | <3 y.o. and >16 y.o. | Infants, young children, and adults | Children of all ages and adults |
| Male:female ratio | 1:1 | 1:1 | 2:1 | 3:1 |
| Location | Cerebellar peduncle, cerebellopontine angle cistern | Cerebellar hemispheres, midline vermis, fourth ventricle | Midline vermis, fourth ventricle | Midline vermis, fourth ventricle |
| Metastasis | Rare (5–10%) | Uncommon (15–20%) | Very frequent (40–45%) | Frequent (35–40%) |
| Prognosis | Low-risk | Low-risk in infants, standard risk in others | High-risk tumor | Standard risk |
| Survival | >90% | 75% | 50% | 75% |
| Histological subgroup | Classic LC/A | Classic LC/A DN MBEN | Classic LC/A | Classic LC/A |
| Carcinogenesis pathway | WNT | SHH | Photoreceptor/ GABA | Neuronal/ glutamatergic |
| Single nucleotide variants | CTNNB1 DDX3X SMARCA4 TP53 | SMO PTCH1 SUFU TERT TP53 | SMARCA4 KBTBD4 KMT2D CTDNEP1 | KDM6A KTM2C ZMYM3 KBTBD4 |
| Gene amplification | NA | GLI1 GLI2 MYCN | OTX2 MYC MYCN | CDK6 OTX2 MYCN SNCAIP |
| miRNA | Upregulation (↑) or Downregulation (↓) | Functional Target | Ref. |
|---|---|---|---|
| miR-17/92 | ↑ | SHH pathway, MYCN/MYC, Gli 1 | [49] |
| Let-7g | RAS, STAT3 | [50] | |
| miR-199-5p | HES 1, Notch pathway, ErbB2 | [51] | |
| miR-214 | SHH pathway, Gli 1 | [52] | |
| miR-100 | BTG2 | [53] | |
| miR-106b | - | [53] | |
| miR-9 | ↓ | REST/NRSF, t-Trk-C | [52] |
| miR-125a | [54] | ||
| miR-124a | CDK6 | [55] | |
| miR-125b | Smo, Gli 1 | [56] | |
| miR-324-5p | [57] | ||
| miR-326 | [45] | ||
| miR-218 | EGFR, Bcl-2 | [58] | |
| miR-584-5p | eIF4E3v, HDAC1 | [59] | |
| miR-34a | TP53 | [60] | |
| miR-128a | BMI1 | [61] |
| Epigenetic Mechanism | Enzyme/ Molecule | Type of Dysregulation | Action | Effect in MB |
|---|---|---|---|---|
| Histone methylation | Histone demethylase KDM1A | Overexpression | Hypo- methylation of H3K4 | Poor prognosis |
| Histone Acetylation | Deacetylases | - | Silences DKK1 | Promotes the WNT signaling pathway |
| miRNA | miR-326 | Downregulation | Promotes mRNA translation | Promotes the SHH signaling pathway |
| DNA methylation | Methyl- transferases | Overexpression | Hyper- methylation of anti-oncogenes promotors | Silences anti-oncogenes |
| Histone Deacetylase Inhibitors | Chemical Structure | Target Histone Deacetylases (HDACs) | Function | Combined Treatment | Ref. |
|---|---|---|---|---|---|
| Suberoylanilide hydroxamic acid (SAHA, Vorinostat) |  | Classes I and II | Activation of the p21WAF1, decline of REST, REST-dependent repression of cell growth | Chemotherapeutics (Doxorubicin, Etoposide, and Cisplatin), → caspase-dependent apoptosis | [109,110,111,112] |
| Panobinostat (LBH-589) |  | Pan-HDACi | Increase of the FOXO1 target genes, down-regulation of ID3 | PI3Ks inhibitors, → inhibition of growth of MYC-driven MB | [113,114] |
| Trichostatin A (TSA) |  | Classes I and II | Upregulation of DKK1, acetylation of Gli1 and Gli2 | Cul3–REN E3 ubiquitin ligase complex, → suppression of MB growth | [38,115,116] |
| Valproic acid (VPA) |  | Classes I and II | Activation of p21; suppression of TP53, CDK4, and c-MYC expression | DAC → prevention of Ptch-associated tumor formation | [117,118] |
| Sodium butyrate (NaB) |  | Classes I and IIa | Downregulation of CD133, BMI1, and ERK activity | (MAPK)/ERK inhibition, → antiproliferative effect | [113,114,115] |
| Quisinostat (JNJ-26481585) |  | Pan-HDACi | Down-regulation of the SHH target genes, especially Gli1; induction of caspase-3 and PARP cleavage | - | [116,117,118] |
| Entinostat (MS-275) |  | Class I | Acetylation of the Ku70 protein | Chemotherapeutics (Doxorubicin, Etoposide, and Cisplatin), → caspase-dependent apoptosis | [112] |
| Drug Combinations | Results | Ref. |
|---|---|---|
| DAC + VPA | Reduction of tumor cell viability in Daoy and D283med lines | [97,110] |
| DAC + SAHA | Induction of apoptosis in Daoy and D283med cells | [110] |
| DAC + parthenolide | ||
| DAC + abacavir + irradiation | Reduction of tumor cells in Daoy, MEB-Med8a, and D283med cell lines | [164] |
| DAC + 4-phenylbutyrate + imatinib | Induction of apoptosis in Daoy and UW228 in group 3 MB cells | [159] |
| JQ1 + Milciclib or Palbociclib | Induction of apoptosis and cell-cycle arrest in MYC, MYCN-derived, and Daoy cell lines | [160] |
| JQ1 + THZ1 | Reduction of Gli1 and Gli2 transcription and proliferation with increased apoptosis in SMB21, SMB21-shSufu, SmoD477G-MB, A673, and ATRT-03 cell lines | [161] |
| MK-8628 + CT7001 | Reduction of Gli expression and viability | [162] |
| MK-8628 + Volasertib or GSK461364A | Reduction of proliferation, tumor cell viability, and expansion, as well as the induction of apoptosis in HD-MB03, Daoy, Uw228, and ONS-76 cell lines | [163] |
| UMB103 or UMB160 * | Reduction of MYC and MYCN transcription, proliferation, and viability with increased apoptosis in HD-MB03 cells | [163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strejczek, A.; Woszczyk, D.; Urbaniak, H.; Różańska, M.; Robak, M.; Matuszewska, Z.; Barciszewska, A.-M. Epigenetic-Based Therapy—A Prospective Chance for Medulloblastoma Patients’ Recovery. Int. J. Mol. Sci. 2021, 22, 4925. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094925
Strejczek A, Woszczyk D, Urbaniak H, Różańska M, Robak M, Matuszewska Z, Barciszewska A-M. Epigenetic-Based Therapy—A Prospective Chance for Medulloblastoma Patients’ Recovery. International Journal of Molecular Sciences. 2021; 22(9):4925. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094925
Chicago/Turabian StyleStrejczek, Agata, Dawid Woszczyk, Helena Urbaniak, Martyna Różańska, Michał Robak, Zofia Matuszewska, and Anna-Maria Barciszewska. 2021. "Epigenetic-Based Therapy—A Prospective Chance for Medulloblastoma Patients’ Recovery" International Journal of Molecular Sciences 22, no. 9: 4925. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094925