
Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer


Centre de Recherche en Cancérologie de Marseille (CRCM), INSERM U1068, CNRS UMR 7258, Aix-Marseille Université and Institut Paoli-Calmettes, Parc Scientifique et Technologique de Luminy, 163 Avenue de Luminy, 13288 Marseille, France
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(9), 4970; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094970
Submission received: 8 April 2021
/
Revised: 4 May 2021
/
Accepted: 5 May 2021
/
Published: 7 May 2021
(This article belongs to the Special Issue Pancreatic Fibrosis in Inflammation and Cancer)
{kind=link}
Abstract
:Pancreatic fibrosis is caused by the excessive deposits of extracellular matrix (ECM) and collagen fibers during repeated necrosis to repair damaged pancreatic tissue. Pancreatic fibrosis is frequently present in chronic pancreatitis (CP) and pancreatic cancer (PC). Clinically, pancreatic fibrosis is a pathological feature of pancreatitis and pancreatic cancer. However, many new studies have found that pancreatic fibrosis is involved in the transformation from pancreatitis to pancreatic cancer. Thus, the role of fibrosis in the crosstalk between pancreatitis and pancreatic cancer is critical and still elusive; therefore, it deserves more attention. Here, we review the development of pancreatic fibrosis in inflammation and cancer, and we discuss the therapeutic strategies for alleviating pancreatic fibrosis. We further propose that cellular stress response might be a key driver that links fibrosis to cancer initiation and progression. Therefore, targeting stress proteins, such as nuclear protein 1 (NUPR1), could be an interesting strategy for pancreatic fibrosis and PC treatment.
1. Introduction
Pancreatitis is triggered by a variety of factors including the activation of pancreatic enzymes, resulting in pancreatic tissue self-digestion accompanied by pancreatic tissue edema, bleeding, and inflammatory necrosis [1,2,3]. Patients with pancreatitis suffer fever, nausea, vomiting, abdominal distension, abdominal pain, and other symptoms [4]. Pancreatitis can be divided into two types: acute pancreatitis (AP) and CP. AP is an acute pancreatic disease with strong pain, which has greater occurrence in middle-aged adults [5,6,7]. On the contrary, most CP patients develop mild disease, and the symptoms are usually not noticeable [8]. In the course of CP, patients often suffer pain and exocrine and endocrine insufficiency [9]. Most patients with AP recover completely after receiving the right treatment. Adjusting one’s diet and stopping smoking and alcohol ingestion can completely restore pancreatic homeostasis [10]. However, without the correct treatment, AP may deteriorate into CP, and the pathological changes caused by CP are often irreversible [11].
PC is a kind of gastrointestinal tumor with high malignancy, which is difficult to diagnose and treat [12]. Pancreatic ductal adenocarcinoma (PDAC) represents 90% of PC, with a poor prognosis (the 5-year survival rate of PDAC is less than 10%) [13]. Most PDAC patients have metastases after diagnosis, which cannot be treated by surgery [14]. In addition, the etiology of PDAC is complex, and both genetic and environmental factors are involved in the disease progression. Specific mutations in genes, such as tumor protein P53 (TP53), V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS), cyclin-dependent kinase inhibitor 2A (CDKN2A), or SMAD family member 4 (SMAD4) increase the risk of developing PDAC [15]. Other significant risk factors for PDAC development that have been associated with this disease are smoking, diabetes, alcoholism, and obesity [16].
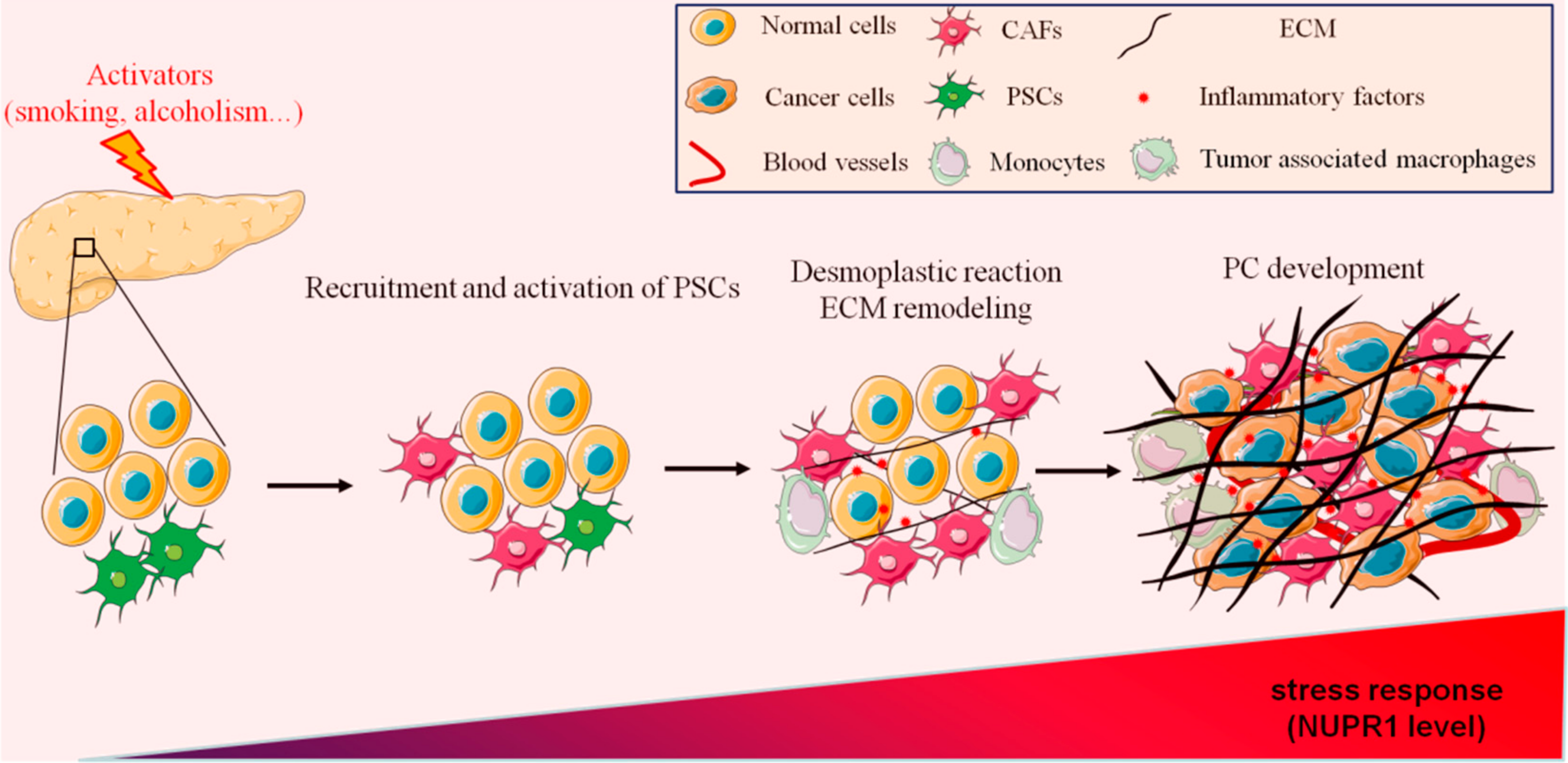
Pancreatitis and PC are two diseases of varying degrees in the pancreas, with similar symptoms. It is frequently necessary to exclude PC in the diagnosis of pancreatitis. From an imaging perspective, pancreatitis and PC are easily confused on magnetic resonance imaging (MRI) or computerized tomography (CT), as only specific imaging features allow one to discriminate between these diseases in the differential diagnosis [17]. Therefore, the screening for PC should combine biochemical tests and pathological diagnosis of tumor markers, such as carbohydrate antigen 199 (CA199), cancer antigen 125 (CA125), carcinoembryonic antigen (CEA), and carbohydrate antigen 50 (CA50) [18]. Most PC patients have a history of CP, which shows that there is a high correlation between PC and CP [19]. Although PC has high genetic factors, pancreatitis and PC have a variety of common pathogenic factors, such as long-term smoking, alcohol abuse, or high protein and high-fat diets [20,21]. In recent years, different works have demonstrated that the processes of wound healing and tumor fibrosis have strong similarities. Interestingly, pancreatic fibrosis is also one of the main pathological features of CP, suggesting a strong relationship between PC and CP [22]. However, whether CP increases the risk of PC and promotes the occurrence and development of PC through tissue fibrosis remains an open question. Thus, in this work, we discuss the role of pancreatic fibrosis in pancreatitis and PC development as well as the recent findings targeting this process (Figure 1).
2. The Role of Fibrosis in Pancreatitis
CP is a pancreatic fibrotic syndrome associated with genetic, environmental, and/or other risk factors [23]. Clinically, CP patients have recurrent abdominal pain, nausea, dyspepsia symptoms, and different complications including fat-soluble vitamin deficiency, exocrine dysfunction, metabolic bone disease, and diabetes [24]. The pathological features of CP include pancreatic fibrosis, acinar injury, pancreatic calcification, and exocrine and endocrine dysfunction [25]. CP is a persistent pathological response to substantial injury or stress. Irreversible fibrosis is one of the most typical pathological features of CP and deeply affects the physiological function of the pancreas [26]. Thus far, there is no clinical treatment that can reverse the inflammatory damage associated with CP. Therefore, CP treatment is focused on relieving the symptoms and screening and treating disease-related complications [27].
2.1. Pancreatic Fibrosis Promotes Inflammation
Numerous studies have shown that pancreatic fibrosis not only is a feature of pancreatitis disease but also has an active role in CP development [28,29,30]. For instance, alcohol consumption triggers pathological changes in the pancreas, leading to pancreatic fibrosis, causing alcoholic CP [31]. Interestingly, increased trypsinogen content in the pancreas is a pivotal event in the initiation of alcoholic CP, although the mechanism remains elusive [32,33]. Serine protease inhibitor Kazal type 1 (SPINK1) acts as a trypsin inhibitor, and its mutation dramatically increases the risk of alcoholic CP [34]. Recent evidence shows that SPINK1-associated pancreatitis or alcohol-induced CP can be characterized by progressive parenchymal fibrosis [35,36]. In addition, a variety of immune cell types, such as monocytes and macrophages, have been detected in the dense fibrotic areas in pancreatic cancer. Indeed, as part of the innate immune response, these immune cells can be recruited by inflammatory signals [37,38,39,40]. However, previous studies have shown that monocytes are recruited into damaged tissues, subsequently differentiating into macrophages, stimulating the synthesis of collagen and fibronectin (FN), and participating in the process of pancreatic fibrosis [41]. Furthermore, macrophages interact with neighboring cells in a cytokine-dependent manner to accelerate the formation of pancreatic fibrosis during pancreatitis [42,43]. Recent studies have shown that the oral administration of camostat mesilate (CM), a drug for CP treatment, reduces pancreatic fibrosis and subsequent inflammation by inhibiting the activity of monocytes [44]. In sum, macrophages, as the major infiltrating immune cells in the tumor microenvironment (TME), play an important role in pancreatic fibrosis and inflammation.
2.2. Pancreatic Stellate Cells (PSCs) Are Key Mediators of Fibrosis in Pancreatitis
Based on the close relationship between pancreatitis and fibrosis, some studies have shown that pro-inflammatory cytokines induce PSCs activation [45]. The activated PSCs further secrete more inflammatory factors, such as monocyte chemotactic protein 1 (MCP-1), which regulates fibrosis through its cognate CC chemokine receptor 2 (CCR2) [46]. Therefore, PSCs activation is considered to be the core event in the development of pancreatic fibrosis, suggesting that targeting PSCs is a potential strategy for CP therapy. Hydrogen peroxide-inducible clone-5 (Hic-5) is a member of the paxillin family, which acts as a molecular scaffold, and its expression leads to a poor prognosis in PC patients [47]. In caerulein-induced CP, the expression of Hic-5 was strongly up-regulated in activated PSCs in the fibrotic tissue [48]. As such, decreasing the expression of Hic-5 significantly attenuated pancreatic fibrosis and PSCs activation in experimental CP mice. Therefore, Hic-5 is an important therapeutic target to reduce pancreatic fibrosis and delay CP [48]. Vitamin deficiency is usually present in patients with pancreatitis and PC and may result from pancreatic insufficiency [49,50]. Interestingly, dietary interventions, such as long-term consumption of vitamin-rich vegetables and fruits, also slow down the CP caused by pancreatic fibrosis [27]. Vitamins C and E act as classical antioxidants, and both have shown a potent anti-fibrotic and anti-inflammatory action by preventing oxidative damage in several organs, including the pancreas [51,52,53,54]. In some studies, vitamins A, D, and K also exert a protective role in the inflammatory response, probably through their antioxidant properties, implying the importance of oxidative stress in inflammation [55,56,57]. Fibrosis is usually considered irreversible in CP, but some studies have shown that pancreatic fibrosis can be reversed in the early stage [20,26,58,59]. However, fibrosis prevention is an effective strategy to reduce CP either by drug treatment or dietary adjustments [60].
3. The Role of Fibrosis in PC
Currently, there is increasing evidence that PC is a chronic inflammatory disease, as with fibrosis being one of the main pathological characteristics [61]. It is well known that there is a close relationship between pancreatic fibrosis and PC [62]. Many pro-fibrotic cells and cancer-associated fibroblasts (CAFs) are abundantly present in PDAC [63]. Most studies have shown that pancreatic fibrosis level is closely related to the survival rate of the patients after chemotherapy [64,65]. Thus, having a quantitative and reproducible method, evaluating fibrosis might be more accurate than either pathologic regression grade or response evaluation criteria in solid tumors (RECIST) score [66].
3.1. Pancreatic Fibrosis Promotes PC Progression
Pancreatic fibrosis is a defining hallmark of PDAC occurrence and prognosis. Interestingly, a set of genes were recently reported to be involved in the development of pancreatic fibrosis and PC. For example, C-X-C motif chemokine receptor 2 (CXCR2), functionally expressed in leukocytes, such as neutrophils, natural killer cells (NK cells), monocytes, macrophages, and T cells, regulates the migration of neutrophils to inflammatory sites by binding to Interleukin-8 (IL-8) [67,68]. CXCR2 knockout mice showed higher levels of pancreatic fibrosis and increased the malignancy of PDAC in vivo, indicating that CXCR2 played an important role in the transition from pancreatic fibrosis to PC [69]. Besides chemokine receptors, some metabolic enzymes are also involved in regulating pancreatic fibrosis. Long-chain acyl coenzyme A synthase 3 (ACSL3) is a lipid metabolizing enzyme, which is up-regulated in PC and related to the increased fibrosis. Interestingly, Sebastiano and colleagues demonstrated that ACSL3 knockout prevents pancreatic fibrosis and delays the PDAC development in mice [70]. A disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) also correlates with the occurrence and development of PC [71]. Both gene-targeting and drug-targeting ADAM10 reduced radiotherapy-induced pancreatic fibrosis and tissue tension, decreasing the migration and invasion of tumor cells, increasing the tumor sensitivity after radiation, and ultimately prolonging the survival of mice [72]. Furthermore, a recent study confirmed that pancreatic fibrosis reduces the lethality and immunity of immune cells to pancreatic tumor cells, thus promoting PDAC progression [73]. Altogether, pancreatic fibrosis is not only a marker in the formation, development, and prognosis of PC, but also has active participation in cancer disease.
3.2. CAFs Contributes to Drug Resistance
Currently, treatment of PC is a big challenge; thus, patients face a poor prognosis, and drug resistance is a major problem in PC therapy [74]. The tumor tissues in PC are composed of a small proportion of cancer cells. In fact, there is an extensive amount of proliferative matrix surrounding the cancer cells, that represents up to 90% of the tumor mass [75]. These proliferative matrices include ECM, CAFs, endothelial cells, and invasive immune cells [76]. These abnormally rich matrices act as a tight blockade to prevent chemotherapeutic drugs from penetrating into the tumor and playing their anti-cancer role, which is one of the important factors that endow cancer cells with chemotherapeutic resistance. Among them, CAFs are the most critical part of TME regulation.
3.2.1. Therapeutic Targeting of the Crosstalk between CAFs and PC
FN assembled by CAFs is an ECM integrin-binding protein. FN promotes fiber formation, provides a track for the migration of cancer cells, and mediates the directional migration of cancer cells [77,78]. Moreover, many signaling molecules produced by CAFs directly participate in regulating nearby cancer cells, thereby stimulating proliferation, invasion, and chemical resistance, which promotes PC malignancy [79]. The consumption of matrix in PDAC blocks some signaling pathways, so it significantly improves the effect of chemotherapy [80]. In vivo studies have shown that the Hedgehog receptor Smoothened (SMO) overexpression in CAFs is an important mechanism of Hedgehog (Hh) signal transduction in pancreatic stromal cells, and the Hh signaling pathway has a close interaction with the tumor matrix [81]. N-myc downstream-regulated gene 1 (NDRG1) is considered to be a potential anticancer gene, and its expression correlates with the differentiation of tumors [82]. Recent studies showed that targeting NDRG1 blocks the crosstalk between PC cells and matrix [83]. The TGF-β/Smad4 signaling axis plays an important role in regulating the TME and mediating tumor-stroma crosstalk [84]. The Met/HGF pathway not only involves the interaction between cancer cells and activated PSCs but also takes part in the crosstalk between tumor and matrix [85]. The complex NT-S100A8/TGF-β1 is also involved in the crosstalk between PDAC and stromal cells in some specific PDAC cell lines [86]. In addition, many studies have confirmed that microRNAs (MIRs) related to epigenetic regulation is a key factor in the formation of the TME, because MIRs are involved in the transformation of normal fibroblasts (NFs) to CAFs, and MIRs released from CAFs affect various features of cancer cells such as tumor migration, tumor invasion, metastasis, and drug resistance [87,88]. Pasireotide (Som230) is a novel multireceptor-targeted somatostatin analog, which inhibits the secretion of symbiotic sulfate transporter 1 (SST1) in CAFs, thus eliminating the interaction between CAFs and PDAC [89]. Insulin/IGF-1R signal is also involved in the crosstalk between cancer cells and matrix, and research on compounds targeting Insulin/IGF signal in the treatment of PDAC entered into clinical trials in phase II [90].
3.2.2. Targeting CAFs in Combination with Chemotherapy, a Field to Explore
Besides the heterogeneity of PDAC itself, the complex matrix crosstalk of tumor cells in the TME also endows cancer cells with resistance to anticancer drugs, which makes the current targeted therapy for some oncogenes weak [91]. Therefore, depletion of dense matrix or destruction of its crosstalk with tumor tissue overcomes the resistance of cancer cells to chemotherapy agents and enhances the anti-cancer effect.
For instance, IL-1β/IRAK4 is the feedforward signal of the tumor matrix with a very high expression level in cancer development, and disrupting the tumor-stroma IL-1β/IRAK4 feedforward circuitry improves the chemotherapy in PDAC [92]. Reducing perlecan in the matrix decreases the contact and communication between the matrix and cancer cells. Depleting perlecan in the stroma and combining with chemotherapy drugs such as gemcitabine (GEM) or Abraxane can prolong the survival rate of PDAC mice [93]. Erdafitinib, a selective pan-fibroblast growth factor receptor (FGFR) inhibitor approved by FDA, reduces the drug resistance of PC cells by targeting tumor fibroblast receptors to prevent the crosstalk between CAFs and cancer cells [94]. In clinical studies, Vismodegib, an orally bioavailable small molecule, has been found to inhibit Shh pathway and to reduce the production of stromal cells, thereby enhancing the anti-cancer effect of GEM on PC [95]. The combination of the least toxic anti-cancer drugs and anti-matrix drugs has gradually become a promising new cancer treatment [96].
3.2.3. CAFs Activation Suppresses Tumor Immune Response
Recent studies showed that the matrix in the tumor stroma also participates in the immune response [97]. For example, high expression of Caveolin-1 (CAV1), a membrane-associated scaffold protein, enhances the secretion of Interleukin-6 (IL-6) and IL-8 in CAFs and promotes PC invasion, while the down-regulation of CAV1 slows down the proliferation of PC cells [98]. Netrin-G1 (NETG1), a lipid anchored protein promotes CAFs to secrete glutamine, glutamate, and cytokines through p38/Fra-1 and Akt/4E-BP1 pathways, thus supporting the survival of PDAC cells under low nutritional conditions and reducing the antitumoral effect of NK cells on PDAC cells [99]. Hypoxia-induced fibrosis can inhibit the infiltration of T cells into the tumor, and the continuous activation of hypoxia-inducible factor 1 alpha (HIF-1α) can negatively regulate the signal transduction function of T cell receptors [100]. In human pancreatic fibrosis, macrophages are closely linked to PSCs, which may promote the activation of CAFs during chronic pancreatitis [43]. Tumor-associated macrophages promote cancer fibrosis by regulating ECM [101]. Monocytes can be recruited by CAFs via the IL-8/CXCR2 pathway and differentiate into macrophages that support tumor growth [102]. Therefore, CAFs can not only directly contact PC cells and secrete metabolites but also participate in the immune regulation of the TME. The strategy of targeting the interaction between CAFs, the immune system, and cancer cells can enhance the anti-tumor activity. Recently, it has been reported that knockout of the adhesion molecule, cadherin 11 (CDH11), which is mainly produced by CAFs, inhibits the growth of the pancreatic tumor, increases the response to GEM, reverses the immunosuppression of CAFs, and ultimately significantly prolongs the survival of mice [63].
3.2.4. CAFs Heterogeneity Is a Challenge in Cancer Therapy
Several subtypes of CAFs have been identified, including myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs) [103]. MyCAFs with a high expression of actin alpha 2 (ACTA2) were first identified in PC. For a long time, MyCAFs were considered to be the only CAF population, because α-SMA is widely expressed in CAFs [104,105]. MyCAFs play a major role in regulating the deposition and remodeling of ECM, highlighting the important role of myCAFs in promoting pancreatic fibrosis and pancreatitis [106]. ICAF is a subtype of CAFs with a high expression of Ly6C. ICAFs are driven by tumor secretory factors, such as Interleukin-1 alpha (IL-1α) and Interleukin 1 beta (IL-1β), and gather at the edge of the tumor [107]. IL-1α signaling also drives the autocrine signaling in iCAFs, which helps to maintain the inflammatory phenotype. ICAFs produce a variety of cytokines and chemokines (CCL2, CCL7, IL-6, and CXCL12) and may have a stronger tumor-promoting effect than myCAFs [108,109]. ICAF stimulates the proliferation and angiogenesis of PC cells and promotes PC development [110]. Furthermore, iCAFs inhibit the immune response through recruit regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) [111]. ApCAFs are the CAFs with antigen-presenting function, and the expression of major histocompatibility II (MHC II) is the major feature of apCAFs [110]. ApCAFs present antigens to T cells and affect T cell immunity [110]. ApCAFs can induce CD4+ Tregs differentiation through antigen-dependent T cell antigen receptor (TCR) ligation, reduce the anti-tumor immune response by changing the ratio of CD8+ T cells to Tregs and prevent PC cells from being monitored by immune cells [110,112,113]. According to the latest research, all three of these CAFs can transform into each other, which emphasizes the dynamic process of TME [114,115].
The majority of studies suggested that CAFs promote the development of PDAC. Recently, some unexpected results showed that myCAFs depletion may also promote PDAC development and metastasis [116]. It possibly depends on whether these CAFs are invasive (carcinogenic) or tumor suppressor CAFs because they might play different or even opposite roles in PDAC development [117]. Considering these studies, it would be necessary to distinguish the subtypes of CAFs, identify different markers, and explore the reasons for the high CAFs heterogeneity.
4. Cellular Stress Response Led to Pancreatic Fibrosis
When healthy cells suffer constant damage such as genotoxicity, protein, or lipid damage, cellular stress response confers the cellular adaptation that prevents cell death and promotes the transformation of healthy cells into tumor cells. Previous studies have clearly shown that stress proteins play an important role in maintaining the homeostatic microenvironment in both healthy and tumor cells [118,119,120]. Moreover, oxidative stress is required for driving metabolic reprogramming and the re-establishment of antioxidant systems in cancer cells [121]. Therefore, it is necessary to address how to target pancreatic fibrosis by reducing reactive oxidative species (ROS) or targeting important stress proteins.
4.1. ROS Scavengers for Treating Pancreatic Fibrosis
A large number of studies support oxidative stress as a triggering factor for pancreatic fibrosis. Oxidative stress directly promotes the activation of quiescent PSCs and the formation of an extensive amount of ECM, and subsequently promotes excessive fibrosis [122,123]. Meanwhile, oxidative stress also aggravates the damage of pancreatic cells in pancreatitis [124]. During the inflammatory phase, CAFs are recruited and activated under oxidative stress, which induces changes in the morphology and the functions of CAFs. However, this activated phenotype was prevented by several antioxidants [125]. For example, the ROS induced by H2O2 promotes the activation of PSCs, while resveratrol prevents the activation of PSCs by reducing the production of ROS [126]. Moreover, melatonin, at pharmacological concentrations, has shown a concentration-dependent decrease in cell viability in rat [127] and human [128] PSCs by modifying the redox state of the cells. Dimethyl fumarate (DMF) promotes the activation of nuclear factor erythroid 2-related factor 2 (NRF2) and the expression of downstream antioxidant genes, eliminating intracellular ROS, inhibiting the activation of PSCs, and reducing the pancreatic fibrosis level [129]. ROS-induced inflammation caused pancreatic cell death through RF2/NF-kB and SAPK/JNK pathways. The antioxidant N-acetyl cysteine (NAC) rescues cell viability by decreasing oxidative stress and inflammation in primary pancreatic cells [130]. Diethyldithiocarbamate is a kind of superoxide dismutase (SOD) inhibitor, was able to induce pancreatic fibrosis by increasing ROS in treated rats [131]. Vitamin E reduces oxidative stress and collagen deposition during CP, thereby reducing pancreatic fibrosis in cerulein-treated mice [124]. Theobromine and scoparone reduce the oxidative stress of pancreatic cells, inhibiting the activation of PSCs and attenuating pancreatic fibrosis through the TGF-β/Smad signaling pathway [132]. In mice, long-term administration of antioxidants prevents PSCs activation (by high glucose-diet) and subsequent fibrosis cascade. Coenzyme Q10 (CoQ10) reduces oxidative stress response, blocks ROS-induced PI3K/Akt/mTOR signaling pathway, decreases pancreatic fibrosis, and prevents the activation of PSCs [133]. Therefore, CoQ10 may be a drug candidate to treat pancreatic fibrosis [134].
Clinical research data show that antioxidant supplementation reduces the level of oxidative stress in patients with idiopathic CP and alcoholic pancreatitis and then weakens the process of pancreatic fibrosis [135]. More evidence supports that oxidative stress has an essential role in the progression of pancreatic fibrosis. Compared with other human organs, such as the liver or the kidney, the pancreas is more sensitive to long-term oxidative stress, triggering inflammation [123]. It has been suggested that ROS-induced oxidative stress can cause persistent damage to the biomacromolecules, such as DNA, RNA and proteins in pancreatic cells, promoting metabolic reprogramming and antioxidant system remodeling. Therefore, the use of antioxidants can reduce oxidative stress response, inhibit pancreatic fibrosis, and reduce the transformation of cells, compromising tumor development.
4.2. Targeting Stress-Inducible Protein NUPR1 for Treating Pancreatic Fibrosis and PC
NUPR1 is a stress-induced protein, which is over-activated in the damaged pancreas cells in AP and CP and plays an important role in PC development [136,137,138]. In addition, NUPR1 plays a crucial role in the fibrosis of multiple organs and tissues. For example, NUPR1 activated in the fibroblasts and the renal tubular epithelial cells promotes renal interstitial fibrosis [139]. Similarly, type I collagen and FN promote glioma progression by the activation of NUPR1 [140]. A recent study also found that knockout of NUPR1 decreases cardiac fibrosis and partially restores cardiac function [141]. In a spontaneous mouse model of CP, the oral protease inhibitor CM inhibits CP and pancreatic fibrosis by reducing the expression of NUPR1 [142]. Therefore, we proposed that NUPR1 plays an indispensable role in the progression of fibrosis, and inactivation of NUPR1 is a promising strategy for preventing fibrosis.
In this line, our recent studies have shown that ZZW-115, a powerful inhibitor of NUPR1, is able to kill cancer cells from different tumors, including PC. ZZW-115 is extremely effective in every subtype of PC, but also enhances the sensitivity of cancer cells to chemotherapeutic drugs [143,144]. Importantly, ZZW-115 cannot improve the sensitivity of the untransformed fibroblasts to chemotherapeutic drugs [145]. Interestingly, NUPR1 as a transcriptional factor is not only activated in oxidative stress but also activated in response to other cellular stress, such as endoplasmic reticulum stress (ER stress) and metabolic stress [146,147,148]. Our recent research shows that ZZW-115 treatment triggers ROS production, thus highlighting the role of NUPR1 in the oxidative stress response [143]. In conclusion, NUPR1 inhibitors have a variety of interesting effects in the treatment of PC, including attenuating fibrosis to slow the PC progression, killing PC cells directly through a variety of ways of death, and fighting drug resistance of cancer cells [149].
5. Conclusions
CP is characterized by persistent and permanent damage in pancreatic tissue [23]. The endocrine and exocrine compartments of the damaged pancreas are gradually lost and replaced by atrophy or fibrosis [23]. The CP development leads to organ dysfunction and increases the risk of PC development. Pancreatic fibrosis is also a typical feature of PC, which promotes the recruitment and activation of CAFs [150]. Pancreatic fibrosis can be used as a diagnostic marker of PC. Besides the traditional imaging methods, evaluating the level of pancreatic fibrosis could improve the diagnosis. Moreover, detecting inflammatory and oxidative stress indicators will contribute in the future to a better understanding of PC development, but also the diagnosis, prevention, and prognosis of the disease [151]. Furthermore, the development and application of the new generation of histology needles provide the possibility to analyze the TME of PC via endoscopic ultrasound [152,153,154], collecting the tumor tissue and allowing the analysis of the tumor matrix.
In addition, pancreatic fibrosis leads to hypoxia in the pancreatic tumor, which causes more oxidative stress response, promoting tumor aggressiveness, increasing drug resistance in cancer cells, and thereby causing higher patient mortality rates [155]. Interestingly, NUPR1, a stress protein activated in pancreatitis, promotes fibrosis, inflammation, and cancer initiation and development, indicating that NUPR1 is essential for TME. Collectively, cellular stress response drives fibrosis, playing a vital role in the transformation from CP to PC. Therefore, prevent fibrosis or targeting stress proteins, such as NUPR1, could be a promising therapeutic strategy for PC and CP therapy.
Author Contributions
Writing—Original Draft Preparation, C.H., J.I and P.S.-C.; Writing—Review & Editing, C.H., J.I. and P.S.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by China Scholarship Council to CH; La Ligue Contre le Cancer, INCa, Canceropole PACA, and INSERM to JI; Fondation de France and INSERM to PSC.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| TP53 | Tumor protein p53 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| KRAS | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| MRI | Magnetic resonance imaging |
| CT | Computerized tomography |
| CA199 | Carbohydrate antigen 199 |
| PC | Pancreatic cancer |
| CA125 | Cancer antigen 125 |
| GEM | Gemcitabine |
| CEA | Carcinoembryonic antigen |
| CA50 | Carbohydrate antigen 50 |
| CP | Chronic pancreatitis |
| AP | Acute pancreatitis |
| RECIST | response evaluation criteria in solid tumors |
| SPINK1 | Serine protease inhibitor Kazal-type 1 |
| PSCs | Pancreatic stellate cells |
| SST1 | symbiotic sulfate transporter 1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| Hic-5 | Hydrogen peroxide-inducible clone-5 |
| PDAC | Pancreatic ductal adenocarcinoma |
| CXCR2 | C-X-C motif chemokine receptor 2 |
| ACSL3 | Long-chain acyl coenzyme A synthase 3 |
| ADAM10 | A Disintegrin and metalloproteinase domain-containing protein 10 |
| CAFs | Cancer-related fibroblasts |
| FN | Fibronectin |
| SMO | Smoothened |
| FGFR | Fibroblast growth factor receptor |
| Hh | Hedgehog |
| ER stress | Endoplasmic reticulum stress |
| CAV1 | Caveolin-1 |
| ACTA2 | Actin alpha 2 |
| Tregs | Regulatory T cells |
| MDSCs | Myeloid-derived suppressor cells |
| myCAFs | Myofibroblastic CAFs |
| iCAFs | Inflammatory CAFs |
| apCAFs | Antigen-presenting CAFs |
| NK cells | Natural killer cells |
| CDH11 | Cadherin 11 |
| SMAD4 | SMAD family member 4 |
| TME | Tumor microenvironment |
| NDRG1 | N-myc downstream regulated gene-1 |
| CM | Camostat mesilate |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| MIR | MicroRNA |
| ECM | Extracellular matrix |
| NFs | Normal fibroblasts |
| MEFs | Mouse embryonic fibroblasts |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| IL-1α | Interleukin-1 alpha |
| IL-1β | Interleukin 1 beta |
| TCR | T cell antigen receptor |
| NET-G1 | Grade 1 neuroendocrine tumor |
| 4E-BP1 | Eukaryotic translation initiation factor 4E binding protein 1 |
| MHC II | Major histocompatibility complex II |
| ROS | Reactive oxidative species |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| NUPR1 | Nuclear protein 1 |
| NAC | N-acetyl cysteine |
| SOD | Superoxide dismutase |
| CoQ10 | Coenzyme Q10 |
References
- Xiao, B.; Zhang, X.-M. Magnetic resonance imaging for acute pancreatitis. World J. Radiol. 2010, 2, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Waldo, S.W.; Rosario, H.S.; Penn, A.H.; Schmid-Schönbein, G.W. Pancreatic digestive enzymes are potent generators of mediators for leukocyte activation and mortality. Shock 2003, 20, 138–143. [Google Scholar] [CrossRef]
- Watanabe, T.; Kudo, M.; Strober, W. Immunopathogenesis of pancreatitis. Mucosal Immunol. 2017, 10, 283–298. [Google Scholar] [CrossRef] [Green Version]
- Grigorian, A.; Lin, M.Y.C.; de Virgilio, C. Severe Epigastric Pain with Nausea and Vomiting. Surgery 2019, 227–237. [Google Scholar]
- Restrepo, R.; Hagerott, H.E.; Kulkarni, S.; Yasrebi, M.; Lee, E.Y. Acute Pancreatitis in Pediatric Patients: Demographics, Etiology, and Diagnostic Imaging. Am. J. Roentgenol. 2016, 206, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, E.V.F.; Guerrero-Lozano, R. Acute pancreatitis and recurrent acute pancreatitis: An exploration of clinical and etiologic factors and outcomes. J. Pediatr. 2019, 95, 713–719. [Google Scholar] [CrossRef]
- Barry, K. Chronic Pancreatitis: Diagnosis and Treatment. Am. Fam. Physician 2018, 97, 385–393. [Google Scholar]
- Pham, A.; Forsmark, C. Chronic pancreatitis: Review and update of etiology, risk factors, and management. F1000Research 2018, 7, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegyi, P.J.; Soós, A.; Tóth, E.; Ébert, A.; Venglovecz, V.; Márta, K.; Mátrai, P.; Mikó, A.; Bajor, J.; Sarlós, P.; et al. Evidence for diagnosis of early chronic pancreatitis after three episodes of acute pancreatitis: A cross-sectional multicentre international study with experimental animal model. Sci. Rep. 2021, 11, 1367. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Pandol, S.J.; Porcel, J.; Wilkens, L.R.; Le Marchand, L.; Pike, M.C.; Monroe, K.R. Prospective Study of Alcohol Drinking, Smoking, and Pancreatitis: The Multiethnic Cohort. Pancreas 2016, 45, 819–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, U.A.; Issa, Y.; Hagenaars, J.C.; Bakker, O.J.; van Goor, H.; Nieuwenhuijs, V.B.; Bollen, T.L.; van Ramshorst, B.; Witteman, B.J.; Brink, M.A.; et al. Risk of Recurrent Pancreatitis and Progression to Chronic Pancreatitis After a First Episode of Acute Pancreatitis. Clin. Gastroenterol. Hepatol. 2016, 14, 738–746. [Google Scholar]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Jungmann, F.; Kaissis, G.A.; Ziegelmayer, S.; Harder, F.; Schilling, C.; Yen, H.Y.; Steiger, K.; Weichert, W.; Schirren, R.; Demir, I.E.; et al. Prediction of Tumor Cellularity in Resectable PDAC from Preoperative Computed Tomography Imaging. Cancers 2021, 13. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Satoi, S.; Yamamoto, T.; Yamaki, S.; Sekimoto, M. The past, present, and future status of multimodality treatment for resectable/borderline resectable pancreatic ductal adenocarcinoma. Surg. Today 2020, 50, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Srisajjakul, S.; Prapaisilp, P.; Bangchokdee, S. CT and MR features that can help to differentiate between focal chronic pancreatitis and pancreatic cancer. Radiol. Med. 2020, 125, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Benini, L.; Cavallini, G.; Zordan, D.; Rizzotti, P.; Rigo, L.; Brocco, G.; Perobelli, L.; Zanchetta, M.; Pederzoli, P.; Scuro, L.A. A clinical evaluation of monoclonal (CA19-9, CA50, CA12-5) and polyclonal (CEA, TPA) antibody-defined antigens for the diagnosis of pancreatic cancer. Pancreas 1988, 3, 61–66. [Google Scholar] [CrossRef]
- Goulden, M.R. The pain of chronic pancreatitis: A persistent clinical challenge. Br. J. Pain 2013, 7, 8–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kichler, A.; Jang, S. Chronic Pancreatitis: Epidemiology, Diagnosis, and Management Updates. Drugs 2020, 80, 1155–1168. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Prim. 2017, 3, 17060. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.L.; Conwell, D.L.; Hart, P.A. Complications of Chronic Pancreatitis. Dig. Dis. Sci. 2017, 62, 1745–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, G.; Habtezion, A.; Werner, J.; Lerch, M.M.; Mayerle, J. Chronic pancreatitis. Lancet 2020, 396, 499–512. [Google Scholar] [CrossRef]
- Apte, M.; Pirola, R.; Wilson, J. The Fibrosis of Chronic Pancreatitis: New Insights into the Role of Pancreatic Stellate Cells. Antioxid. Redox Signal. 2011, 15, 2711–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, D.; DiVincenzo, M.J.; Torok, M.; Choueiry, F.; Kumar, R.J.; Deems, A.; Miller, J.L.; Hinton, A.; Geraghty, C.; Maranon, J.A.; et al. Soy-tomato enriched diet reduces inflammation and disease severity in a pre-clinical model of chronic pancreatitis. Sci. Rep. 2020, 10, 21824. [Google Scholar] [CrossRef]
- Di Magno, M.J.; Di Magno, E.P. Chronic pancreatitis. Curr. Opin. Gastroenterol. 2012, 28, 523–531. [Google Scholar] [CrossRef]
- Kanikovskiy, O.E.; Pavlyk, I.V.; Oliinyk, I.V.; Mosondz, V.V. The key role of pancreatic fibrosis severity in the surgical treatment algorithm of patients with chronic pancreatitis. Wiad. Lek. 2020, 73, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Witt, H. Chronic pancreatitis and cystic fibrosis. Gut 2003, 52, ii31–ii41. [Google Scholar] [CrossRef]
- Chowdhury, P.; Gupta, P. Pathophysiology of alcoholic pancreatitis: An overview. World J. Gastroenterol. 2006, 12, 7421–7427. [Google Scholar] [CrossRef]
- Lee, S.Y.; Goh, B.K.P.; Chan, C.Y. Chapter 55—Etiology, pathogenesis, and diagnostic assessment of acute pancreatitis. In Blumgart’s Surgery of the Liver, Biliary Tract and Pancreas; Jarnagin, W.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 883–896. [Google Scholar]
- Clemens, D.L.; Wells, M.A.; Schneider, K.J.; Singh, S. Molecular mechanisms of alcohol associated pancreatitis. World J. Gastrointest. Pathophysiol. 2014, 5, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, B.S.; Vasioukhin, V. Chapter 1—Mechanisms of Prostate Cancer Initiation and Progression. In Advances in Cancer Research; Woude, G.F.V., Klein, G., Eds.; Academic Press: London, UK, 2010; pp. 1–50. [Google Scholar]
- Jones, T.E.; Bellin, M.D.; Yadav, D.; Freeman, M.L.; Schwarzenberg, S.J.; Slivka, A.; Chennat, J.S.; Beilman, G.J.; Chinnakotla, S.; Pruett, T.L.; et al. The histopathology of SPINK1-associated chronic pancreatitis. Pancreatology 2020, 20, 1648–1655. [Google Scholar] [CrossRef]
- Schneider, A.; Larusch, J.; Sun, X.; Aloe, A.; Lamb, J.; Hawes, R.; Cotton, P.; Brand, R.E.; Anderson, M.A.; Money, M.E.; et al. Combined Bicarbonate Conductance-Impairing Variants in CFTR and SPINK1 Variants are Associated with Chronic Pancreatitis in Patients without Cystic Fibrosis. Gastroenterology 2011, 140, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhakaran, P.R.; Radhika, A.; Jacob, S.S. Monocyte macrophage differentiation in vitro: Fibronectin-dependent upregulation of certain macrophage-specific activities. Glycoconj. J. 2007, 24, 49–55. [Google Scholar] [CrossRef]
- Mescher, A.L. Macrophages and fibroblasts during inflammation and tissue repair in models of organ regeneration. Regeneration 2017, 4, 39–53. [Google Scholar] [CrossRef]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of Immune Cells and Immune-Based Therapies in Pancreatitis and Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158-7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.; Lou, N.; Jiao, J.; Guo, F.; Xiang, H.; Shang, D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. Biomed. Pharmacother. 2020, 131, 110693. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Duan, L.; Wu, N.; Xu, X.; Xin, J.; Jiang, S.; Zhang, C.; Zhang, H. Baicalin Ameliorates Pancreatic Fibrosis by Inhibiting the Activation of Pancreatic Stellate Cells in Mice with Chronic Pancreatitis. Front. Pharmacol. 2020, 11, 607133. [Google Scholar] [CrossRef]
- Alpha, K.M.; Xu, W.; Turner, C.E. Chapter One—Paxillin family of focal adhesion adaptor proteins and regulation of cancer cell invasion. In International Review of Cell and Molecular Biology; Thomas, C., Galluzzi, L., Eds.; Academic Press: London, UK, 2020; pp. 1–52. [Google Scholar]
- Gao, L.; Lei, X.-F.; Miyauchi, A.; Noguchi, M.; Omoto, T.; Haraguchi, S.; Miyazaki, T.; Miyazaki, A.; Kim-Kaneyama, J.-R. Hic-5 is required for activation of pancreatic stellate cells and development of pancreatic fibrosis in chronic pancreatitis. Sci. Rep. 2020, 10, 19105. [Google Scholar] [CrossRef]
- Rasmussen, H.H.; Irtun, O.; Olesen, S.S.; Drewes, A.M.; Holst, M. Nutrition in chronic pancreatitis. World J. Gastroenterol. 2013, 19, 7267–7275. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moneo, E.; Stigliano, S.; Hedstrom, A.; Kaczka, A.; Malvik, M.; Waldthaler, A.; Maisonneuve, P.; Simon, P.; Capurso, G. Deficiency of fat-soluble vitamins in chronic pancreatitis: A systematic review and meta-analysis. Pancreatology 2016, 16. [Google Scholar] [CrossRef]
- Du, W.D.; Yuan, Z.R.; Sun, J.; Tang, J.X.; Cheng, A.Q.; Shen, D.M.; Huang, C.J.; Song, X.H.; Yu, X.F.; Zheng, S.B. Therapeutic efficacy of high-dose vitamin C on acute pancreatitis and its potential mechanisms. World J. Gastroenterol. 2003, 9, 2565–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharer, N.M.; Taylor, P.M.; Linaker, B.D.; Gutteridge, J.M.C.; Braganza, J.M. Safe and Successful Use of Vitamin C to Treat Painful Calcific Chronic Pancreatitis Despite Iron Overload from Primary Haemochromatosis. Clin. Drug Investig. 1995, 10, 310–315. [Google Scholar] [CrossRef]
- Lu, X.L.; Song, Y.H.; Fu, Y.B.; Si, J.M.; Qian, K.D. Ascorbic Acid Alleviates Pancreatic Damage Induced by Dibutyltin Dichloride (DBTC) in Rats. Yonsei Med. J. 2007, 48, 1028–1034. [Google Scholar] [CrossRef]
- Al-Hashem, F.; Ellatif, M.A.; Shamseldeen, A.M.; Kamar, S.S.; Al-Ani, B.; Haidara, M.A. Vitamin E protects against the modulation of TNF-α-AMPK axis and inhibits pancreas injury in a rat model of L-arginine-induced acute necrotising pancreatitis. Arch. Physiol. Biochem. 2020, 1–9. [Google Scholar] [CrossRef]
- Reifen, R. Vitamin A as an anti-inflammatory agent. Proc. Nutr. Soc. 2002, 61, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Chen, H.; Qin, S.; Wang, M.; Wang, X.; Zhang, X.; Liu, F.; Zhang, S. The association between dietary vitamin A intake and pancreatic cancer risk: A meta-analysis of 11 studies. Biosci. Rep. 2016, 36, e00414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siener, R.; Machaka, I.; Alteheld, B.; Bitterlich, N.; Metzner, C. Effect of Fat-Soluble Vitamins A, D, E and K on Vitamin Status and Metabolic Profile in Patients with Fat Malabsorption with and without Urolithiasis. Nutrients 2020, 12. [Google Scholar] [CrossRef]
- Miyauchi, M.; Suda, K.; Kuwayama, C.; Abe, H.; Kakinuma, C. Role of fibrosis-related genes and pancreatic duct obstruction in rat pancreatitis models: Implications for chronic pancreatitis. Histol. Histopathol. 2007, 22, 1119–1127. [Google Scholar]
- Tseng, J.; Choi, E.A.; Matthews, J.B. Chapter 92—Chronic Pancreatitis. In Shackelford’s Surgery of the Alimentary Tract; Yeo, C.J., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1085–1096. [Google Scholar]
- Ramakrishnan, P.; Loh, W.M.; Gopinath, S.C.B.; Bonam, S.R.; Fareez, I.M.; Mac Guad, R.; Sim, M.S.; Wu, Y.S. Selective phytochemicals targeting pancreatic stellate cells as new anti-fibrotic agents for chronic pancreatitis and pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Diazzi, S.; Tartare-Deckert, S.; Deckert, M. Bad Neighborhood: Fibrotic Stroma as a New Player in Melanoma Resistance to Targeted Therapies. Cancers 2020, 12, 1364. [Google Scholar] [CrossRef] [PubMed]
- Peran, I.; Dakshanamurthy, S.; McCoy, M.D.; Mavropoulos, A.; Allo, B.; Sebastian, A.; Hum, N.R.; Sprague, S.C.; Martin, K.A.; Pishvaian, M.J.; et al. Cadherin 11 Promotes Immunosuppression and Extracellular Matrix Deposition to Support Growth of Pancreatic Tumors and Resistance to Gemcitabine in Mice. Gastroenterology 2021, 160. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Inoue, Y.; Hiratsuka, M.; Kawakatsu, S.; Arai, T.; Matsueda, K.; Saiura, A.; Takazawa, Y. Encapsulating fibrosis following neoadjuvant chemotherapy is correlated with outcomes in patients with pancreatic cancer. PLoS ONE 2019, 14, e0222155. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erstad, D.J.; Sojoodi, M.; Taylor, M.S.; Jordan, V.C.; Farrar, C.T.; Axtell, A.L.; Rotile, N.J.; Jones, C.; Graham-O’Regan, K.A.; Ferreira, D.S.; et al. Fibrotic Response to Neoadjuvant Therapy Predicts Survival in Pancreatic Cancer and Is Measurable with Collagen-Targeted Molecular MRI. Clin. Cancer Res. 2020, 26, 5007–5018. [Google Scholar] [CrossRef]
- Palomino, D.C.T.; Marti, L.C. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Admyre, C.; Axelsson, L.-G.; von Stein, O.; Zargari, A. Immunomodulatory oligonucleotides inhibit neutrophil migration by decreasing the surface expression of interleukin-8 and leukotriene B4 receptors. Immunology 2015, 144, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Pozzato, C.; Saliakoura, M.; Yang, Z.; Peng, R.-W.; Galiè, M.; Oberson, K.; Simon, H.-U.; Karamitopoulou, E.; Konstantinidou, G. ACSL3-PAI-1 signaling axis mediates tumor-stroma cross-talk promoting pancreatic cancer progression. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Fàbrega, A.; Guyonnet, B.; Dacheux, J.L.; Gatti, J.L.; Puigmulé, M.; Bonet, S.; Pinart, E. Expression, immunolocalization and processing of fertilins ADAM-1 and ADAM-2 in the boar (Sus domesticus) spermatozoa during epididymal maturation. Reprod. Biol. Endocrinol. 2011, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.C.; Piper, M.; Goodspeed, A.; Bhuvane, S.; Williams, J.S.; Bhatia, S.; Phan, A.V.; Van Court, B.; Zolman, K.L.; Peña, B.; et al. Induction of ADAM10 by RT drives fibrosis, resistance, and EMT in pancreatic cancer. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; De Nardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. 2017, 66, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [Green Version]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Araki, K.; Yokobori, T.; Hagiwara, K.; Gantumur, D.; Yamanaka, T.; Handa, T.; Tsukagoshi, M.; Igarashi, T.; Watanabe, A.; et al. Conophylline suppresses pancreatic cancer desmoplasia and cancer-promoting cytokines produced by cancer-associated fibroblasts. Cancer Sci. 2019, 110, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife 2019, 8. [Google Scholar] [CrossRef]
- Walter, K.; Omura, N.; Hong, S.-M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of Smoothened Activates the Sonic Hedgehog Signaling Pathway in Pancreatic Cancer-Associated Fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Ellen, T.P.; Ke, Q.; Zhang, P.; Costa, M. NDRG1, a growth and cancer related gene: Regulation of gene expression and function in normal and disease states. Carcinogenesis 2008, 29, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geleta, B.; Park, K.C.; Jansson, P.J.; Sahni, S.; Maleki, S.; Xu, Z.; Murakami, T.; Pajic, M.; Apte, M.V.; Richardson, D.R.; et al. Breaking the cycle: Targeting of NDRG1 to inhibit bi-directional oncogenic cross-talk between pancreatic cancer and stroma. FASEB J. 2021, 35, e21347. [Google Scholar] [CrossRef]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Modica, C.; Tortarolo, D.; Comoglio, P.M.; Basilico, C.; Vigna, E. MET/HGF Co-Targeting in Pancreatic Cancer: A Tool to Provide Insight into the Tumor/Stroma Crosstalk. Int. J. Mol. Sci. 2018, 19, 3920. [Google Scholar] [CrossRef] [Green Version]
- Basso, D.; Bozzato, D.; Padoan, A.; Moz, S.; Zambon, C.-F.; Fogar, P.; Greco, E.; Scorzeto, M.; Simonato, F.; Navaglia, F.; et al. Inflammation and pancreatic cancer: Molecular and functional interactions between S100A8, S100A9, NT-S100A8 and TGFβ1. Cell Commun. Signal. 2014, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Schoepp, M.; Ströse, A.J.; Haier, J. Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers 2017, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Ning, Z.; Ma, L.; Liu, W.; Shao, C.; Shu, Y.; Shen, H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol. Cancer 2017, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Samain, R.; Brunel, A.; Douché, T.; Fanjul, M.; Cassant-Sourdy, S.; Rochotte, J.; Cros, J.; Neuzillet, C.; Raffenne, J.; Duluc, C.; et al. Pharmacological Normalization of Pancreatic Cancer-Associated Fibroblast Secretome Impairs Pro-metastatic Cross-Talk with Macrophages. Cell. Mol. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Mutgan, A.C.; Besikcioglu, H.E.; Wang, S.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol. Cancer 2018, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Shi, S.; Meng, Q.; Liang, D.; Ji, S.; Zhang, B.; Qin, Y.; Xu, J.; Ni, Q.; Yu, X. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 2017, 49, e406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.-H. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef] [Green Version]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Cui, J.Y.; Gao, H.F.; Yu, H.; Gao, F.F.; Chen, J.L.; Chen, L. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol. 2020, 16, 2619–2633. [Google Scholar] [CrossRef]
- Qu, C.; Wang, Q.; Meng, Z.; Wang, P. Cancer-Associated Fibroblasts in Pancreatic Cancer: Should They Be Deleted or Reeducated? Integr. Cancer Ther. 2018, 17, 1016–1019. [Google Scholar] [CrossRef]
- Dauer, P.; Zhao, X.; Gupta, V.K.; Sharma, N.; Kesh, K.; Gnamlin, P.; Dudeja, V.; Vickers, S.M.; Banerjee, S.; Saluja, A. Inactivation of Cancer-Associated-Fibroblasts Disrupts Oncogenic Signaling in Pancreatic Cancer Cells and Promotes Its Regression. Cancer Res. 2018, 78, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Yamao, T.; Yamashita, Y.I.; Yamamura, K.; Nakao, Y.; Tsukamoto, M.; Nakagawa, S.; Okabe, H.; Hayashi, H.; Imai, K.; Baba, H. Cellular Senescence, Represented by Expression of Caveolin-1, in Cancer-Associated Fibroblasts Promotes Tumor Invasion in Pancreatic Cancer. Ann. Surg. Oncol. 2019, 26, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Vendramini-Costa, D.B.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. Netrin G1 Promotes Pancreatic Tumorigenesis through Cancer-Associated Fibroblast-Driven Nutritional Support and Immunosuppression. Cancer Discov. 2021, 11, 446–479. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Johnson, D.K.; Homann, D.; Clambey, E.T. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 2013, 55, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.B.; Steele, N.G.; Carpenter, E.S.; Donahue, K.L.; Bushnell, G.G.; Morris, A.H.; The, S.; Orbach, S.M.; Sirihorachai, V.R.; Nwosu, Z.C.; et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Sci. Alliance 2021, 4, e202000935. [Google Scholar] [CrossRef]
- Huang, H.; Brekken, R.A. Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am. J. Physiol. Physiol. 2020, 319, C233–C243. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Louault, K.; Li, R.-R.; DeClerck, Y.A. Cancer-Associated Fibroblasts: Understanding Their Heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef] [PubMed]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef]
- Colombo, M.; Mirandola, L.; Chiriva-Internati, M.; Basile, A.; Locati, M.; Lesma, E.; Chiaramonte, R.; Platonova, N. Cancer Cells Exploit Notch Signaling to Redefine a Supportive Cytokine Milieu. Front. Immunol. 2018, 9, 1823-1823. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Zheng, L. Epigenetics in modulating immune functions of stromal and immune cells in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 940–953. [Google Scholar] [CrossRef]
- Freeman, P.; Mielgo, A. Cancer-Associated Fibroblast Mediated Inhibition of CD8+ Cytotoxic T Cell Accumulation in Tumours: Mechanisms and Therapeutic Opportunities. Cancers 2020, 12, 2687. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Z.; Zhang, Y.; Brekken, R. Mesothelial Cell-Derived Antigen-Presenting Cancer-Associated Fibroblasts Induce Expansion of Regulatory T Cells in Pancreatic Cancer. bioRxiv 2021. bioRxiv:10.2139/ssrn.3790948. [Google Scholar]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Schuck, K.; Friess, H.; Kong, B. Targeting Aggressive Fibroblasts to Enhance the Treatment of Pancreatic Cancer. Expert Opin. Ther. Targets 2021, 25, 5–13. [Google Scholar] [CrossRef]
- Chovatiya, R.; Medzhitov, R. Stress, Inflammation, and Defense of Homeostasis. Mol. Cell 2014, 54, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031-16031. [Google Scholar] [CrossRef]
- Fiaschi, T.; Chiarugi, P. Oxidative Stress, Tumor Microenvironment, and Metabolic Reprogramming: A Diabolic Liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, B.-M.; Oh, T.-Y.; Kim, Y.-B.; Yeo, M.; Lee, J.-S.; Surh, Y.J.; Ahn, B.-O.; Kim, W.-H.; Sohn, S.; Kim, J.-H.; et al. Novel antioxidant ameliorates the fibrosis and inflammation of cerulein-induced chronic pancreatitis in a mouse model. Pancreatology 2005, 5, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Heras-Castaño, G.D.L.; García-Unzueta, M.T.; Domínguez-Diez, A.; Fernández-González, M.D.; la Paz, A.M.G.D.; Mayorga-Fernández, M.; Fernández-Fernández, F. Pancreatic fibrosis in rats and its response to antioxidant treatment. JOP 2005, 6, 316–324. [Google Scholar]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 5486. [Google Scholar] [CrossRef]
- Yan, B.; Cheng, L.; Jiang, Z.; Chen, K.; Zhou, C.; Sun, L.; Cao, J.; Qian, W.; Li, J.; Shan, T.; et al. Resveratrol Inhibits ROS-Promoted Activation and Glycolysis of Pancreatic Stellate Cells via Suppression of miR-21. Oxidative Med. Cell. Longev. 2018, 2018, 1346958. [Google Scholar] [CrossRef]
- Gonzalez, A.; Estaras, M.; Martinez-Morcillo, S.; Martinez, R.; García, A.; Estévez, M.; Santofimia-Castaño, P.; Tapia, J.A.; Moreno, N.; Pérez-López, M.; et al. Melatonin modulates red-ox state and decreases viability of rat pancreatic stellate cells. Sci. Rep. 2020, 10, 6352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estaras, M.; Moreno, N.; Santofimia-Castaño, P.; Martinez-Morcillo, S.; Roncero, V.; Blanco, G.; Lopez, D.; Fernandez-Bermejo, M.; Mateos, J.M.; Iovanna, J.L.; et al. Melatonin induces reactive oxygen species generation and changes in glutathione levels and reduces viability in human pancreatic stellate cells. J. Physiol. Biochem. 2019, 75, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Kojayan, G.G.; Alizadeh, R.F.; Li, S.; Ichii, H. Reducing Pancreatic Fibrosis Using Antioxidant Therapy Targeting Nrf2 Antioxidant Pathway: A Possible Treatment for Chronic Pancreatitis. Pancreas 2019, 48, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Ghosh, S.; Gupta, P.; Mukherjee, S.; Chattopadhyay, S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic. Res. 2015, 49, 1371–1383. [Google Scholar] [CrossRef]
- Matsumura, N.; Ochi, K.; Ichimura, M.; Mizushima, T.; Harada, H.; Harada, M. Study on free radicals and pancreatic fibrosis--pancreatic fibrosis induced by repeated injections of superoxide dismutase inhibitor. Pancreas 2001, 22, 53–57. [Google Scholar] [CrossRef]
- Xu, M.; Cai, J.; Wei, H.; Zhou, M.; Xu, P.; Huang, H.; Peng, W.; Du, F.; Gong, A.; Zhang, Y. Scoparone Protects Against Pancreatic Fibrosis via TGF-β/Smad Signaling in Rats. Cell. Physiol. Biochem. 2016, 40, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Yang, J.; Wu, J.; Meng, Q.; Hao, J. Coenzyme Q10 inhibits the activation of pancreatic stellate cells through PI3K/AKT/mTOR signaling pathway. Oncotarget 2017, 8. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Xue, R.; Wang, J.; Yang, L.; Liu, X.; Gao, Y.; Pang, Y.; Wang, Y.; Hao, J. Coenzyme Q10 Ameliorates Pancreatic Fibrosis via the ROS-Triggered mTOR Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 8039694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallo, G.V.; Fiedler, F.; Calvo, E.L.; Ortiz, E.M.; Vasseur, S.; Keim, V.; Morisset, J.; Iovanna, J.L. Cloning and Expression of the Rat p8 cDNA, a New Gene Activated in Pancreas during the Acute Phase of Pancreatitis, Pancreatic Development, and Regeneration, and Which Promotes Cellular Growth. J. Biol. Chem. 1997, 272, 32360–32369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motoo, Y.; Iovanna, J.L.; Mallo, G.V.; Su, S.B.; Xie, M.J.; Sawabu, N. P8 expression is induced in acinar cells during chronic pancreatitis. Dig. Dis. Sci. 2001, 46, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Su, S.B.; Motoo, Y.; Iovanna, J.L.; Berthézène, P.; Xie, M.J.; Mouri, H.; Ohtsubo, K.; Matsubara, F.; Sawabu, N. Overexpression of p8 is inversely correlated with apoptosis in pancreatic cancer. Clin. Cancer Res. 2001, 7, 1320–1324. [Google Scholar] [PubMed]
- Zhou, R.; Liao, J.; Cai, D.; Tian, Q.; Huang, E.; Lü, T.; Chen, S.Y.; Xie, W.B. Nupr1 mediates renal fibrosis via activating fibroblast and promoting epithelial-mesenchymal transition. FASEB J. 2021, 35, e21381. [Google Scholar] [CrossRef]
- Zhong, C.; Tao, B.; Tang, F.; Yang, X.; Peng, T.; You, J.; Xia, K.; Xia, X.; Chen, L.; Peng, L. Remodeling cancer stemness by collagen/fibronectin via the AKT and CDC42 signaling pathway crosstalk in glioma. Theranostics 2021, 11, 1991–2005. [Google Scholar] [CrossRef]
- Georgescu, S.P.; Aronovitz, M.J.; Iovanna, J.L.; Patten, R.D.; Kyriakis, J.M.; Goruppi, S. Decreased metalloprotease 9 induction, cardiac fibrosis, and higher autophagy after pressure overload in mice lacking the transcriptional regulator p8. Am. J. Physiol. Physiol. 2011, 301, C1046–C1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.-B.; Motoo, Y.; Iovanna, J.L.; Xie, M.-J.; Sawabu, N. Effect of Camostat Mesilate on the Expression of Pancreatitis-Associated Protein (PAP), p8, and Cytokines in Rat Spontaneous Chronic Pancreatitis. Pancreas 2001, 23, 134–140. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abián, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef]
- Lan, W.; Santofimia-Castaño, P.; Xia, Y.; Zhou, Z.; Huang, C.; Fraunhoffer, N.; Barea, D.; Cervello, M.; Giannitrapani, L.; Montalto, G.; et al. Targeting NUPR1 with the small compound ZZW-115 is an efficient strategy to treat hepatocellular carcinoma. Cancer Lett. 2020, 486, 8–17. [Google Scholar] [CrossRef]
- Lan, W.; Santofimia-Castaño, P.; Swayden, M.; Xia, Y.; Zhou, Z.; Audebert, S.; Camoin, L.; Huang, C.; Peng, L.; Jiménez-Alesanco, A.; et al. ZZW-115-dependent inhibition of NUPR1 nuclear translocation sensitizes cancer cells to genotoxic agents. JCI Insight 2020. [Google Scholar] [CrossRef] [PubMed]
- Cano, C.E.; Hamidi, T.; Sandi, M.J.; Iovanna, J.L. Nupr1: The Swiss-knife of cancer. J. Cell. Physiol. 2011, 226, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castaño, P.; Lan, W.; Bintz, J.; Gayet, O.; Carrier, A.; Lomberk, G.; Neira, J.L.; González, A.; Urrutia, R.; Soubeyran, P.; et al. Inactivation of NUPR1 promotes cell death by coupling ER-stress responses with necrosis. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, T.; Cano, C.E.; Grasso, D.; Garcia, M.N.; Sandi, M.J.; Calvo, E.L.; Dagorn, J.C.; Lomberk, G.; Goruppi, S.; Urrutia, R.; et al. NUPR1 works against the metabolic stress-induced autophagy-associated cell death in pancreatic cancer cells. Autophagy 2013, 9, 95–97. [Google Scholar] [CrossRef] [Green Version]
- Santofimia-Castaño, P.; Iovanna, J. Combating pancreatic cancer chemoresistance by triggering multiple cell death pathways. Pancreatology 2021. [Google Scholar] [CrossRef]
- Adam, M.G.; Beyer, G.; Christiansen, N.; Kamlage, B.; Pilarsky, C.; Distler, M.; Falbusch, T.; Chromik, A.; Klein, F.; Bahra, M.; et al. Identification and validation of a multivariable prediction model based on blood plasma and serum metabolomics for the distinction of chronic pancreatitis subjects from non-pancreas disease control subjects. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Novovic, S.; Borch, A.; Werge, M.; Karran, D.; Gluud, L.; Schmidt, P.N.; Hansen, E.F.; Nøjgaard, C.; Jensen, A.B.; Jensen, F.K.; et al. Characterisation of the fibroinflammatory process involved in progression from acute to chronic pancreatitis: Study protocol for a multicentre, prospective cohort study. BMJ Open 2019, 9, e028999. [Google Scholar] [CrossRef] [Green Version]
- Oppong, K.W.; Bekkali, N.L.H.; Leeds, J.S.; Johnson, S.J.; Nayar, M.K.; Darné, A.; Egan, M.; Bassett, P.; Haugk, B. Fork-tip needle biopsy versus fine-needle aspiration in endoscopic ultrasound-guided sampling of solid pancreatic masses: A randomized crossover study. Endoscopy 2020, 52, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Crinò, S.F.; Le Grazie, M.; Manfrin, E.; Bellocchi, M.C.C.; Bernardoni, L.; Granato, A.; Locatelli, F.; Parisi, A.; Di Stefano, S.; Frulloni, L.; et al. Randomized trial comparing fork-tip and side-fenestrated needles for EUS-guided fine-needle biopsy of solid pancreatic lesions. Gastrointest. Endosc. 2020, 92, 648–658. [Google Scholar] [CrossRef]
- Kandel, P.; Nassar, A.; Gomez, V.; Raimondo, M.; Woodward, T.A.; Crook, J.E.; Fares, N.S.; Wallace, M.B. Comparison of endoscopic ultrasound-guided fine-needle biopsy versus fine-needle aspiration for genomic profiling and DNA yield in pancreatic cancer: A randomized crossover trial. Endoscopy 2021, 53, 376–382. [Google Scholar]
- Hollinshead, K.E.R.; Parker, S.J.; Eapen, V.V.; Encarnacion-Rosado, J.; Sohn, A.; Oncu, T.; Cammer, M.; Mancias, J.D.; Kimmelman, A.C. Respiratory Supercomplexes Promote Mitochondrial Efficiency and Growth in Severely Hypoxic Pancreatic Cancer. Cell Rep. 2020, 33, 108231. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the mechanisms involved in PC initiation.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, C.; Iovanna, J.; Santofimia-Castaño, P. Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 4970. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094970
AMA Style
Huang C, Iovanna J, Santofimia-Castaño P. Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer. International Journal of Molecular Sciences. 2021; 22(9):4970. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094970
Chicago/Turabian StyleHuang, Can, Juan Iovanna, and Patricia Santofimia-Castaño. 2021. "Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer" International Journal of Molecular Sciences 22, no. 9: 4970. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094970
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.