
Natural and Designed Toxins for Precise Therapy: Modern Approaches in Experimental Oncology


Abstract
:1. Introduction
2. Soluble Targeted Toxins
2.1. Targeting and Toxic Modules Coupling Strategies
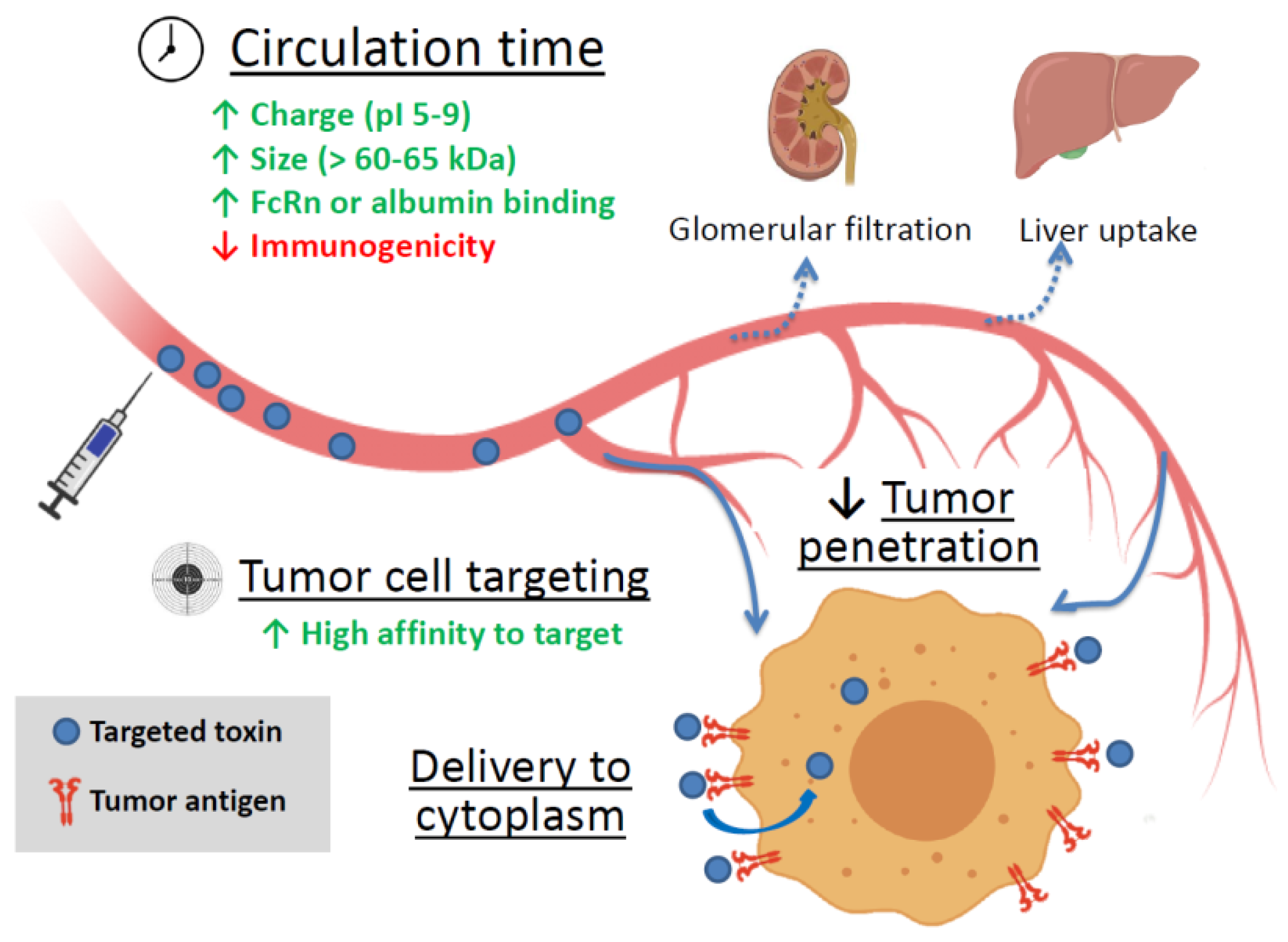
2.2. Factors Affecting a Targeted Toxin Efficiency
3. Targeted Toxins as Components of Nanoagents

4. Cytotoxic Mechanisms of Natural Toxins
4.1. Toxins Inhibiting Protein Synthesis
4.2. Toxins Disrupting Cell Signaling
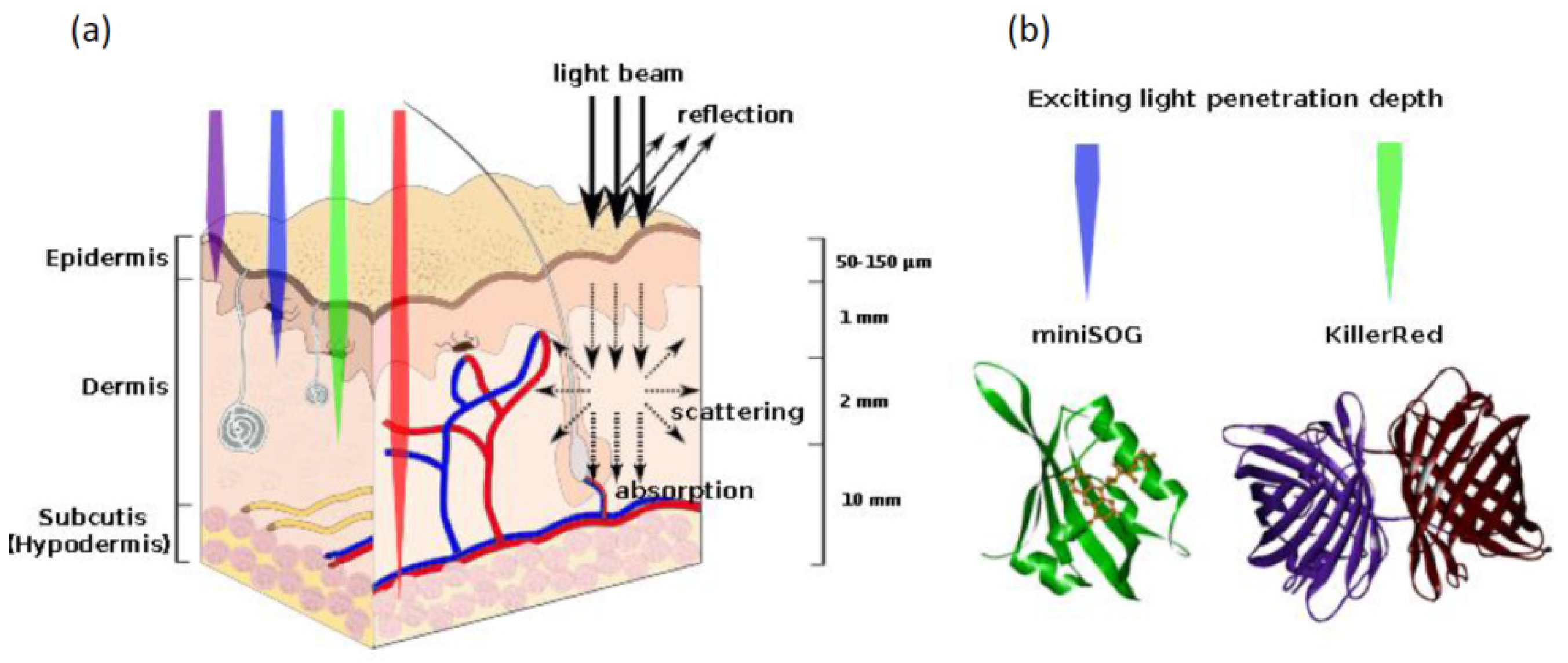
4.3. Proteins Inducing Oxidative Stress
4.4. Direct Apoptosis Induction
4.5. Enhanced Diffusion of Other Anticancer Drug
5. Reducing Protein Toxins Side Toxicity
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| DT | Diphtheria toxin |
| PE | Pseudomonas aeruginosa exotoxin A |
| RIT | Ribosome inactivating toxin |
| Stx | Shiga toxin |
| HER2 | Human epidermal growth factor receptor 2 |
| EpCAM | Epithelial cell adhesion molecule |
| FcRn | the neonatal immunoglobulin Fc receptor |
| MMP | Matrix metalloprotease |
| uPA | Urokinase plasminogen activator |
References
- Deyev, S.M.; Lebedenko, E.N. Modern Technologies for Creating Synthetic Antibodies for Clinical Application. Acta Nat. 2009, 1, 32–50. [Google Scholar] [CrossRef]
- Shilova, O.N.; Deyev, S.M. DARPins: Promising scaffolds for theranostics. Acta Nat. 2019, 11, 42–53. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Serna, N.; Sánchez-García, L.; Unzueta, U.; Díaz, R.; Vázquez, E.; Mangues, R.; Villaverde, A. Protein-Based Therapeutic Killing for Cancer Therapies. Trends Biotechnol. 2018, 36, 318–335. [Google Scholar] [CrossRef]
- Pastan, I.; FitzGerald, D. Recombinant toxins for cancer treatment. Science 1991, 254, 1173–1177. [Google Scholar] [CrossRef]
- Souslova, E.A.; Mironova, K.E.; Deyev, S.M. Applications of genetically encoded photosensitizer miniSOG: From correlative light electron microscopy to immunophotosensitizing. J. Biophotonics 2017, 10, 338–352. [Google Scholar] [CrossRef]
- Sokolova, E.; Guryev, E.; Yudintsev, A.; Vodeneev, V.; Deyev, S.; Balalaeva, I. HER2-specific recombinant immunotoxin 4D5scFv-PE40 passes through retrograde trafficking route and forces cells to enter apoptosis. Oncotarget 2017, 8, 22048–22058. [Google Scholar] [CrossRef] [Green Version]
- Bulina, M.E.; Chudakov, D.M.; Britanova, O.V.; Yanushevich, Y.G.; Staroverov, D.B.; Chepurnykh, T.V.; Merzlyak, E.M.; Shkrob, M.A.; Lukyanov, S.; Lukyanov, K.A. A genetically encoded photosensitizer. Nat. Biotechnol. 2006, 24, 95–99. [Google Scholar] [CrossRef]
- Mironova, K.E.; Proshkina, G.M.; Ryabova, A.V.; Stremovskiy, O.A.; Lukyanov, S.A.; Petrov, R.V.; Deyev, S.M. Genetically encoded immunophotosensitizer 4D5scFv-miniSOG is a highly selective agent for targeted photokilling of tumor cells in vitro. Theranostics 2013, 3, 831–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proshkina, G.M.; Shilova, O.N.; Ryabova, A.V.; Stremovskiy, O.A.; Deyev, S.M. A new anticancer toxin based on HER2/neu-specific DARPin and photoactive flavoprotein miniSOG. Biochimie 2015, 118, 116–122. [Google Scholar] [CrossRef]
- Sarkisyan, K.S.; Zlobovskaya, O.A.; Gorbachev, D.A.; Bozhanova, N.G.; Sharonov, G.V.; Staroverov, D.B.; Egorov, E.S.; Ryabova, A.V.; Solntsev, K.M.; Mishin, A.S.; et al. KillerOrange, a Genetically Encoded Photosensitizer Activated by Blue and Green Light. PLoS ONE 2015, 10, e0145287. [Google Scholar] [CrossRef] [PubMed]
- Ross, W.C.J.; Thorpe, P.E.; Cumber, A.J.; Edwards, D.C.; Hinson, C.A.; Davies, A.J.S. Increased Toxicity of Diphtheria Toxin for Human Lymphoblastoid Cells following Covalent Linkage to Anti-(human lymphocyte) Globulin or Its F(ab′)2 Fragment. Eur. J. Biochem. 1980, 104, 381–390. [Google Scholar] [CrossRef]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment a introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Antignani, A.; FitzGerald, D. Immunotoxins: The role of the toxin. Toxins (Basel) 2013, 5, 1486–1502. [Google Scholar] [CrossRef] [Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Blythman, H.E.; Casellas, P.; Gros, O.; Gros, P.; Jansen, F.K.; Paolucci, F.; Pau, B.; Vidal, H. Immunotoxins: Hybrid molecules of monoclonal antibodies and a toxin subunit specifically kill tumour cells. Nature 1981, 290, 145–146. [Google Scholar] [CrossRef]
- Pastan, I.; Willingham, M.C.; FitzGerald, D.J.P. Immunotoxins. Cell 1986, 47, 641–648. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Uhr, J.W. Immunotoxins: Redirecting nature’s poisons. Cell 1985, 41, 653–654. [Google Scholar] [CrossRef]
- FitzGerald, D.; Pastan, I. Redirecting Pseudomonas exotoxin. Semin. Cell Biol. 1991, 2, 31–37. [Google Scholar]
- Simeon, R.; Chen, Z. In vitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Weidle, U.H.; Tiefenthaler, G.; Schiller, C.; Weiss, E.H.; Georges, G.; Brinkmann, U. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genom. Proteom. 2014, 11, 25–38. [Google Scholar]
- Sokolova, E.A.; Stremovskiy, O.A.; Zdobnova, T.A.; Balalaeva, I.V.; Deyev, S.M. Recombinant immunotoxin 4D5scFv-PE40 for targeted therapy of HER2-positive tumors. Acta Nat. 2015, 7, 93–96. [Google Scholar] [CrossRef]
- Amoozadeh, S.; Hemmati, M.; Farajollahi, M.M.; Akbari, N.; Tarighi, P. Preparation of Diphtheria and Pseudomonas Exotoxin A Immunotoxins and Evaluation of Their Cytotoxicity Effect on SK-BR-3, BT-474, and MDA-MB-231 Breast Cancer Cell Lines. Cancer Invest. 2019, 37, 546–557. [Google Scholar] [CrossRef]
- Lakayan, D.; Haselberg, R.; Gahoual, R.; Somsen, G.W.; Kool, J. Affinity profiling of monoclonal antibody and antibody-drug-conjugate preparations by coupled liquid chromatography-surface plasmon resonance biosensing. Anal. Bioanal. Chem. 2018, 410, 7837–7848. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, E.A.; Zdobnova, T.A.; Stremovskiy, O.A.; Balalaeva, I.V.; Deyev, S.M. Novel recombinant anti-HER2/neu immunotoxin: Design and antitumor efficiency. Biochemistry 2014, 79, 1376–1381. [Google Scholar] [CrossRef]
- Lee, B.S.; Lee, Y.; Park, J.; Jeong, B.S.; Jo, M.; Jung, S.T.; Yoo, T.H. Construction of an immunotoxin via site-specific conjugation of anti-Her2 IgG and engineered Pseudomonas exotoxin A. J. Biol. Eng. 2019, 13. [Google Scholar] [CrossRef]
- Hoffmann, R.M.; Mele, S.; Cheung, A.; Larcombe-Young, D.; Bucaite, G.; Sachouli, E.; Zlatareva, I.; Morad, H.O.J.; Marlow, R.; McDonnell, J.M.; et al. Rapid conjugation of antibodies to toxins to select candidates for the development of anticancer Antibody-Drug Conjugates (ADCs). Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Sapozhnikov, A.M.; Klinkova, A.V.; Shustova, O.A.; Grechikhina, M.V.; Kilyachus, M.S.; Stremovskiy, O.A.; Kovalenko, E.I.; Deyev, S.M. A Novel Approach to Anticancer Therapy: Molecular Modules Based on the Barnase:Barstar Pair for Targeted Delivery of HSP70 to Tumor Cells. Acta Nat. 2018, 10, 85–91. [Google Scholar] [CrossRef]
- Johannes, L.; Römer, W. Shiga toxins from cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef]
- Engedal, N.; Skotland, T.; Torgersen, M.L.; Sandvig, K. Shiga toxin and its use in targeted cancer therapy and imaging. Microb. Biotechnol. 2011, 4, 32–46. [Google Scholar] [CrossRef]
- Viel, T.; Dransart, E.; Nemati, F.; Henry, E.; Thézé, B.; Decaudin, D.; Lewandowski, D.; Boisgard, R.; Johannes, L.; Tavitian, B. In Vivo Tumor Targeting by the B-Subunit of Shiga Toxin. Mol. Imaging 2008, 7, 7290.2008.00022. [Google Scholar] [CrossRef]
- St. Croix, B.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montegomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef]
- Peters, D.E.; Hoover, B.; Cloud, L.G.; Liu, S.; Molinolo, A.A.; Leppla, S.H.; Bugge, T.H. Comparative toxicity and efficacy of engineered anthrax lethal toxin variants with broad anti-tumor activities. Toxicol. Appl. Pharmacol. 2014, 279, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Scobie, H.M.; Rainey, G.J.A.; Bradley, K.A.; Young, J.A.T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.H.; Liu, S.; Bankston, L.A.; Liddington, R.C.; Leppla, S.H. Selection of anthrax toxin protective antigen variants that discriminate between the cellular receptors TEM8 and CMG2 and achieve targeting of tumor cells. J. Biol. Chem. 2007, 282, 9834–9845. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.; Henri, J.; Roy, K.; Hays, E.; Bauer, M.; Veedu, R.N.; Pouliot, N.; Shigdar, S. EpCAM immunotherapy versus specific targeted delivery of drugs. Cancers 2018, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Melkko, S.; Halin, C.; Borsi, L.; Zardi, L.; Neri, D. An antibody-calmodulin fusion protein reveals a functional dependence between macromolecular isoelectric point and tumor targeting performance. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 1485–1490. [Google Scholar] [CrossRef]
- Gage, E.; Hernandez, M.O.; O’Hara, J.M.; McCarthy, E.A.; Mantis, N.J. Role of the mannose receptor (CD206) in innate immunity to ricin toxin. Toxins 2011, 3, 1131–1145. [Google Scholar] [CrossRef] [Green Version]
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.; Watson, G.J.; Blakey, D.C.; Newell, D.R. Improved Antitumor Effects of Immunotoxins Prepared with Deglycosylated Ricin A-Chain and Hindered Disulfide Linkages. Cancer Res. 1988, 48, 6396–6403. [Google Scholar]
- Trejtnar, F.; Laznicek, M. Analysis of renal handling of radiopharmaceuticals. Q. J. Nucl. Med. 2002, 46, 181–194. [Google Scholar]
- Deyev, S.M.; Waibel, R.; Lebedenko, E.N.; Schubiger, A.P.; Plückthun, A. Design of multivalent complexes using the barnase·barstar module. Nat. Biotechnol. 2003, 21, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef]
- Steiner, D.; Merz, F.W.; Sonderegger, I.; Gulotti-Georgieva, M.; Villemagne, D.; Phillips, D.J.; Forrer, P.; Stumpp, M.T.; Zitt, C.; Binz, H.K. Half-life extension using serum albumin-binding DARPin® domains. Protein Eng. Des. Sel. 2017, 30, 583–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—Recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [Green Version]
- Simmons, B.M.; Stahl, P.D.; Russell, J.H. Mannose receptor-mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for a chain translocation. J. Biol. Chem. 1986, 261, 7912–7920. [Google Scholar] [CrossRef]
- Mantis, N.J.; Farrant, S.A.; Mehta, S. Oligosaccharide Side Chains on Human Secretory IgA Serve as Receptors for Ricin. J. Immunol. 2004, 172, 6838–6845. [Google Scholar] [CrossRef] [Green Version]
- Blakey, D.C.; Watson, G.J.; Knowles, P.P.; Thorpe, P.E. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987, 47, 947–952. [Google Scholar]
- Schmohl, J.U.; Todhunter, D.; Oh, S.; Vallera, D.A. Mutagenic deimmunization of diphtheria toxin for use in biologic drug development. Toxins 2015, 7, 4067–4082. [Google Scholar] [CrossRef] [Green Version]
- Onda, M.; Nagata, S.; FitzGerald, D.J.; Beers, R.; Fisher, R.J.; Vincent, J.J.; Lee, B.; Nakamura, M.; Hwang, J.; Kreitman, R.J.; et al. Characterization of the B Cell Epitopes Associated with a Truncated Form of Pseudomonas Exotoxin (PE38) Used to Make Immunotoxins for the Treatment of Cancer Patients. J. Immunol. 2006, 177, 8822–8834. [Google Scholar] [CrossRef] [Green Version]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar] [CrossRef] [Green Version]
- Onda, M.; Beers, R.; Xiang, L.; Lee, B.; Weldon, J.E.; Kreitman, R.J.; Pastan, I. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc. Natl. Acad. Sci. USA 2011, 108, 5742–5747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Choi, I.K.; Strauss, R.; Richter, M.; Yun, C.O.; Lieber, A. Strategies to increase drug penetration in solid tumors. Front. Oncol. 2013, 3, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilova, O.N.; Shilov, E.S.; Lieber, A.; Deyev, S.M. Disassembling a cancer puzzle: Cell junctions and plasma membrane as targets for anticancer therapy. J. Control. Release 2018, 286, 125–136. [Google Scholar] [CrossRef]
- Ansiaux, R.; Gallez, B. Use of botulinum toxins in cancer therapy. Expert Opin. Investig. Drugs 2007, 16, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.F.; Gallez, B. Surrogate MR markers of response to chemo- or radiotherapy in association with co-treatments: A retrospective analysis of multi-modal studies. Contrast Media Mol. Imaging 2010, 5, 323–332. [Google Scholar] [CrossRef]
- Shilova, O.N.; Proshkina, G.M.; Lebedenko, E.N.; Deyev, S.M. Internalization and Recycling of the HER2 Receptor on Human Breast Adenocarcinoma Cells Treated with Targeted Phototoxic Protein DARPinminiSOG. Acta Nat. 2015, 7, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Shilova, O.N.; Proshkina, G.M.; Ryabova, A.V.; Deyev, S.M.; Petrov, R.V. Cytotoxicity of targeted HER2-specific phototoxins based on flavoprotein miniSOG is determined by the rate of their internalization. Dokl. Biochem. Biophys. 2017, 475, 256–258. [Google Scholar] [CrossRef]
- Kuzichkina, E.O.; Shilova, O.N.; Deyev, S.M. The mechanism of fluorescence quenching of protein photosensitizers based on miniSOG during internalization of the HER2 receptor. Acta Nat. 2018, 10, 87–94. [Google Scholar] [CrossRef]
- Collier, R.J.; Gilliland, D.G.; Lory, S. Structure-activity relationships in diphtheria toxin and exotoxin A from Pseudomonas aeruginosa. Prog. Clin. Biol. Res. 1979, 31, 751–759. [Google Scholar]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. [Google Scholar] [CrossRef]
- Luginbuehl, V.; Meier, N.; Kovar, K.; Rohrer, J. Intracellular drug delivery: Potential usefulness of engineered Shiga toxin subunit B for targeted cancer therapy. Biotechnol. Adv. 2018, 36, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ryou, J.H.; Sohn, Y.K.; Hwang, D.E.; Park, W.Y.; Kim, N.; Heo, W.D.o.; Kim, M.Y.; Kim, H.S. Engineering of bacterial exotoxins for highly efficient and receptor-specific intracellular delivery of diverse cargos. Biotechnol. Bioeng. 2016, 113, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Feni, L.; Neundorf, I. The current role of cell-penetrating peptides in cancer therapy. Adv. Exp. Med. Biol. 2017, 1030, 279–295. [Google Scholar]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Pirie, C.M.; Liu, D.V.; Dane Wittrup, K. Targeted cytolysins synergistically potentiate cytoplasmic delivery of gelonin immunotoxin. Mol. Cancer Ther. 2013, 12, 1774–1782. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.M.; Egorin, M.J. Limited Penetration of Anticancer Drugs through Tumor Tissue. Clin. Cancer Res. 2002, 8, 878–884. [Google Scholar] [PubMed]
- Sokolova, E.; Kutova, O.; Grishina, A.; Pospelov, A.; Guryev, E.; Schulga, A.; Deyev, S.; Balalaeva, I. Penetration efficiency of antitumor agents in ovarian cancer spheroids: The case of recombinant targeted toxin DARPin-LoPE and the chemotherapy drug, doxorubicin. Pharmaceutics 2019, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Tahmasbi Rad, A.; Chen, C.W.; Aresh, W.; Xia, Y.; Lai, P.S.; Nieh, M.P. Combinational Effects of Active Targeting, Shape, and Enhanced Permeability and Retention for Cancer Theranostic Nanocarriers. ACS Appl. Mater. Interfaces 2019, 11, 10505–10519. [Google Scholar] [CrossRef]
- Yan, W.; Leung, S.S.Y.; To, K.K.W. Updates on the use of liposomes for active tumor targeting in cancer therapy. Nanomedicine 2019, 15, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Palazzolo, S.; Bayda, S.; Hadla, M.; Caligiuri, I.; Corona, G.; Toffoli, G.; Rizzolio, F. The Clinical Translation of Organic Nanomaterials for Cancer Therapy: A Focus on Polymeric Nanoparticles, Micelles, Liposomes and Exosomes. Curr. Med. Chem. 2017, 25, 4224–4268. [Google Scholar] [CrossRef]
- van Elk, M.; Murphy, B.P.; Eufrásio-da-Silva, T.; O’Reilly, D.P.; Vermonden, T.; Hennink, P.W.E.; Duffy, G.P.; Ruiz-Hernández, E. Nanomedicines for advanced cancer treatments: Transitioning towards responsive systems. Int. J. Pharm. 2016, 515, 132–164. [Google Scholar] [CrossRef]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: Nanosized vehicles for drug delivery in cancer. Trends Pharmacol. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Zelepukin, I.V.; Yaremenko, A.V.; Shipunova, V.O.; Babenyshev, A.V.; Balalaeva, I.V.; Nikitin, P.I.; Deyev, S.M.; Nikitin, M.P. Nanoparticle-based drug delivery: Via RBC-hitchhiking for the inhibition of lung metastases growth. Nanoscale 2019, 11, 1636–1646. [Google Scholar] [CrossRef]
- Nikitin, M.P.; Zelepukin, I.V.; Shipunova, V.O.; Sokolov, I.L.; Deyev, S.M.; Nikitin, P.I. Enhancement of the blood-circulation time and performance of nanomedicines via the forced clearance of erythrocytes. Nat. Biomed. Eng. 2020, 4, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, D.P.; Heath, T.D. Liposome-mediated delivery of ribosome inactivating proteins to cells in vitro. BBA Biomembr. 1982, 690, 224–230. [Google Scholar] [CrossRef]
- Jansons, V.K.; Panzner, E.A. Liposomes as a means to introduce fragment A of diphtheria toxin into cells. BBA Biomembr. 1983, 735, 433–437. [Google Scholar] [CrossRef]
- Collins, D.; Huang, L. Cytotoxicity of Diphtheria Toxin A Fragment to Toxin-resistant Murine Cells Delivered by pH-sensitive Immunoliposomes. Cancer Res. 1987, 47, 735–739. [Google Scholar] [PubMed]
- Vingerhoeds, M.H.; Steerenberg, P.A.; Hendriks, J.J.G.W.; Crommelin, D.J.A.; Storm, G. Targeted delivery of diphtheria toxin via immunoliposomes: Efficient antitumor activity in the presence of inactivating anti-diphtheria toxin antibodies. FEBS Lett. 1996, 395, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Pai, L.H.; Bookman, M.A.; Ozols, R.F.; Young, R.C.; Smith, J.W.; Longo, D.L.; Gould, B.; Frankel, A.; McClay, E.F.; Howell, S.; et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J. Clin. Oncol. 1991, 9, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Provoda, C.J.; Stier, E.M.; Lee, K.D. Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 2003, 278, 35102–35108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janicki, J.; Chancellor, M.; Kaufman, J.; Gruber, M.; Chancellor, D. Potential Effect of Liposomes and Liposome-Encapsulated Botulinum Toxin and Tacrolimus in the Treatment of Bladder Dysfunction. Toxins 2016, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-C.; Tyagi, P.; Huang, C.-C.; Yoshimura, N.; Wu, M.; Kaufman, J.; Chancellor, M.B.; Rd, T.P.; Hsiang, N.S.; Hsien, K. Urodynamic and Immunohistochemical Evaluation of Intravesical Botulinum Toxin A Delivery Using Liposomes. J. Urol. 2009, 182, 786–792. [Google Scholar] [CrossRef]
- Kuo, H.C.; Liu, H.T.; Chuang, Y.C.; Birder, L.A.; Chancellor, M.B. Pilot study of liposome-encapsulated onabotulinumtoxinA for patients with overactive bladder: A single-center study. Eur. Urol. 2014, 65, 1117–1124. [Google Scholar] [CrossRef]
- Yaghini, E.; Dondi, R.; Edler, K.J.; Loizidou, M.; Macrobert, A.J.; Eggleston, I.M. Codelivery of a cytotoxin and photosensitiser: Via a liposomal nanocarrier: A novel strategy for light-triggered cytosolic release. Nanoscale 2018, 10, 20366–20376. [Google Scholar] [CrossRef] [Green Version]
- de Matos, M.B.C.; Beztsinna, N.; Heyder, C.; Fens, M.H.A.M.; Mastrobattista, E.; Schiffelers, R.M.; Leneweit, G.; Kok, R.J. Thermosensitive liposomes for triggered release of cytotoxic proteins. Eur. J. Pharm. Biopharm. 2018, 132, 211–221. [Google Scholar] [CrossRef]
- Gao, J.; Zhong, W.; He, J.; Li, H.; Zhang, H.; Zhou, G.; Li, B.; Lu, Y.; Zou, H.; Kou, G.; et al. Tumor-targeted PE38KDEL delivery via PEGylated anti-HER2 immunoliposomes. Int. J. Pharm. 2009, 374, 145–152. [Google Scholar] [CrossRef]
- Gao, J.; Kou, G.; Wang, H.; Chen, H.; Li, B.; Lu, Y.; Zhang, D.; Wang, S.; Hou, S.; Qian, W.; et al. PE38KDEL-loaded anti-HER2 nanoparticles inhibit breast tumor progression with reduced toxicity and immunogenicity. Breast Cancer Res. Treat. 2009, 115, 29–41. [Google Scholar] [CrossRef]
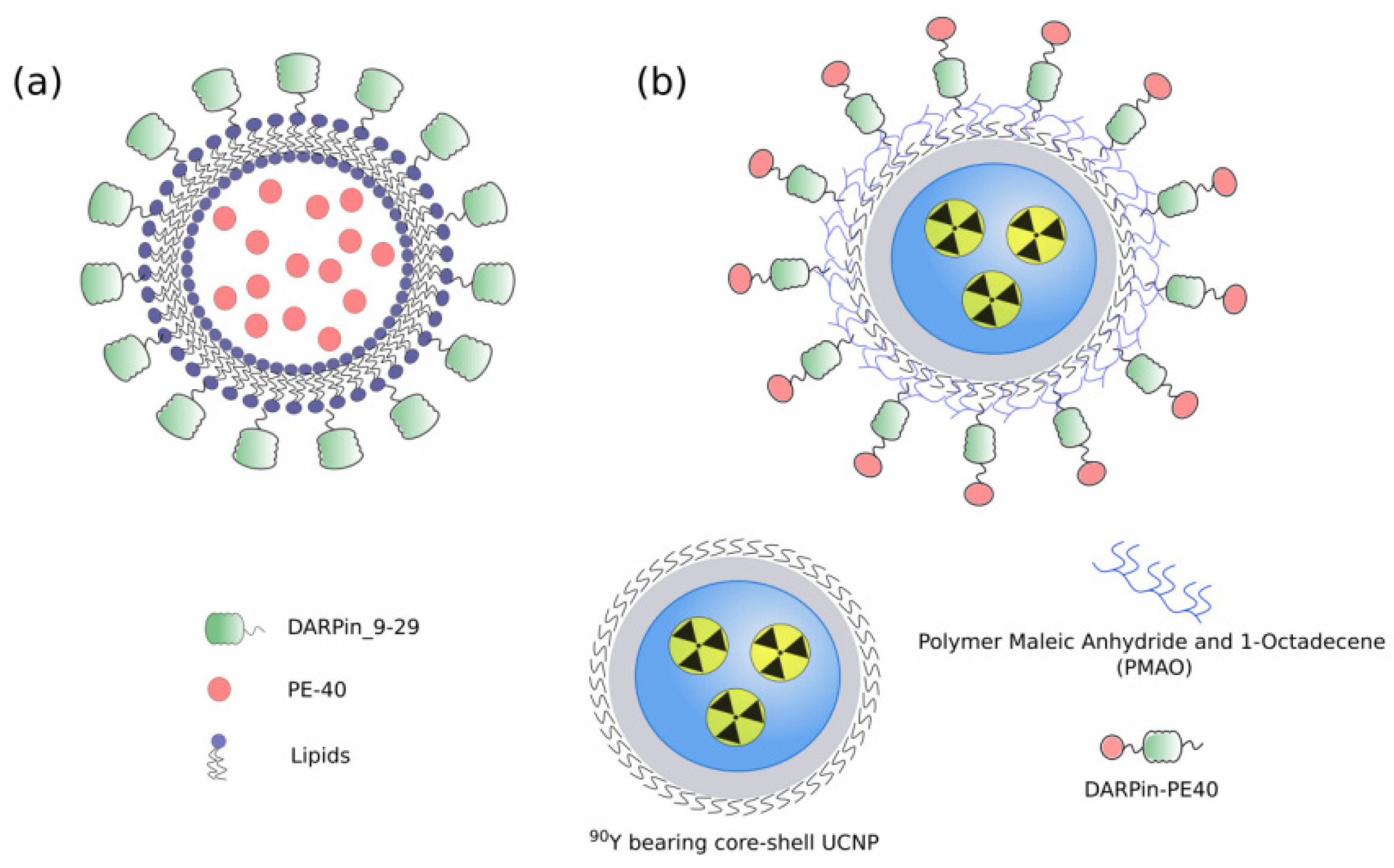
- Deyev, S.; Proshkina, G.; Baryshnikova, O.; Ryabova, A.; Avishai, G.; Katrivas, L.; Giannini, C.; Levi-Kalisman, Y.; Kotlyar, A. Selective staining and eradication of cancer cells by protein-carrying DARPin-functionalized liposomes. Eur. J. Pharm. Biopharm. 2018, 130, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, E.; Proshkina, G.; Kutova, O.; Shilova, O.; Ryabova, A.; Schulga, A.; Stremovskiy, O.; Zdobnova, T.; Balalaeva, I.; Deyev, S. Recombinant targeted toxin based on HER2-specific DARPin possesses a strong selective cytotoxic effect in vitro and a potent antitumor activity in vivo. J. Control. Release 2016, 233, 48–56. [Google Scholar] [CrossRef]
- Shramova, E.; Proshkina, G.; Shipunova, V.; Ryabova, A.; Kamyshinsky, R.; Konevega, A.; Schulga, A.; Konovalova, E.; Telegin, G.; Deyev, S. Dual targeting of cancer cells with darpin-based toxins for overcoming tumor escape. Cancers 2020, 12, 3014. [Google Scholar] [CrossRef] [PubMed]
- Shipunova, V.O.; Shramova, E.I.; Shulga, A.A.; Shilova, M.V.; Deyev, S.M.; Proshkina, G.M. Delivery of barnase to cells in liposomes functionalized by HER2-specific DARPin module. Russ. J. Biorgan. Chem. 2020, 46, 1156–1161. [Google Scholar] [CrossRef]
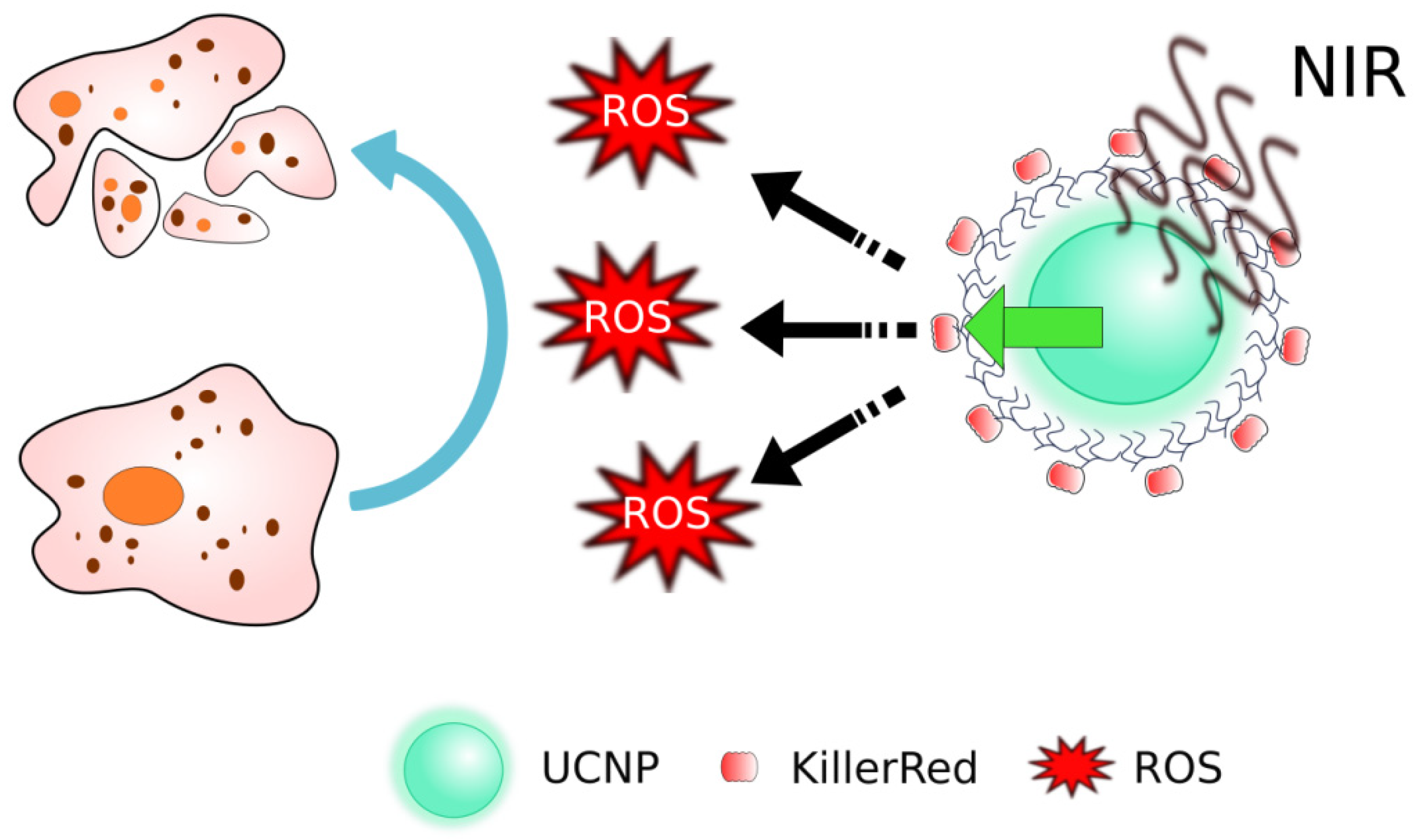
- Guryev, E.L.; Volodina, N.O.; Shilyagina, N.Y.; Gudkov, S.V.; Balalaeva, I.V.; Volovetskiy, A.B.; Lyubeshkin, A.V.; Sen’, A.V.; Ermilov, S.A.; Vodeneev, V.A.; et al. Radioactive (90Y) upconversion nanoparticles conjugated with recombinant targeted toxin for synergistic nanotheranostics of cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 9690–9695. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Gao, J.; Lu, Y.; Kou, G.; Zhang, H.; Fan, L.; Sun, Z.; Guo, Y.; Zhong, Y. Preparation and characterization of PE38KDEL-loaded anti-HER2 nanoparticles for targeted cancer therapy. J. Control. Release 2008, 128, 209–216. [Google Scholar] [CrossRef]
- Gholami, N.; Cohan, R.A.; Razavi, A.; Bigdeli, R.; Dashbolaghi, A.; Asgary, V. Cytotoxic and apoptotic properties of a novel nano-toxin formulation based on biologically synthesized silver nanoparticle loaded with recombinant truncated pseudomonas exotoxin A. J. Cell. Physiol. 2020, 235, 3711–3720. [Google Scholar] [CrossRef]
- Bhowmik, T.; Saha, P.P.; Dasgupta, A.; Gomes, A. Antileukemic potential of PEGylated gold nanoparticle conjugated with protein toxin (NKCT1) isolated from Indian cobra (Naja kaouthia) venom. Cancer Nanotechnol. 2013, 4, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Bhowmik, T.; Saha, P.P.; Sarkar, A.; Gomes, A. Evaluation of cytotoxicity of a purified venom protein from Naja kaouthia (NKCT1) using gold nanoparticles for targeted delivery to cancer cell. Chem. Biol. Interact. 2017, 261, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Shipunova, V.O.; Komedchikova, E.N.; Kotelnikova, P.A.; Zelepukin, I.V.; Schulga, A.A.; Proshkina, G.M.; Shramova, E.I.; Kutscher, H.L.; Telegin, G.B.; Kabashin, A.V.; et al. Dual Regioselective Targeting the Same Receptor in Nanoparticle-Mediated Combination Immuno/Chemotherapy for Enhanced Image-Guided Cancer Treatment. ACS Nano 2020, 14, 12781–12795. [Google Scholar] [CrossRef]
- Sánchez-García, L.; Serna, N.; Álamo, P.; Sala, R.; Céspedes, M.V.; Roldan, M.; Sánchez-Chardi, A.; Unzueta, U.; Casanova, I.; Mangues, R.; et al. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release 2018, 274, 81–92. [Google Scholar] [CrossRef]
- Serna, N.; Álamo, P.; Ramesh, P.; Vinokurova, D.; Sánchez-García, L.; Unzueta, U.; Gallardo, A.; Céspedes, M.V.; Vázquez, E.; Villaverde, A.; et al. Nanostructured toxins for the selective destruction of drug-resistant human CXCR4+ colorectal cancer stem cells. J. Control. Release 2020, 320, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Falgàs, A.; Pallarès, V.; Serna, N.; Sánchez-García, L.; Sierra, J.; Gallardo, A.; Alba-Castellón, L.; Álamo, P.; Unzueta, U.; Villaverde, A.; et al. Selective delivery of T22-PE24-H6 to CXCR4+ diffuse large B-cell lymphoma cells leads to wide therapeutic index in a disseminated mouse model. Theranostics 2020, 10, 5169–5180. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef] [Green Version]
- Fodstad, Ø.; Kvalheim, G.; Godal, A.; Lotsberg, J.; Aamdal, S.; HOst, H.; Pihl, A. Phase I Study of the Plant Protein Ricin. Cancer Res. 1984, 44, 862–865. [Google Scholar]
- Li, C.; Yan, R.; Yang, Z.; Wang, H.; Zhang, R.; Chen, H.; Wang, J. BCMab1-Ra, a novel immunotoxin that BCMab1 antibody coupled to Ricin A chain, can eliminate bladder tumor. Oncotarget 2017, 8, 46704–46705. [Google Scholar] [CrossRef] [Green Version]
- Godal, A.; Fodstad, O.Y.; Pihl, A. Studies On The Mechanism Of Action Of Abrin-9.2.27 Immunotoxin In Human Melanoma Cell Lines. Cancer Res. 1987, 47, 6243–6247. [Google Scholar]
- Wawrzynczak, E.J.; Zangemeister-Wittke, U.; Waibel, R.; Henry, R.V.; Parnell, G.D.; Cumber, A.J.; Jones, M.; Stahel, R.A. Molecular and biological properties of an abrin a chain immunotoxin designed for therapy of human small cell lung cancer. Br. J. Cancer 1992, 66, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J. Pathol. 1987, 151, 221–229. [Google Scholar] [CrossRef]
- Edelweiss, E.; Balandin, T.G.; Ivanova, J.L.; Lutsenko, G.V.; Leonova, O.G.; Popenko, V.I.; Sapozhnikov, A.M.; Deyev, S.M. Barnase as a new therapeutic agent triggering apoptosis in human cancer cells. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balandin, T.G.; Edelweiss, E.; Andronova, N.V.; Treshalina, E.M.; Sapozhnikov, A.M.; Deyev, S.M. Antitumor activity and toxicity of anti-HER2 immunoRNase scFv 4D5-dibarnase in mice bearing human breast cancer xenografts. Invest. New Drugs 2011, 29, 22–32. [Google Scholar] [CrossRef]
- Mironova, N.L.; Petrushanko, I.Y.; Patutina, O.A.; Sen’kova, A.V.; Simonenko, O.V.; Mitkevich, V.A.; Markov, O.V.; Zenkova, M.A.; Makarov, A.A. Ribonuclease binase inhibits primary tumor growth and metastases via apoptosis induction in tumor cells. Cell Cycle 2013, 12, 2120–2131. [Google Scholar] [CrossRef]
- Bachran, C.; Leppla, S.H. Tumor targeting and drug delivery by anthrax toxin. Toxins 2016, 8, 197. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Lu, Y.; Zhang, R.; Care, A.; Ortega, T.A.; Deyev, S.M.; Qian, Y.; Zvyagin, A.V. Deep-penetrating photodynamic therapy with KillerRed mediated by upconversion nanoparticles. Acta Biomater. 2017, 51, 461–470. [Google Scholar] [CrossRef]
- Yuan, M.; Liu, C.; Li, J.; Ma, W.; Yu, X.; Zhang, P.; Ji, Y. The effects of photodynamic therapy on leukemia cells mediated by KillerRed, a genetically encoded fluorescent protein photosensitizer. BMC Cancer 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Kurschus, F.C.; Jenne, D.E. Delivery and therapeutic potential of human granzyme B. Immunol. Rev. 2010, 235, 159–171. [Google Scholar] [CrossRef]
- Walev, I.; Bhakdi, S.C.; Hofmann, F.; Djonder, N.; Valeva, A.; Aktories, K.; Bhakdi, S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA 2001, 98, 3185–3190. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, A.; Tsukuda, M.; Kondo, N.; Ishiguro, Y.; Kimura, M.; Fujita, K.; Takahashi, H.; Matsuda, H. Examination of the optimal condition on the in vitro sensitivity to telomelysin in head and neck cancer cell lines. Auris Nasus Larynx 2011, 38, 589–599. [Google Scholar] [CrossRef]
- Shafiee, F.; Aucoin, M.G.; Jahanian-Najafabadi, A. Targeted Diphtheria Toxin-Based Therapy: A Review Article. Front. Microbiol. 2019, 10, 2340. [Google Scholar] [CrossRef]
- Soler-Rodriguez, A.M.; Uhr, J.W.; Richardson, J.; Vitetta, E.S. The toxicity of chemically deglycosylated ricin A-chain in mice. Int. J. Immunopharmacol. 1992, 14, 281–291. [Google Scholar] [CrossRef]
- Proulx, F.Ç.; Seidman, E.G.; Karpman, D. Pathogenesis of Shiga toxin—Associated hemolytic uremic syndrome. Pediatr. Res. 2001, 50, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Palermo, M.S.; Exeni, R.A.; Fernández, G.C. Hemolytic uremic syndrome: Pathogenesis and update of interventions. Expert Rev. Anti. Infect. Ther. 2009, 7, 697–707. [Google Scholar] [CrossRef]
- Newton, D.L.; Ilercil, O.; Laske, D.W.; Oldfield, E.; Rybak, S.M.; Youle, R.J. Cytotoxic ribonuclease chimeras. Targeted tumoricidal activity in vitro and in vivo. J. Biol. Chem. 1992, 267, 19572–19578. [Google Scholar] [CrossRef]
- Haigis, M.C.; Kurten, E.L.; Raines, R.T. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 2003, 31, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Schubert, R.L.; Bugge, T.H.; Leppla, S.H. Anthrax toxin: Structures, functions and tumour targeting. Expert Opin. Biol. Ther. 2003, 3, 843–853. [Google Scholar] [CrossRef]
- Feld, G.K.; Brown, M.J.; Krantz, B.A. Ratcheting up protein translocation with anthrax toxin. Protein Sci. 2012, 21, 606–624. [Google Scholar] [CrossRef] [Green Version]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; Van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003, 160, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Feld, G.K.; Thoren, K.L.; Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Greenberg, S.G.; Williams, E.R.; Krantz, B.A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations in eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [Green Version]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Herlyn, M.; Smalley, K.S.; Bromberg-White, J.L.; Duesbery, N.S.; Frankel, A.E. Cytotoxicity of the matrix metalloproteinase-activated anthrax lethal toxin is dependent on gelatinase expression and B-RAF status in human melanoma cells. Mol. Cancer Ther. 2008, 7, 1218–1226. [Google Scholar] [CrossRef] [Green Version]
- Abi-Habib, R.J.; Urieto, J.O.; Liu, S.; Leppla, S.H.; Duesbery, N.S.; Frankel, A.E. BRAF status and mitogen-activated protein/extracellular signal regulated kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol. Cancer Ther. 2005, 4, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Abi-Habib, R.J.; Singh, R.; Leppla, S.H.; Greene, J.J.; Ding, Y.; Berghuis, B.; Duesbery, N.S.; Frankel, A.E. Systemic anthrax lethal toxin therapy produces regressions of subcutaneous human melanoma tumors in athymic nude mice. Clin. Cancer Res. 2006, 12, 7437–7443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wang, H.; Currie, B.M.; Molinolo, A.; Leung, H.J.; Moayeri, M.; Basile, J.R.; Alfano, R.W.; Gutkind, J.S.; Frankel, A.E.; et al. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J. Biol. Chem. 2008, 283, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor Cell-selective Cytotoxicity of Matrix Metalloproteinase-activated Anthrax Toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar] [PubMed]
- Liu, S.; Bugge, T.H.; Leppla, S.H. Targeting of Tumor Cells by Cell Surface Urokinase Plasminogen Activator-dependent Anthrax Toxin. J. Biol. Chem. 2001, 276, 17976–17984. [Google Scholar] [CrossRef] [Green Version]
- Phillips, D.D.; Fattah, R.J.; Crown, D.; Zhang, Y.; Liu, S.; Moayeri, M.; Fischer, E.R.; Hansen, B.T.; Ghirlando, R.; Nestorovich, E.M.; et al. Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J. Biol. Chem. 2013, 288, 9058–9065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroras, N.; Williamson, L.C.; Lepplas, S.H.; Halpernnll, J.L. Cytotoxic Effects of a Chimeric Protein Consisting of Tetanus Toxin Light Chain and Anthrax Toxin Lethal Factor in Non-neuronal Cells*. J. Biol. Chem. 1994, 269, 26165–26171. [Google Scholar] [CrossRef]
- Arora, N.; Lepplas, S.H. Residues 1-254 of Anthrax Toxin Lethal Factor Are Sufficient to Cause Cellular Uptake of Fused Polypeptides*. J. Biol. Chem. 1993, 268, 3334–3341. [Google Scholar] [CrossRef]
- Milne, J.C.; Blanket, S.R.; Hanna, P.C.; Collier, R.J. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol. Microbiol. 1995, 15, 661–666. [Google Scholar] [CrossRef]
- Arora, N.; Leppla, S.H. Fusions of anthrax toxin lethal factor with Shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect. Immun. 1994, 62, 4955–4961. [Google Scholar] [CrossRef] [Green Version]
- Mechaly, A.; McCluskey, A.J.; John Collier, R. Changing the receptor specificity of anthrax toxin. MBio 2012, 3, e00088-12. [Google Scholar] [CrossRef] [Green Version]
- McCluskey, A.J.; Olive, A.J.; Starnbach, M.N.; Collier, R.J. Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol. 2013, 7, 440–451. [Google Scholar] [CrossRef]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Lovell, J.F.; Liu, T.W.B.; Chen, J.; Zheng, G. Activatable photosensitizers for imaging and therapy. Chem. Rev. 2010, 110, 2839–2857. [Google Scholar] [CrossRef] [PubMed]
- Theodossiou, T.A.; Gonçalves, A.R.; Yannakopoulou, K.; Skarpen, E.; Berg, K. Photochemical internalization of tamoxifens transported by a “trojan-horse” nanoconjugate into breast-cancer cell lines. Angew. Chemie 2015, 54, 4885–4889. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2012, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Mallidi, S.; Anbil, S.; Bulin, A.L.; Obaid, G.; Ichikawa, M.; Hasan, T. Beyond the barriers of light penetration: Strategies, perspectives and possibilities for photodynamic therapy. Theranostics 2016, 6, 2458–2487. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, K.; Matsuda, T.; Sakai, N.; Fu, D.; Noda, M.; Uchiyama, S.; Kotera, I.; Arai, Y.; Horiuchi, M.; Fukui, K.; et al. SuperNova, a monomeric photosensitizing fluorescent protein for chromophore-assisted light inactivation. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, X.; Lev-ram, V.; Deerinck, T.J.; Qi, Y.; Ramko, E.B.; Michael, W.; Jin, Y.; Ellisman, M.H.; Tsien, R.Y. A Genetically Encoded Tag for Correlated Light and Electron Microscopy of Intact Cells, Tissues, and Organisms. PLoS Biol. 2011, 9, e1001041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-González, R.; Cortajarena, A.L.; Mejias, S.H.; Agut, M.; Nonell, S.; Flors, C. Singlet oxygen generation by the genetically encoded tag minisog. J. Am. Chem. Soc. 2013, 135, 9564–9567. [Google Scholar] [CrossRef]
- Pimenta, F.M.; Jensen, R.L.; Breitenbach, T.; Etzerodt, M.; Ogilby, P.R. Oxygen-dependent photochemistry and photophysics of “miniSOG,” a protein-encased flavin. Photochem. Photobiol. 2013, 89, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Bulina, M.E.; Lukyanov, K.A.; Britanova, O.V.; Onichtchouk, D.; Lukyanov, S.; Chudakov, D.M. Chromophore-assisted light inactivation (CALI) using the phototoxic fluorescent protein KillerRed. Nat. Protoc. 2006, 1, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Ryumina, A.P.; Serebrovskaya, E.O.; Shirmanova, M.V.; Snopova, L.B.; Kuznetsova, M.M.; Turchin, I.V.; Ignatova, N.I.; Klementieva, N.V.; Fradkov, A.F.; Shakhov, B.E.; et al. Flavoprotein miniSOG as a genetically encoded photosensitizer for cancer cells. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5059–5067. [Google Scholar] [CrossRef]
- Ryumina, A.P.; Serebrovskaya, E.O.; Staroverov, D.B.; Zlobovskaya, O.A.; Shcheglov, A.S.; Lukyanov, S.A.; Lukyanov, K.A. Lysosome-associated minisog as a photosensitizer for Mammalian cells. Biotechniques 2016, 61, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.-X.; Li, Y.-C.; Lu, H.-M.; Sung, H.-W. A genetically-encoded KillerRed protein as an intrinsically generated photosensitizer for photodynamic therapy. Biomaterials 2014, 35, 500–508. [Google Scholar] [CrossRef]
- Serebrovskaya, E.O.; Ryumina, A.P.; Boulina, M.E.; Shirmanova, M.V.; Zagaynova, E.V.; Bogdanova, E.A.; Lukyanov, S.A.; Lukyanov, K.A. Phototoxic effects of lysosome-associated genetically encoded photosensitizer KillerRed. J. Biomed. Opt. 2013, 19, 071403. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.B.; Garren, E.J.; Shu, X.; Tsien, R.Y.; Jin, Y. Photo-inducible cell ablation in Caenorhabditis elegans using the genetically encoded singlet oxygen generating protein miniSOG. Proc. Natl. Acad. Sci. USA 2012, 109, 7499–7504. [Google Scholar] [CrossRef] [Green Version]
- Leinwand, S.G.; Chalasani, S.H. Neuropeptide signaling remodels chemosensory circuit composition in Caenorhabditis elegans. Nat. Neurosci. 2013, 16, 1461–1467. [Google Scholar] [CrossRef] [Green Version]
- Fry, A.L.; Laboy, J.T.; Norman, K.R. VAV-1 acts in a single interneuron to inhibit motor circuit activity in Caenorhabditis elegans. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Chisholm, A.D. Highly efficient optogenetic cell ablation in C. Elegans using membrane-targeted miniSOG. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
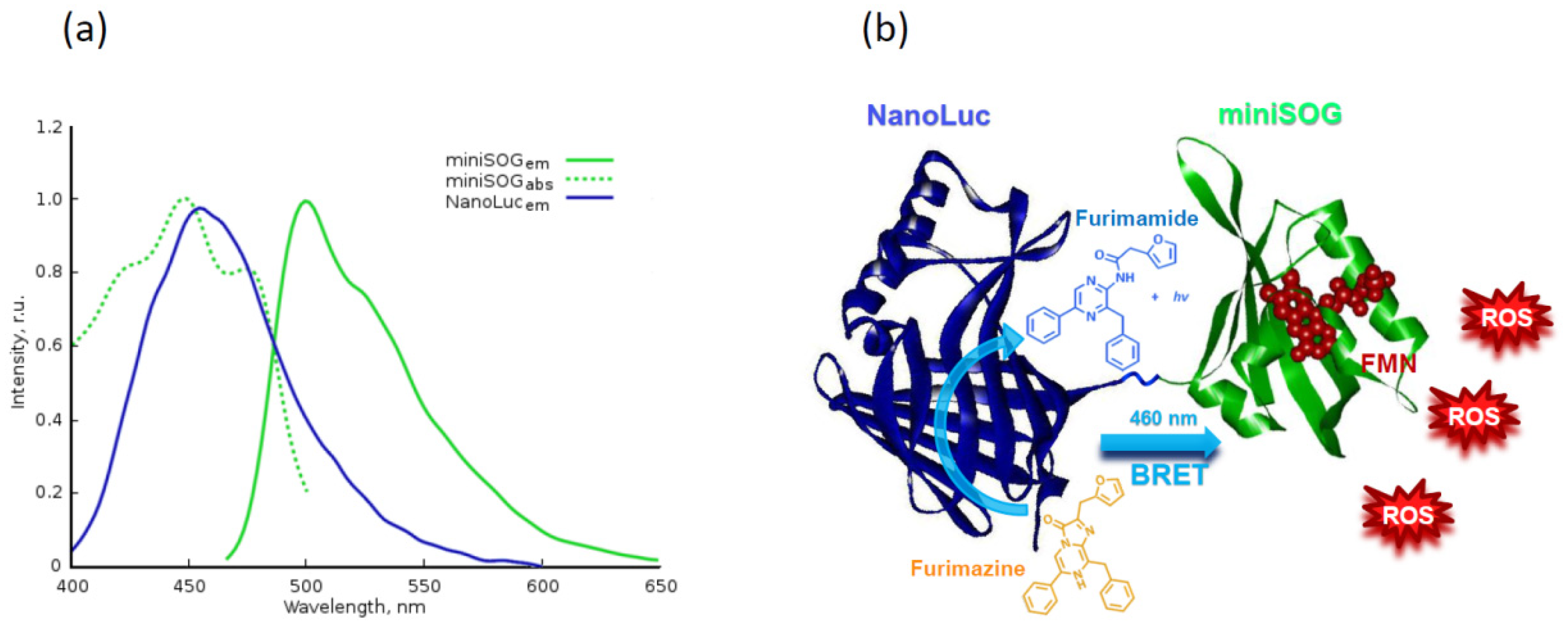
- Shramova, E.I.; Proshkina, G.M.; Chumakov, S.P.; Khodarovich, Y.M.; Deyev, S.M. Flavoprotein miniSOG Cytotoxisity Can Be Induced By Bioluminescence Resonance Energy Transfer. Acta Nat. 2016, 8, 118–125. [Google Scholar] [CrossRef]
- Proshkina, G.M.; Shramova, E.I.; Shilova, O.N.; Ryabova, A.V.; Deyev, S.M. Phototoxicity of flavoprotein miniSOG induced by bioluminescence resonance energy transfer in genetically encoded system NanoLuc-miniSOG is comparable with its LED-excited phototoxicity. J. Photochem. Photobiol. B Biol. 2018, 188, 107–115. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; MacHleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef]
- Shramova, E.I.; Proshkina, G.M.; Deyev, S.M. Bioluminescence system based on luciferase NanoLuc and flavoprotein miniSOG for photodynamic therapy of deep tissues. In Abstract Book, Proceedings of the 45th Annual Congress of the International Society of Oncology and Biomarkers, Hambrug, Germany, 24–27 November 2018; SAGE: Newbury Park, CA, USA, 2019; p. 52. [Google Scholar]
- Cullen, S.P.; Martin, S.J. Mechanisms of granule-dependent killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, D.; Lieberman, J. Death by a thousand cuts: Granzyme pathways of programmed cell death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef] [Green Version]
- Dälken, B.; Giesübel, U.; Knauer, S.K.; Wels, W.S. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006, 13, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderluh, G.; Kisovec, M.; Krasevec, N.; Gilbert, R.J.C. Distribution of MACPF/CDC proteins. Subcell. Biochem. 2014, 80, 7–10. [Google Scholar] [CrossRef]
- Schnupf, P.; Portnoy, D.A. Listeriolysin O: A phagosome-specific lysin. Microbes Infect. 2007, 9, 1176–1187. [Google Scholar] [CrossRef]
- Geoffroy, C.; Gaillard, J.L.; Alouf, J.E.; Berche, P. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect. Immun. 1987, 55, 1641–1646. [Google Scholar] [CrossRef] [Green Version]
- Ansiaux, R.; Baudelet, C.; Cron, G.O.; Segers, J.; Dessy, C.; Martinive, P.; De Wever, J.; Verrax, J.; Wauthier, V.; Beghein, N.; et al. Botulinum toxin potentiates cancer radiotherapy and chemotherapy. Clin. Cancer Res. 2006, 12, 1276–1283. [Google Scholar] [CrossRef] [Green Version]
- Cron, G.O.; Beghein, N.; Ansiaux, R.; Martinive, P.; Feron, O.; Gallez, B. 19F NMR in vivo spectroscopy reflects the effectiveness of perfusion-enhancing vascular modifiers for improving gemcitabine chemotherapy. Magn. Reson. Med. 2008, 59, 19–27. [Google Scholar] [CrossRef]
- Alipour, M.; Pucaj, K.; Smith, M.G.; Suntres, Z.E. Toxicity of ricin toxin a chain in rats. Drug Chem. Toxicol. 2013, 36, 224–230. [Google Scholar] [CrossRef]
- Sokolova, E.A.; Shilova, O.N.; Kiseleva, D.V.; Schulga, A.A.; Balalaeva, I.V.; Deyev, S.M. HER2-Specific Targeted Toxin DARPin-LoPE: Immunogenicity and Antitumor Effect on Intraperitoneal Ovarian Cancer Xenograft Model. Int. J. Mol. Sci. 2019, 20, 2399. [Google Scholar] [CrossRef] [Green Version]
- Mirkasymov, A.B.; Zelepukin, I.V.; Nikitin, P.I.; Nikitin, M.P.; Deyev, S.M. In vivo blockade of mononuclear phagocyte system with solid nanoparticles: Efficiency and affecting factors. J. Control. Release 2021, 330, 111–118. [Google Scholar] [CrossRef]
- Kondo, T.; FitzGerald, D.; Chaudhary, V.K.; Adhya, S.; Pastan, I. Activity of immunotoxins constructed with modified Pseudomonas exotoxin A lacking the cell recognition domain. J. Biol. Chem. 1988, 263, 9470–9475. [Google Scholar] [CrossRef]
- Batra, J.K.; Kasprzyk, P.G.; Bird, R.E.; Pastan, I.; King, C.R. Recombinant anti-erbB2 immunotoxins containing Pseudomonas exotoxin. Proc. Natl. Acad. Sci. USA 1992, 89, 5867–5871. [Google Scholar] [CrossRef] [Green Version]
- Proshkina, G.M.; Kiseleva, D.V.; Shilova, O.N.; Ryabova, A.V.; Shramova, E.I.; Stremovskiy, O.A.; Deyev, S.M. Bifunctional Toxin DARP-LoPE Based on the Her2-Specific Innovative Module of a Non-Immunoglobulin Scaffold as a Promising Agent for Theranostics. Mol. Biol. 2017, 51, 865–873. [Google Scholar] [CrossRef]
- Hassan, R.; Sharon, E.; Thomas, A.; Zhang, J.; Ling, A.; Miettinen, M.; Kreitman, R.J.; Steinberg, S.M.; Hollevoet, K.; Pastan, I. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen. Cancer 2014, 120, 3311–3319. [Google Scholar] [CrossRef]
- FitzGerald, D.J.; Wayne, A.S.; Kreitman, R.J.; Pastan, I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res. 2011, 71, 6300–6309. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [Green Version]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; FitzGerald, D.J.P.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar] [CrossRef] [Green Version]
- Kreitman, R.J.; Pastan, I. Development of recombinant immunotoxins for hairy cell leukemia. Biomolecules 2020, 10, 1140. [Google Scholar] [CrossRef]
- Weldon, J.E.; Xiang, L.; Chertov, O.; Margulies, I.; Kreitman, R.J.; Fitzgerald, D.J.; Pastan, I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009, 113, 3792–3800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahaf, N.I.; Lang, A.E.; Kaiser, L.; Fichter, C.D.; Lassmann, S.; McCluskey, A.; Augspach, A.; Aktories, K.; Schmidt, G. Targeted delivery of an ADP-ribosylating bacterial toxin into cancer cells. Sci. Rep. 2017, 7, 41252. [Google Scholar] [CrossRef] [Green Version]
- Loftis, A.R.; Santos, M.S.; Truex, N.L.; Biancucci, M.; Satchell, K.J.F.; Pentelute, B.L. Anthrax Protective Antigen Retargeted with Single-Chain Variable Fragments Delivers Enzymes to Pancreatic Cancer Cells. ChemBioChem 2020, 21, 2772–2776. [Google Scholar] [CrossRef] [PubMed]
- Allahyari, H.; Heidari, S.; Ghamgosha, M.; Saffarian, P.; Amani, J. Immunotoxin: A new tool for cancer therapy. Tumor Biol. 2017, 39, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antignani, A.; Ho, E.C.H.; Bilotta, M.T.; Qiu, R.; Sarnvosky, R.; Fitzgerald, D.J. Targeting receptors on cancer cells with protein toxins. Biomolecules 2020, 10, 1331. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Details | Examples | References |
|---|---|---|---|
| eEF2 inactivation | ADP-ribosylates elongation factor 2 (eEF2) and halt protein synthesis at the elongation step | Pseudomonas exotoxin A (PE, ETA) | [62,106] |
| Diphtheria toxin (DT) | [12,83] | ||
| Ribosome inactivation | N-glycosidase depurinates a critical adenine in 28S rRNA, which results in the inability of the ribosome to bind elongation factor 2, thereby blocking protein translation | Ricin | [63,107,108] |
| Shiga toxin (Stx) | [30] | ||
| Abrin | [109,110,111] | ||
| RNA degradation | Nonspecific RNA cleavage blocks protein synthesis and leads to apoptosis | Barnase | [112,113] |
| Binase | [114] | ||
| Cell signaling disruption | The cleavages of the MAP kinase family members leading to their inactivation; uncontrolled conversion of ATP to cAMP | Anthrax toxin | [115] |
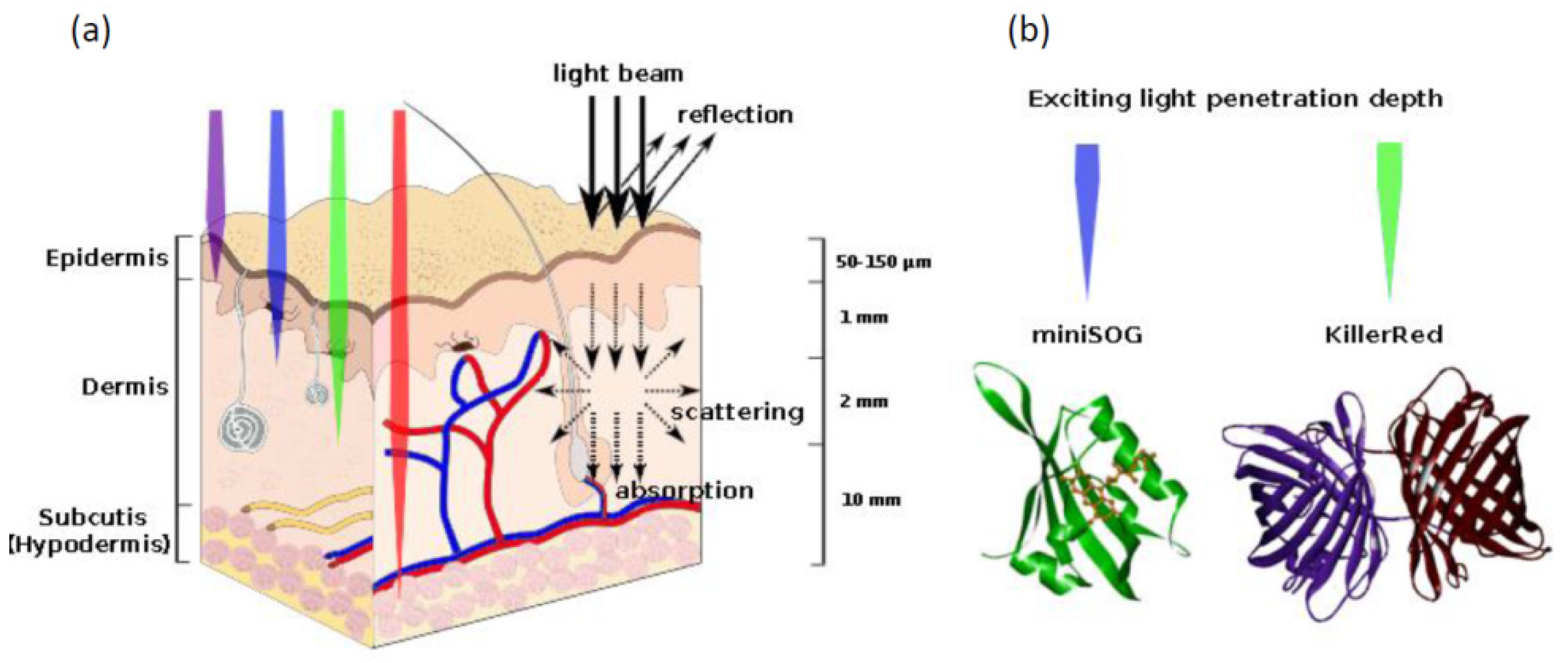
| Photoinduced ROS production | The proteins absorb exciting light and produce reactive oxygen species | KillerRed | [116,117] |
| miniSOG | [6] | ||
| Direct apoptosis induction | Effector caspases cleavage | Granzyme B | [118] |
| Enhanced diffusion of anticancer drug | Vascular network modulation | Botulinum neurotoxin | [57,58] |
| Pore formation for better intracellular delivery | Listeriolysin O | [68,85] | |
| Streptolysin-O | [119,120] |
| Strategy Used for Side Toxicity Reduction | Principle | References |
|---|---|---|
| Impairment of natural tropism | Removing the natural targeting domains of AB toxins | [35] |
| Introduction of point mutations attenuating the target binding | [145] | |
| Construction of miniaturized toxin variants | Deletion of protein parts not directly involved in toxin mechanism of action to reduce any non-specific interaction and immunogenicity | [52,53,178] |
| Tumor-specific activation of a toxin | The replacement of furin cleavage site to tumor-specific proteases cleavage sites (MMP, uPA) | [138,139,140] |
| RES cells inactivation | Macrophages blockade decreasing toxic nanoparticles uptake | [78,179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shilova, O.; Shramova, E.; Proshkina, G.; Deyev, S. Natural and Designed Toxins for Precise Therapy: Modern Approaches in Experimental Oncology. Int. J. Mol. Sci. 2021, 22, 4975. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094975
Shilova O, Shramova E, Proshkina G, Deyev S. Natural and Designed Toxins for Precise Therapy: Modern Approaches in Experimental Oncology. International Journal of Molecular Sciences. 2021; 22(9):4975. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094975
Chicago/Turabian StyleShilova, Olga, Elena Shramova, Galina Proshkina, and Sergey Deyev. 2021. "Natural and Designed Toxins for Precise Therapy: Modern Approaches in Experimental Oncology" International Journal of Molecular Sciences 22, no. 9: 4975. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094975