
Recent Advances in the Use of Molecular Analyses to Inform the Diagnosis and Prognosis of Patients with Polycythaemia Vera


Abstract
:1. Introduction
2. Diagnosis
2.1. Driver Mutations for PV
2.2. Techniques for Detection of the JAK2V617F Variant
2.3. Biological Impact of JAK2 Driver Mutations
2.4. Molecular Differentiation of PV from Other Haematological Neoplasms
2.5. Germline Predisposition for Developing PV
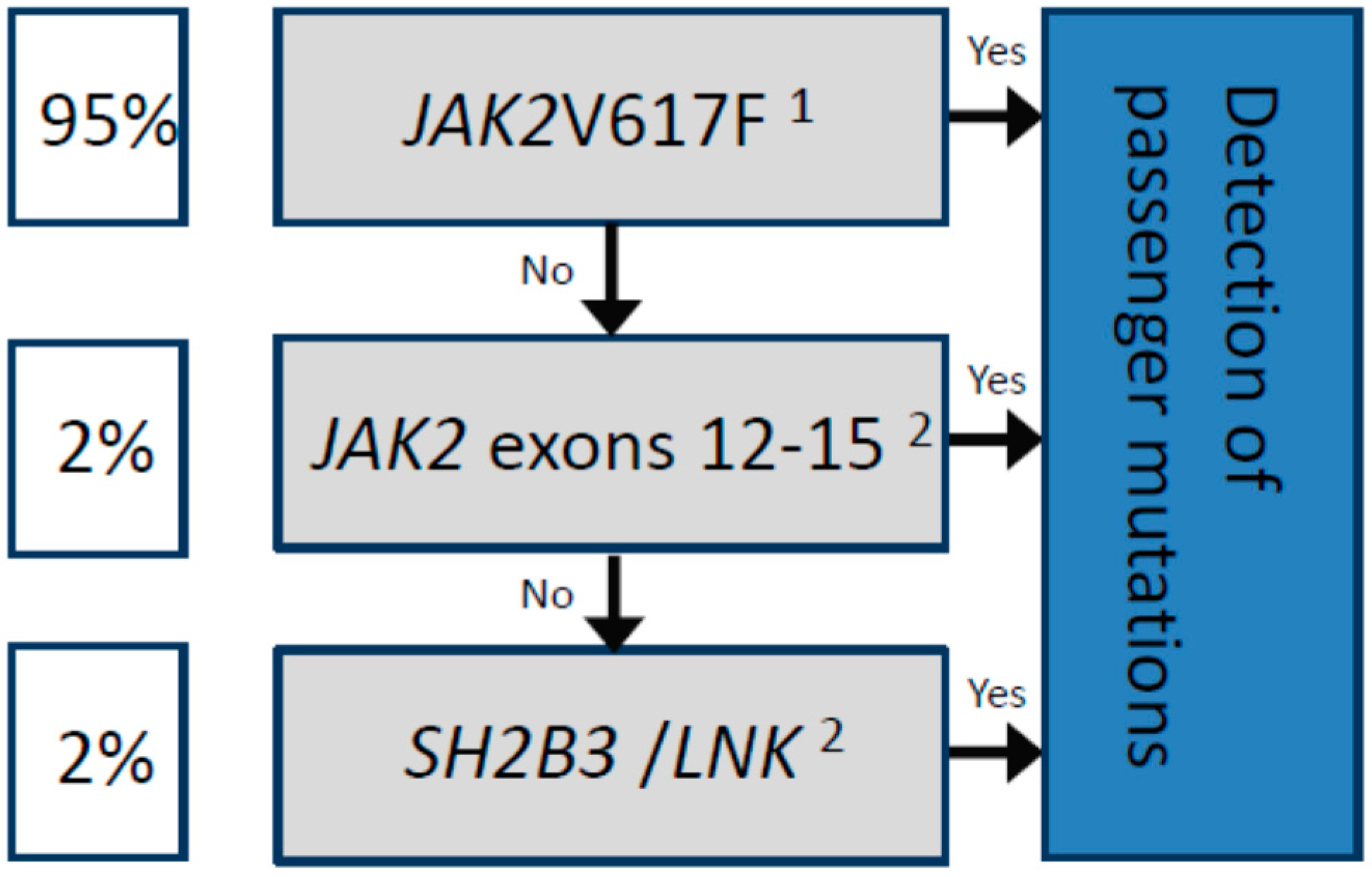
2.6. Passenger Mutations
3. Prognosis
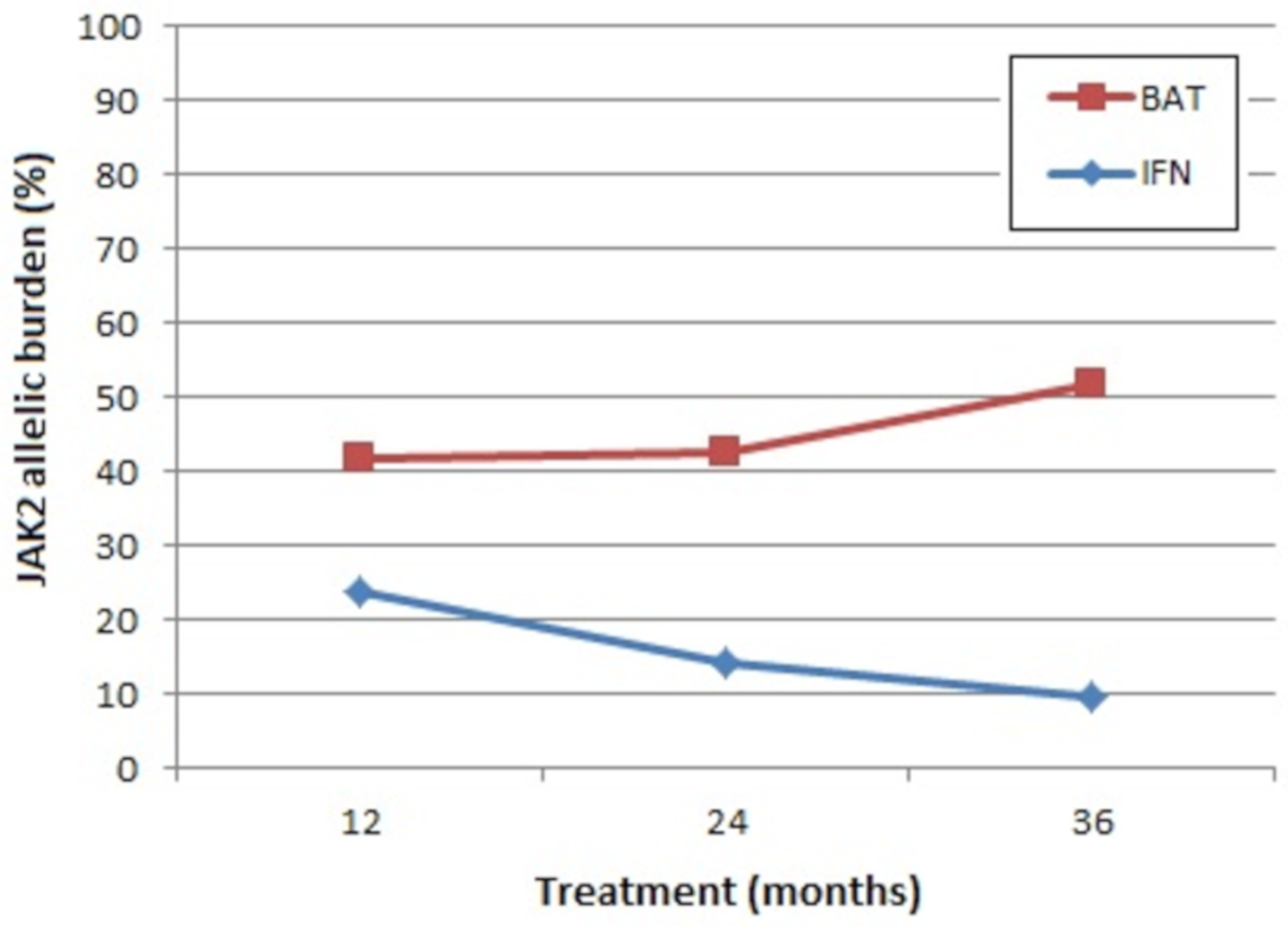
3.1. JAK2V617F Allelic Burden
3.2. Cytogenetics
3.3. Thrombosis
3.4. Molecular Indicators of Transformation
4. Treatment
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. In World Health Organization Classification of Tumours, 4th ed.; Bosman, F.T., Jaffe, E.S., Lakhani, S.R., Ohgaki, H., Eds.; IARC: Lyon, France, 2008; Volume 2. [Google Scholar]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Vanasse, G.; Cartmel, B.; Wang, Y.; Selinger, H.A. Prevalence of polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2008, 83, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.; Miller, C.B.; Thyne, M.; Mangan, J.; Goldberger, S.; Fazal, S.; Ma, X.; Wilson, W.; Paranagama, D.C.; Dubinski, D.G.; et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: The MPN Landmark survey. BMC Cancer 2016, 16, 167. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 2019, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- Raedler, L.A. Diagnosis and Management of Polycythemia Vera: Proceedings from a Multidisciplinary Roundtable. Am. Health Drug Benefits 2014, 7, S36–S47. [Google Scholar] [PubMed]
- Vannucchi, A.M.; Barbui, T.; Cervantes, F.; Harrison, C.; Kiladjian, J.J.; Kröger, N.; Thiele, J.; Buske, C.; ESMO Guidelines Committee. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, 85–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of Myeloproliferative Disorders. Annu Rev. Pathol Mech Dis. 2016, 11, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Rumi, E.; Pietra, D.; Elena, C.; Boveri, E.; Arcaini, L.; Roncoroni, E.; Astori, C.; Merli, M.; Boggi, S.; et al. A prospective study of 338 patients with polycythemia vera: The impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 2010, 24, 1574–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langabeer, S.E.; Andrikovics, H.; Asp, J.; Bellosillo, B.; Carillo, S.; Haslam, K.; Kjaer, L.; Lippert, E.; Mansier, O.; Oppliger Leibundgut, E.; et al. Molecular diagnostics of myeloproliferative neoplasms. Eur. J. Haematol. 2015, 95, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Broséus, J.; Park, J.H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcic Mikic, T.; Pajic, T.; Sever, M. CALR mutations in a cohort of JAK2 V617F negative patients with suspected myeloproliferative neoplasms. Sci. Rep. 2019, 9, 19838. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.Z.; Cook, J.R.; Greiner, T.C.; Hedvat, C.; Hill, C.E.; Lim, M.S.; Longtine, J.A.; Sabath, D.; Wang, Y.L.; Association for Molecular Pathology. Laboratory practice guidelines for detecting and reporting JAK2 and MPL mutations in myeloproliferative neoplasms: A report of the Association for Molecular Pathology. J. Mol. Diagn 2013, 15, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Kralovics, R.; De Libero, G.; Theocharides, A.; Gisslinger, H.; Skoda, R.C. Clonal heterogeneity in polycythemia vera patients with JAK2 exon12 and JAK2-V617F mutations. Blood 2008, 111, 3863–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietra, D.; Li, S.; Brisci, A.; Passamonti, F.; Rumi, E.; Theocharides, A.; Ferrari, M.; Gisslinger, H.; Kralovics, R.; Cremonesi, L.; et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 2008, 111, 1686–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisouard, J.; Li, S.; Kubovcakova, L.; Rao, T.N.; Meyer, S.C.; Lundberg, P.; Hao-Shen, H.; Romanet, V.; Murakami, M.; Radimerski, T.; et al. JAK2 exon 12 mutant mice display isolated erythrocytosis and changes in iron metabolism favoring increased erythropoiesis. Blood 2016, 128, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, J.V.; Ivey, A.; Vannucchi, A.M.; Lippert, E.; Oppliger Leibundgut, E.; Cassinat, B.; Pallisgaard, N.; Maroc, N.; Hermouet, S.; Nickless, G.; et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: A joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia 2013, 2, 2032–2039. [Google Scholar] [CrossRef] [Green Version]
- Link-Lenczowska, D.; Pallisgaard, N.; Cordua, S.; Zawada, M.; Czekalska, S.; Krochmalczyk, D.; Kanduła, Z.; Sacha, T. A comparison of qPCR and ddPCR used for quantification of the JAK2 V617F allele burden in Ph negative MPNs. Ann. Hematol. 2018, 97, 2299–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coccaro, N.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Digital PCR: A Reliable Tool for Analyzing and Monitoring Hematologic Malignancies. Int. J. Mol. Sci. 2020, 21, 3141. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Kantarjian, H.; Zhang, X.; Yeh, C.H.; Zhang, Z.J.; Verstovsek, S.; Albitar, M. Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J. Mol. Diagn 2009, 11, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslah, N.; Verger, E.; Schlageter, M.H.; Miclea, J.M.; Kiladjian, J.J.; Giraudier, S.; Chomienne, C.; Cassinat, B. Next-generation sequencing for JAK2 mutation testing: Advantages and pitfalls. Ann. Hematol. 2018, 98, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R. Next-Generation Sequencing in High-Sensitive Detection of Mutations in Tumors: Challenges, Advances, and Applications. J. Mol. Diagn 2020, 22, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Hocking, J.; Mithraprabhu, S.; Kalff, A.; Spencer, A. Liquid biopsies for liquid tumors: Emerging potential of circulating free nucleic acid evaluation for the management of hematologic malignancies. Cancer Biol. Med. 2016, 13, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gisbert, N.; Fernández-Ibarrondo, L.; Fernández-Rodríguez, C.; Gibert, J.; Andrade-Campos, M.; Arenillas, L.; Camacho, L.; Angona, A.; Longarón, R.; Salar, A.; et al. Circulating cell-free DNA improves the molecular characterisation of Ph-negative myeloproliferative neoplasms. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Lasho, T.L.; Pardanani, A.; Tefferi, A. LNK mutations in JAK2 mutation-negative erythrocytosis. N. Engl. J. Med. 2010, 363, 1189–1190. [Google Scholar] [CrossRef]
- Oh, S.T.; Simonds, E.F.; Jones, C.; Hale, M.B.; Goltsev, Y.; Gibbs, K.D., Jr.; Merker, J.D.; Zehnder, J.L.; Nolan, G.P.; Gotlib, J. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 2010, 116, 988–992. [Google Scholar] [CrossRef] [Green Version]
- Ema, H.; Sudo, K.; Seita, J.; Matsubara, A.; Morita, Y.; Osawa, M.; Takatsu, K.; Takaki, S.; Nakauchi, H. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev. Cell 2005, 8, 907–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersenev, A.; Wu, C.; Balcerek, J.; Tong, W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Invest 2008, 118, 2832–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Pietra, D.; Pane, F.; Pancrazzi, A.; Cazzola, M.; Vannucchi, A.M.; Tura, S.; Barosi, G. Recommendations for molecular testing in classical Ph1-neg myeloproliferative disorders-A consensus project of the Italian Society of Hematology. Leuk Res. 2017, 58, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fang, F.; Hu, Y.; Liu, Q.; Bu, D.; Tan, M.; Wu, L.; Zhu, P. The Polymorphisms in LNK Gene Correlated to the Clinical Type of Myeloproliferative Neoplasms. PLoS ONE 2016, 11, e0154183. [Google Scholar] [CrossRef]
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998, 93, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Quelle, F.W.; Wang, J.; Feng, J.; Wang, D.; Cleveland, J.L.; Ihle, J.N.; Zambetti, G.P. Cytokine rescue of p53-dependent apoptosis and cell cycle arrest is mediated by distinct Jak kinase signaling pathways. Genes Dev. 1998, 12, 1099–1107. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralovics, R.; Guan, Y.; Prchal, J.T. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp. Hematol. 2002, 30, 229–236. [Google Scholar] [CrossRef]
- Jamieson, C.H.; Gotlib, J.; Durocher, J.A.; Chao, M.P.; Mariappan, M.R.; Lay, M.; Jones, C.; Zehnder, J.L.; Lilleberg, S.L.; Weissman, I.L. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 6224–6229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubach, J.P.; Fu, P.; Jiang, X.; Salter, K.H.; Potti, A.; Arcasoy, M.O. Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp. Hematol. 2009, 37, 1411–1422. [Google Scholar] [CrossRef] [Green Version]
- Walz, C.; Crowley, B.J.; Hudon, H.E.; Gramlich, J.L.; Neuberg, D.S.; Podar, K.; Griffin, J.D.; Sattler, M. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J. Biol. Chem. 2006, 281, 18177–18183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plo, I.; Nakatake, M.; Malivert, L.; de Villartay, J.P.; Giraudier, S.; Villeval, J.L.; Wiesmuller, L.; Vainchenker, W. JAK2 stimulates homologous recombination and genetic instability: Potential implication in the heterogeneity of myeloproliferative disorders. Blood 2008, 112, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Ahn, J.S.; Massie, C.E.; Clynes, D.; Godfrey, A.L.; Li, J.; Park, H.J.; Nangalia, J.; Silber, Y.; Mullally, A.; et al. JAK2V617F promotes replication fork stalling with disease-restricted impairment of the intra-S checkpoint response. Proc. Natl. Acad. Sci. USA 2014, 111, 15190–15195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatake, M.; Monte-Mor, B.; Debili, N.; Casadevall, N.; Ribrag, V.; Solary, E.; Vainchenker, W.; Plo, I. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene 2012, 31, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.; Richard, C.; Benito, A.; Sanz, C.; Olalla, I.; Fernández-Luna, J.L. Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. N. Engl. J. Med. 1998, 338, 564–571. [Google Scholar] [CrossRef]
- Zhao, R.; Follows, G.A.; Beer, P.A.; Scott, L.M.; Huntly, B.J.; Green, A.R.; Alexander, D.R. Inhibition of the Bcl-xL deamidation pathway in myeloproliferative disorders. N. Engl. J. Med. 2008, 359, 2778–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherber, R.; Geyer, H.; Dueck, A.; Johnston, C.; Langlais, B.; Padrnos, L.; Palmer, J.; Fleischman, A.; Mesa, R. Nutritional needs and preferences of myeloproliferative neoplasm patients: Phase IA of the Nutrient Study; European Hematology Association Congress: Madrid, Spain, 2017. [Google Scholar]
- Coussens, L.M.; Web, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, M.; Ghirardi, A.; Masciulli, A.; Carobbio, A.; Palandri, F.; Vianelli, N.; Rossi, E.; Betti, S.; Di Veroli, A.; Iurlo, A.; et al. Second cancers in MPN: Survival analysis from an international study. Am. J. Hematol. 2020, 95, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, H.; Knutsen, H.; Holmberg, E.; Andréasson, B. Increased incidence of another cancer in myeloproliferative neoplasms patients at the time of diagnosis. Eur. J. Haematol. 2015, 94, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Ghirardi, A.; Carobbio, A.; Masciulli, A.; Barbui, T. Incidence of solid tumors in polycythemia vera treated with phlebotomy with or without hydroxyurea: ECLAP follow-up data. Blood Cancer J. 2018, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C. Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk Res. 2015, S0145–S2126, 30373–30378. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.M.; Bak, M.; Sørensen, A.L.; Zwisler, A.D.; Ellervik, C.; Larsen, M.K.; Hasselbalch, H.C.; Tolstrup, J.S. Smoking is associated with increased risk of myeloproliferative neoplasms: A general population-based cohort study. Cancer Med. 2018, 7, 5796–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folsom, A.R.; Lutsey, P.L.; Astor, B.C.; Cushman, M. C-reactive protein and venous thromboembolism. A prospective investigation in the ARIC cohort. Thromb Haemost 2009, 102, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Genetics and Pathogenetic Role of Inflammasomes in Philadelphia Negative Chronic Myeloproliferative Neoplasms: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 561. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, J.; Oki, Y.; Gharibyan, V.; Bueso-Ramos, C.; Prchal, J.T.; Verstovsek, S.; Beran, M.; Estey, E.; Kantarjian, H.M.; Issa, J.P.J. JAK2 mutation 1849G > T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood 2005, 106, 3370–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.L.; Loriaux, M.; Huntly, B.J.; Loh, M.L.; Beran, M.; Stoffregen, E.; Berger, R.; Clark, J.J.; Willis, S.G.; Nguyen, K.T.; et al. The JAK2 V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 2005, 106, 3377–3379. [Google Scholar] [CrossRef] [Green Version]
- Sidon, P.; El Housni, H.; Dessars, B.; Heimann, P. The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 2006, 20, 1622. [Google Scholar] [CrossRef] [PubMed]
- Klippel, S.; Strunck, E.; Temerinac, S.; Bench, A.J.; Meinhardt, G.; Mohr, U.; Leichtle, R.; Green, A.R.; Griesshammer, M.; Heimpel, H.; et al. Quantification of PRV-1 mRNA distinguishes polycythemia vera from secondary erythrocytosis. Blood 2003, 102, 3569–3574. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Teofili, L.; Larocca, L.M. Overexpression of PRV-1 gene in polycythemia rubra vera and essential thrombocythemia. Methods Mol. Med. 2006, 125, 265–273. [Google Scholar] [CrossRef]
- Bock, O.; Serinsöz, E.; Neusch, M.; Schlué, J.; Kreipe, H. The polycythaemia rubra vera-1 gene is constitutively expressed by bone marrow cells and does not discriminate polycythaemia vera from reactive and other chronic myeloproliferative disorders. Br. J. Haematol. 2003, 123, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.M.; Correa, P.N.; Axelrad, A.A. Increased basal and induced tyrosine phosphorylation of the insulin-like growth factor I receptor beta subunit in circulating mononuclear cells of patients with polycythemia vera. Blood 1995, 86, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Shi, G.; Baptiste, S.; Yarotska, M.; Sindhu, H.; Wong, C.; Kalavar, M.; Gotlieb, V.; Bandarchuk, A.; Chen, H. Quantification of IGF-1 Receptor May Be Useful in Diagnosing Polycythemia Vera-Suggestion to Be Added to Be One of the Minor Criterion. PLoS ONE 2016, 11, e0165299. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.L.; Safai-Kutti, S.; Kutti, J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: A study of patients with polycythaemia vera and apparent polycythaemia. Br. J. Haematol. 2005, 129, 701–705. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Ancochea, A.; Angona, A.; Pedro, C.; García-Pallarols, F.; Martínez-Avilés, L.; Bellosillo, B.; Besses, C. Red cell mass measurement in patients with clinically suspected diagnosis of polycythemia vera or essential thrombocythemia. Haematologica 2012, 97, 1704–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Thiele, J.; Carobbio, A. Masked polycythemia vera diagnosed according to WHO and BCSH classification. Am. J. Hematol. 2014, 89, 199–202. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Carobbio, A.; Guglielmelli, P.; Rambaldi, A.; Vannucchi, A.M.; Tefferi, A. Discriminating between essential thrombocythemia and masked polycythemia vera in JAK2 mutated patients. Am. J. Hematol. 2014, 89, 588–590. [Google Scholar] [CrossRef]
- Silver, R.T.; Chow, W.; Orazi, A.; Arles, S.P.; Goldsmith, S.J. Evaluation of WHO criteria for diagnosis of polycythemia vera: A prospective analysis. Blood 2013, 122, 1881–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Finazzi, G.; Carobbio, A.; Rumi, E.; Luigia Randi, M.; Bertozzi, I.; Vannucchi, A.M.; Pieri, L.; et al. Masked polycythemia vera (mPV): Results of an international study. Am. J. Hematol. 2014, 89, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Carobbio, A.; Randi, M.L.; Elena, C.; Rumi, E.; Finazzi, G.; Bertozzi, I.; Pieri, L.; Ruggeri, M.; Palandri, F.; et al. A lower intensity of treatment may underlie the increased risk of thrombosis in young patients with masked polycythaemia vera. Br. J. Haematol. 2014, 167, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hussaini, M.; Zhang, H.; <named-content content-type="background:white">Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the mutational profiles of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goerttler, P.S.; Kreutz, C.; Donauer, J.; Faller, D.; Maiwald, T.; März, E.; Rumberger, B.; Sparna, T.; Schmitt-Gräff, A.; Wilpert, J.; et al. Gene expression profiling in polycythaemia vera: Overexpression of transcription factor NF-E2. Br. J. Haematol. 2005, 129, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.; Barrio, S.; Fernandez, M.; Paradela, A.; Arenas, A.; Toldos, O.; Ayala, R.; Albizua, E.; Jimenez, A.; Redondo, S.; et al. Proteomic analysis reveals heat shock protein 70 has a key role in polycythemia Vera. Mol. Cancer 2013, 12, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusson, M.; Cochet, S.; Leduc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; Le Van Kim, C.; Cassinat, B.; Kiladjian, J.J.; et al. Enhanced calreticulin expression in red cells of polycythemia vera patients harboring the JAK2V617F mutation. Haematologica 2017, 102, e241–e244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchova, H.; Merkerova, M.; Prchal, J.T. Aberrant expression of microRNA in polycythemia vera. Haematologica 2008, 93, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Zhan, H.; Cardozo, C.; Yu, W.; Wang, A.; Moliterno, A.R.; Dang, C.V.; Spivak, J.L. MicroRNA deregulation in polycythemia vera and essential thrombocythemia patients. Blood Cells Mol. Dis. 2013, 50, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Scott, L.M.; Buck, G.; Wheatley, K.; East, C.L.; Marsden, J.T.; Duffy, A.; Boyd, E.M.; Bench, A.J.; Scott, M.A.; et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: A prospective study. Lancet 2005, 366, 1945–1953. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Pietra, D.; Della Porta, M.G.; Boveri, E.; Pascutto, C.; Vanelli, L.; Arcaini, L.; Burcheri, S.; Malcovati, L.; et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood 2006, 107, 3676–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshemmari, S.H.; Rajaan, R.; Ameen, R.; Al-Drees, M.A.; Almosailleakh, M.R. JAK2V617F allele burden in patients with myeloproliferative neoplasms. Ann. Hematol. 2014, 93, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Tiedt, R.; Hao-Shen, H.; Sobas, M.A.; Looser, R.; Dirnhofer, S.; Schwaller, J.; Skoda, R.C. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008, 111, 3931–3940. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kent, D.G.; Godfrey, A.L.; Manning, H.; Nangalia, J.; Aziz, A.; Chen, E.; Saeb-Parsy, K.; Fink, J.; Sneade, R.; et al. JAK2V617FJAK2V617F homozygosity drives a phenotypic switch between myeloproliferative neoplasms in a murine model, but is insufficient to sustain disease. Blood 2014, 123, 3139–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaiter, Y.; Brenner, B.; Aghai, E.; Tatarsky, I. High incidence of myeloproliferative disorders in Ashkenazi Jews in northern Israel. Leuk Lymphoma 1992, 7, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Goldin, L.R.; Kristinsson, S.Y.; Helgadottir, E.A.; Samuelsson, J.; Bjorkholm, M. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24577 first-degree relatives of 11039 patients with myeloproliferative neoplasms in Sweden. Blood 2008, 112, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Passamonti, F.; Della Porta, M.G.; Elena, C.; Arcaini, L.; Vanelli, L.; Del Curto, C.; Pietra, D.; Boveri, E.; Pascutto, C.; et al. Familial chronic myeloproliferative disorders: Clinical phenotype and evidence of disease anticipation. J. Clin. Oncol. 2007, 25, 5630–5635. [Google Scholar] [CrossRef]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olcaydu, D.; Harutyunyan, A.; Jäger, R.; Berg, T.; Gisslinger, B.; Pabinger, I.; Gisslinger, H.; Kralovics, R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat. Genet. 2009, 41, 450–454. [Google Scholar] [CrossRef]
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)- positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olcaydu, D.; Skoda, R.C.; Looser, R.; Li, S.; Cazzola, M.; Pietra, D.; Passamonti, F.; Lippert, E.; Carillo, S.; Girodon, F.; et al. The ‘GGCC’ haplotype of JAK2 confers susceptibility to JAK2 exon 12 mutation-positive polycythemia vera. Leukemia 2009, 23, 1924–1926. [Google Scholar] [CrossRef] [PubMed]
- Nussenzveig, R.H.; Swierczek, S.I.; Jelinek, J.; Gaikwad, A.; Liu, E.; Verstovsek, S.; Prchal, J.F.; Prchal, J.T. Polycythemia vera is not initiated by JAK2V617F mutation. Exp. Hematol. 2007, 35, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Teo, S.S.; Li, S.; Theocharides, A.; Buser, A.S.; Tichelli, A.; Skoda, R.C. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006, 108, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Passamonti, F.; Pietra, D.; Della Porta, M.G.; Arcaini, L.; Boggi, S.; Elena, C.; Boveri, E.; Pascutto, C.; Lazzarino, M.; et al. JAK2 (V617F) as an acquired somatic mutation and a secondary genetic event associated with disease progression in familial myeloproliferative disorders. Cancer 2006, 107, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- James, C. The JAK2V617F Mutation in Polycythemia Vera and Other Myeloproliferative Disorders: One Mutation for Three Diseases? Hematology Am. Soc. Hematol. Educ. Program. 2008, 2008, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanikova, L.; Babosova, O.; Swierczek, S.; Wang, L.; Wheeler, D.A.; Divoky, V.; Korinek, V.; Prchal, J.T. Coexistence of gain-of-function JAK2 germ line mutations with JAK2V617F in polycythemia vera. Blood 2016, 128, 2266–2270. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Giambruno, R.; Krendl, C.; Stukalov, A.; Klampfl, T.; Berg, T.; Milosevic, J.D.; Chen, D.; Gisslinger, B.; Gisslinger, H.; et al. Germline RBBP6 Mutations In Myeloproliferative Neoplasms [abstract]. Blood 2013, 122, 267. [Google Scholar] [CrossRef]
- Oddsson, A.; Kristinsson, S.Y.; Helgason, H.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Steingrimsdottir, H.; Vidarsson, B.; et al. The germline sequence variant rs2736100_C in TERT associates with myeloproliferative neoplasms. Leukemia 2014, 28, 1371–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harutyunyan, A.S.; Giambruno, R.; Krendl, C.; Stukalov, A.; Klampfl, T.; Berg, T.; Chen, D.; Milosevic Feenstra, J.D.; Jäger, R.; Gisslinger, B.; et al. Germline RBBP6 mutations in familial myeloproliferative neoplasms. Blood 2016, 127, 362–365. [Google Scholar] [CrossRef] [Green Version]
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775. [Google Scholar] [CrossRef]
- Giaccherini, M.; Macauda, A.; Sgherza, N.; Sainz, J.; Gemignani, F.; Maldonado, J.; Jurado, M.; Tavano, F.; Mazur, G.; Jerez, A.; et al. Genetic polymorphisms associated with telomere length and risk of developing myeloproliferative neoplasms. Blood Cancer J. 2020, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.; Lee, J.; Moore, L.; Baxter, J.E.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; Campbell, P.J.; et al. Driver mutation acquisition in utero and childhood followed by lifelong clonal evolution underlie myeloproliferative neoplasms. 2020 ASH Annual Meeting & Exposition. Abstract LBA-1. Presented December 8, 2020. Available online: https://ash.confex.com/ash/2020/webprogram/Paper143813.html (accessed on 15 February 2021).
- Nangalia, J.; Green, T.R. The evolving genomic landscape of myeloproliferative neoplasms. Hematology Am. Soc. Hematol. Educ. Program. 2014, 2014, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura-Díaz, A.; Stuckey, R.; Florido, Y.; González-Martín, J.M.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; González-Pérez, E.; Sáez Perdomo, M.N.; Perera, M.; de la Iglesia, S.; et al. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers 2020, 12, 934. [Google Scholar] [CrossRef] [PubMed]
- Green, A.; Beer, P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N. Engl. J. Med. 2010, 362, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcault, C.; Zhao, L.P.; Daltro De Oliveira, R.; Soret, J.; Gauthier, N.; Verger, E.; Maslah, N.; Roux, B.; Parquet, N.; Dosquet, C.; et al. NFE2 Mutations Impact AML Transformation and Overall Survival in Patients with Myeloproliferative Neoplasms (MPN). Blood 2020, 136 (Suppl. 1), 36. [Google Scholar] [CrossRef]
- Marinaccio, C.; Suraneni, P.K.; Celik, H.; Volk, A.; Wen, J.Q.; Ling, T.; Lasho, T.; Koche, R.P.; Famulare, C.; Stein, B.L.; et al. Loss of LKB1/STK11 Facilitates Leukemic Progression of the Myeloproliferative Neoplasms. Blood 2020, 136 (Suppl. 1), 1. [Google Scholar] [CrossRef]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavezet, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Shide, K.; Yamaji, T.; Kamiunten, A.; Sekine, M.; Taniguchi, Y.; Hidaka, T.; Kubuki, Y.; Shimoda, H.; Marutsuka, K.; et al. Loss of TET2 has dual roles in murine myeloproliferative neoplasms: Disease sustainer and disease accelerator. Blood 2015, 125, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Schneider, R.K.; Breyfogle, L.J.; Rosen, E.A.; Poveromo, L.; Elf, S.; Ko, A.; Brumme, K.; Levine, R.; Ebert, B.L.; et al. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood 2015, 125, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.E.; Magrini, V.; Demeter, R.; Miller, C.A.; Fulton, R.; Fulton, L.L.; Eades, W.C.; Elliott, K.; Heath, S.; Westervelt, P.; et al. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet. 2014, 10, e1004462. [Google Scholar] [CrossRef] [PubMed]
- Nice, F.L.; Massie, C.E.; Klampfl, T.; Green, A.R. Determination of complex subclonal structures of hematological malignancies by multiplexed genotyping of blood progenitor colonies. Exp. Hematol. 2018, 57, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- McMullin, M.F.; Bareford, D.; Campbell, P.; Green, A.R.; Harrison, C.; Hunt, B.; Oscier, D.; Polkey, M.I.; Reilly, J.T.; Rosenthal, E.; et al. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br. J. Haematol. 2005, 130, 174–195. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. MPD Research Consortium. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007, 21, 1952–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangat, N.; Strand, J.J.; Lasho, T.L.; Li, C.Y.; Pardanani, A.; Tefferi, A. Pruritus in polycythemia vera is associated with a lower risk of arterial thrombosis. Am. J. Hematol. 2008, 83, 451–453. [Google Scholar] [CrossRef]
- Larsen, T.S.; Pallisgaard, N.; Møller, M.B.; Hasselbalch, H.C. The JAK2 V617F allele burden in essential thrombocythemia, polycythemia vera and primary myelofibrosis--impact on disease phenotype. Eur. J. Haematol. 2007, 79, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.M.; Schemionek, M.; Schubert, C.; Chatain, N.; Sontag, S.; Isfort, S.; Ortiz-Brüchle, N.; Schmitt, K.; Krüger, L.; Zerres, K.; et al. Dissecting Genomic Aberrations in Myeloproliferative Neoplasms by Multiplex-PCR and Next Generation Sequencing. PLoS ONE 2015, 10, e0123476. [Google Scholar] [CrossRef] [Green Version]
- Borowczyk, M.; Wojtaszewska, M.; Lewandowski, K.; Gil, L.; Lewandowska, M.; Lehmann-Kopydłowska, A.; Kroll-Balcerzak, R.; Balcerzak, A.; Iwoła, M.; Michalak, M.; et al. The JAK2 V617F mutational status and allele burden may be related with the risk of venous thromboembolic events in patients with Philadelphia-negative myeloproliferative neoplasms. Thromb Res. 2015, 135, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Malak, S.; Labopin, M.; Saint-Martin, C.; Bellanne-Chantelot, C.; Najman, A.; French Group of Familial Myeloproliferative Disorders. Long term follow up of 93 families with myeloproliferative neoplasms: Life expectancy and implications of JAK2V617F in the occurrence of complications. Blood Cells Mol. Dis. 2012, 49, 170–176. [Google Scholar] [CrossRef]
- Kralovics, R.; Teo, S.S.; Buser, A.S.; Brutsche, M.; Tiedt, R.; Tichelli, A.; Passamonti, F.; Pietra, D.; Cazzola, M.; Skoda, R.C. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood 2005, 106, 3374–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainchenker, W.; Constantinescu, S.N. A Unique Activating Mutation in JAK2 (V617F) Is at the Origin of Polycythemia Vera and Allows a New Classification of Myeloproliferative Diseases. Hematology Am. Soc. Hematol. Educ. Program. 2005, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Pardanani, A.; Tefferi, A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: A critical reappraisal. Leukemia 2008, 22, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Antonioli, E.; Guglielmeli, P.; Pancrazzi, A.; Bogani, C.; Pieri, L.; Bosi, A. Influence of the JAK2V617F mutational load at diagnosis on major clinical aspects in patients with Polycythemia vera. Blood 2006, 108, 5. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Barosi, G.; Rimbaldi, A.; Barosi, G.; Rambaldi, A.; Marchioli, R.; Barbui, T.; Gimema, W.P. Clinical significance of JAK2V617F homozygosity in the chronic myeloproliferative disorders. A study of 1306 patients. Blood 2006, 108, 664. [Google Scholar] [CrossRef]
- Spivak, J.L.; Considine, M.; Williams, D.M.; Talbot, C.C., Jr.; Rogers, O.; Moliterno, A.R.; Jie, C.; Ochs, M.F. Two clinical phenotypes in polycythemia vera. N. Engl. J. Med. 2014, 371, 808–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swolin, B.; Weinfeld, A.; Westin, J. A prospective long-term cytogenetic study in polycythemia vera in relation to treatment and clinical course. Blood 1988, 72, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Diez-Martin, J.L.; Graham, D.L.; Petitt, R.M.; Dewald, G.W. Chromosome studies in 104 patients with polycythemia vera. Mayo Clin. Proc. 1991, 66, 287–299. [Google Scholar] [CrossRef]
- Gangat, N.; Strand, J.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Pardanani, A.; Li, C.Y.; Ketterling, R.P.; Tefferi, A. Cytogenetic studies at diagnosis in polycythemia vera: Clinical and JAK2V617F allele burden correlates. Eur. J. Haematol. 2008, 80, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, F.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood 2005, 105, 2664–2670. [Google Scholar] [CrossRef]
- Tang, G.; Hidalgo Lopez, J.E.; Wang, S.A.; Hu, S.; Ma, J.; Pierce, S.; Zuo, W.; Carballo-Zarate, A.A.; Yin, C.C.; Tang, Z.; et al. Characteristics and clinical significance of cytogenetic abnormalities in polycythemia vera. Haematologica 2017, 102, 1511–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sever, M.; Quintás-Cardama, A.; Pierce, S.; Zhou, L.; Kantarjian, H.; Verstovsek, S. Significance of cytogenetic abnormalities in patients with polycythemia vera. Leuk Lymphoma 2013, 54, 2667–2670. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Vannucchi, A.M.; Rodeghiero, F.; Randi, M.L.; Vaidya, R.; Cazzola, M.; Rambaldi, A.; et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: An international study. Leukemia 2013, 27, 1874–1881. [Google Scholar] [CrossRef] [Green Version]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Vannucchi, A.M.; Carobbio, A.; Thiele, J.; Rumi, E.; Gisslinger, H.; Rodeghiero, F.; Randi, M.L.; Rambaldi, A.; Pieri, L.; et al. Patterns of presentation and thrombosis outcome in patients with polycythemia vera strictly defined by WHO-criteria and stratified by calendar period of diagnosis. Am. J. Hematol. 2015, 90, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Barosi, G.; Birgegard, G.; Cervantes, F.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.C.; Hehlmann, R.; Hoffman, R.; et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European LeukemiaNet. J. Clin. Oncol. 2011, 29, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M. JAK2 mutation and thrombosis in the myeloproliferative neoplasms. Curr. Hematol. Malig Rep. 2010, 5, 22–28. [Google Scholar] [CrossRef]
- Chia, Y.C.; Islam, M.A.; Woon, P.Y.; Johan, M.F.; Hassan, R.; Ramli, M. Molecular genetics of thrombotic myeloproliferative neoplasms: Implications in precision oncology. Genes Dis. 2021. [Google Scholar] [CrossRef]
- Ruggeri, M.; Gisslinger, H.; Tosetto, A.; Rintelen, C.; Mannhalter, C.; Pabinger, I.; Heis, N.; Castaman, G.; Missiaglia, E.; Lechner, K.; et al. Leiden mutation carriership and venous thromboembolism in polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2002, 71, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; de Nully Brown, P.; Thorsen, S.; Hasselbalch, H.C. Frequent occurrence of anticardiolipin antibodies, Factor V Leiden mutation, and perturbed endothelial function in chronic myeloproliferative disorders. Am. J. Hematol. 2002, 69, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Trifa, A.P.; Cucuianu, A.; Popp, R.A.; Coadă, C.A.; Costache, R.M.; Militaru, M.S.; Vesa, S.C.; Pop, I.V. The relationship between factor V Leiden, prothrombin G20210A, and MTHFR mutations and the first major thrombotic episode in polycythemia vera and essential thrombocythemia. Ann. Hematol. 2014, 93, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Algahtani, F.H.; Stuckey, R. High factor VIII levels and arterial thrombosis: Illustrative case and literature review. Ther. Adv. Hematol. 2019, 10, 2040620719886685. [Google Scholar] [CrossRef]
- Sacco, M.; Ranalli, P.; Lancellotti, S.; Petrucci, G.; Dragani, A.; Rocca, B.; De Cristofaro, R. Increased von Willebrand factor levels in polycythemia vera and phenotypic differences with essential thrombocythemia. Res. Pract Thromb Haemost. 2020, 4, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Micò, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Leukocytosis is a risk factor for recurrent arterial thrombosis in young patients with polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2010, 85, 97–100. [Google Scholar] [CrossRef]
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737. [Google Scholar] [CrossRef]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133. [Google Scholar] [CrossRef] [Green Version]
- Carobbio, A.; Finazzi, G.; Antonioli, E.; Guglielmelli, P.; Vannucchi, A.M.; Dellacasa, C.M.; Salmoiraghi, S.; Delaini, F.; Rambaldi, A.; Barbui, T. JAK2V617F allele burden and thrombosis: A direct comparison in essential thrombocythemia and polycythemia vera. Exp. Hematol. 2009, 37, 1016–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvat, I.; Boban, A.; Zadro, R.; Antolic, M.R.; Serventi-Seiwerth, R.; Roncevic, P.; Radman, I.; Sertic, D.; Vodanovic, M.; Pulanic, D.; et al. Influence of Blood Count, Cardiovascular Risks, Inherited Thrombophilia, and JAK2 V617F Burden Allele on Type of Thrombosis in Patients with Philadelphia Chromosome Negative Myeloproliferative Neoplasms. Clin. Lymphoma Myeloma Leuk 2019, 19, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2011, 96, 315–318. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1b inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2019, 41, 2153–2163. [Google Scholar] [CrossRef] [Green Version]
- CARDIoGRAMplusC4D Consortium; Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; et al. Large--scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, Y.; Wang, Y.; Tascau, L.; Balcerek, J.; Tong, W.; Levine, R.L.; Welch, C.; Tall, A.R.; Wang, N. LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis. Circ Res. 2016, 119, e91–e103. [Google Scholar] [CrossRef] [Green Version]
- Kaplar, J.K.; Kiss, A.K.; Szabo, M.; Udvardy, M. Increased leukocyte-platelet adhesion in chronic myeloproliferative disorders with high platelet counts. Platelets 2000, 11, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; de Nully Brown, P.; Lund, B.V.; Nielsen, O.J.; Hasselbalch, H.C. Increased circulating platelet–leukocyte aggregates in myeloproliferative disorders is correlated to previous thrombosis, platelet activation and platelet count. Eur. J. Haematol. 2001, 66, 143–151. [Google Scholar] [CrossRef]
- Griesshammer, M.; Klippel, S.; Strunck, E.; Temerinac, S.; Mohr, U.; Heimpel, H.; Pahl, H.L. PRV-1 mRNA expression discriminates two types of essential thrombocythemia. Ann. Hematol. 2004, 83, 364–370. [Google Scholar] [CrossRef]
- Shahrabi, S.; Ehsanpour, A.; Heidary, S.; Shahjahani, M.; Behzad, M.M. Expression of CD markers in JAK2V617F positive myeloproliferative neoplasms: Prognostic significance. Oncol. Rev. 2018, 12, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Marín, F.; Cuenca-Zamora, E.J.; Guijarro-Carrillo, P.J.; Teruel-Montoya, R. Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 1143. [Google Scholar] [CrossRef] [PubMed]
- Benton, C.B.; Tanaka, M.; Wilson, C.; Pierce, S.; Zhou, L.; Cortes, J.; Kantarjian, H.; Verstovsek, S. Increased likelihood of post-polycythemia vera myelofibrosis in Ph-negative MPN patients with chromosome 12 abnormalities. Leuk Res. 2015, 39, 419–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotunno, G.; Pacilli, A.; Artusi, V.; Rumi, E.; Maffioli, M.; Delaini, F.; Brogi, G.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: A study on 359 patients of the AGIMM group. Am. J. Hematol. 2016, 91, 681–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Gowin, K.; Coakley, M.; Kosiorek, H.; Mesa, R. Discrepancies of applying primary myelofibrosis prognostic scores for patients with post polycythemia vera/essential thrombocytosis myelofibrosis. Haematologica 2016, 101, e405–e406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef] [PubMed]
- Palandri, F.; Palumbo, G.A.; Iurlo, A.; Polverelli, N.; Benevolo, G.; Breccia, M.; Abruzzese, E.; Tiribelli, M.; Bonifacio, M.; Tieghi, A.; et al. Differences in presenting features, outcome and prognostic models in patients with primary myelofibrosis and post-polycythemia vera and/or post-essential thrombocythemia myelofibrosis treated with ruxolitinib. New perspective of the MYSEC-PM in a large multicenter study. Semin Hematol. 2018, 55, 248–255. [Google Scholar] [CrossRef]
- Mora, B.; Giorgino, T.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.J.; et al. Value of cytogenetic abnormalities in post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A study of the MYSEC project. Haematologica 2018, 103, e392–e394. [Google Scholar] [CrossRef] [PubMed]
- Theocharides, A.; Boissinot, M.; Girodon, F.; Garand, R.; Teo, S.S.; Lippert, E.; Talmant, P.; Tichelli, A.; Hermouet, S.; Skoda, R.C. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 2007, 110, 375–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, P.A.; Delhommeau, F.; LeCouédic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque Paz, D.; Jouanneau-Courville, R.; Riou, J.; Ianotto, J.C.; Boyer, F.; Chauveau, A.; Renard, M.; Chomel, J.C.; Cayssials, E.; Gallego-Hernanz, M.P.; et al. Leukemic evolution of polycythemia vera and essential thrombocythemia: Genomic profiles predict time to transformation. Blood Adv. 2020, 4, 4887–4897. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [Green Version]
- Senín, A.; Fernández-Rodríguez, C.; Bellosillo, B.; Camacho, L.; Longarón, R.; Angona, A.; Besses, C.; Álvarez-Larrán, A. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018, 97, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, J.L. How I treat polycythemia vera. Blood 2019, 134, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, E.; Guglielmelli, P.; Pieri, L.; Finazzi, M.; Rumi, E.; Martinelli, V.; Vianelli, N.; Luigia Randi, M.; Bertozzi, I.; De Stefano, V.; et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am. J. Hematol. 2012, 87, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larrán, A.; Kerguelen, A.; Hernández-Boluda, J.C.; Pérez-Encinas, M.; Ferrer-Marín, F.; Bárez, A.; Martínez-López, J.; Cuevas, B.; Mata, M.I.; García-Gutiérrez, V.; et al. Frequency and prognostic value of resistance/intolerance to hydroxycarbamide in 890 patients with polycythaemia vera. Br. J. Haematol. 2016, 172, 786–793. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Díaz-González, A.; Such, E.; Mora, E.; Andrade-Campos, M.; García-Hernández, C.; Gómez-Casares, M.T.; García-Gutiérrez, V.; Carreño-Tarragona, G.; Garrote, M.; et al. Genomic characterization of patients with polycythemia vera developing resistance to hydroxyurea. Leukemia 2021, 35, 623–627. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. JAK2 inhibitors for myeloproliferative neoplasms: What is next? Blood 2017, 130, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rosen, P.J.; Rumi, E.; Gattoni, E.; Pieri, L.; Guglielmelli, P.; Elena, C.; et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014, 120, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.M.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet Oncol. 2017, 18, 88–99. [Google Scholar] [CrossRef]
- Verstovsek, S.; Courby, S.; Griesshammer, M.; Mesa, R.A.; Brachmann, C.B.; Kawashima, J.; Maltzman, J.D.; Shao, L.; Xin, Y.; Huang, D.; et al. A phase 2 study of momelotinib, a potent JAK1 and JAK2 inhibitor, in patients with polycythemia vera or essential thrombocythemia. Leuk Res. 2017, 60, 11–17. [Google Scholar] [CrossRef]
- Samuelson, B.T.; Vesely, S.K.; Chai-Adisaksopha, C.; Scott, B.L.; Crowther, M.; Garcia, D. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: A meta-analysis. Blood Coagul Fibrinolysis 2016, 27, 648–652. [Google Scholar] [CrossRef]
- Besremi: EPAR—Public Assessment report., European Medicines Agency, December 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/besremi-epar-public-assessment-report_en.pdf (accessed on 25 February 2021).
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; De Stefano, V.; Masciulli, A.; Carobbio, A.; Ferrari, A.; Ghirardi, A.; Rossi, E.; Ciceri, F.; Bonifacio, M.; et al. Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): A multicentre, randomised phase 2 trial. Lancet Haematol. 2021, 8, e175–e184. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.-F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: Role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef]
- Hansen, I.O.; Sørensen, A.L.; Hasselbalch, H.C. Second malignancies in hydroxyurea and interferon-treated Philadelphia-negative myeloproliferative neoplasms. Eur. J. Haematol. 2017, 98, 75–84. [Google Scholar] [CrossRef]
- Faille, D.; Lamrani, L.; Loyau, S.; Huisse, M.-G.; Bourrienne, M.-C.; Alkhaier, S.; Cassinat, B.; Boulaftali, Y.; Debus, J.; Jandrot-Perrus, M.; et al. Interferon Alpha Therapy Increases Pro-Thrombotic Biomarkers in Patients with Myeloproliferative Neoplasms. Cancers 2020, 12, 992. [Google Scholar] [CrossRef] [Green Version]
- Iurlo, A.; Cattaneo, D.; Bucelli, C.; Baldini, L. New Perspectives on Polycythemia Vera: From Diagnosis to Therapy. Int. J. Mol. Sci. 2020, 21, 5805. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Panagiota, V.; Badbaran, A.; Zabelina, T.; Triviai, I.; Araujo Cruz, M.M.; Shahswar, R.; Ayuk, F.; Gehlhaar, M.; Wolschke, C.; et al. Impact of Molecular Genetics on Outcome in Myelofibrosis Patients after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1095–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintás-Cardama, A.; Abdel-Wahab, O.; Manshouri, T.; Kilpivaara, O.; Cortes, J.; Roupie, A.L.; Zhang, S.-J.; Harris, D.; Estrov, Z.; Kantarjian, H.; et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon α-2a. Blood 2013, 122, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Molecular Marker | PV | ET |
|---|---|---|
| Homozygosity (or high allelic frequency) of JAK2V617F | More common | Less common |
| Mutations in KMT2A and/or TP53 | More common | Less common |
| NFE2 expression | Higher | Lower |
| HSP70 and calreticulin protein expression | Higher | Lower |
| miR-145 and/or miR-451 expression | Higher | Normal |
| miR-222 expression | Lower | Higher |
| Risk Factor | Score |
|---|---|
| Age ≥ 67 years | 5 |
| Age 57–66 years | 2 |
| Leukocytes ≥ 15 × 109/L | 1 |
| Venous thrombosis | 1 |
| Risk Factor | Score |
|---|---|
| Age ≥ 67 years | 2 |
| Leukocytes ≥ 15 × 109/L | 1 |
| Thrombosis history | 1 |
| SRSF2 mutation | 3 |
| Risk Factor | Score |
|---|---|
| Haemoglobin < 11 g/dL | 2 |
| Circulating blasts ≥3% | 2 |
| CALR-unmutated 1 | 2 |
| Platelets < 150 × 109/L | 1 |
| Constitutional symptoms | 1 |
| Age | 0.15 per year |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stuckey, R.; Gómez-Casares, M.T. Recent Advances in the Use of Molecular Analyses to Inform the Diagnosis and Prognosis of Patients with Polycythaemia Vera. Int. J. Mol. Sci. 2021, 22, 5042. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095042
Stuckey R, Gómez-Casares MT. Recent Advances in the Use of Molecular Analyses to Inform the Diagnosis and Prognosis of Patients with Polycythaemia Vera. International Journal of Molecular Sciences. 2021; 22(9):5042. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095042
Chicago/Turabian StyleStuckey, Ruth, and María Teresa Gómez-Casares. 2021. "Recent Advances in the Use of Molecular Analyses to Inform the Diagnosis and Prognosis of Patients with Polycythaemia Vera" International Journal of Molecular Sciences 22, no. 9: 5042. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095042