
Proteomic Identification of Bombyx mori Organelles Using the Engineered Ascorbate Peroxidase APEX and Development of Silkworm Organelle Proteome Database (SilkOrganPDB)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
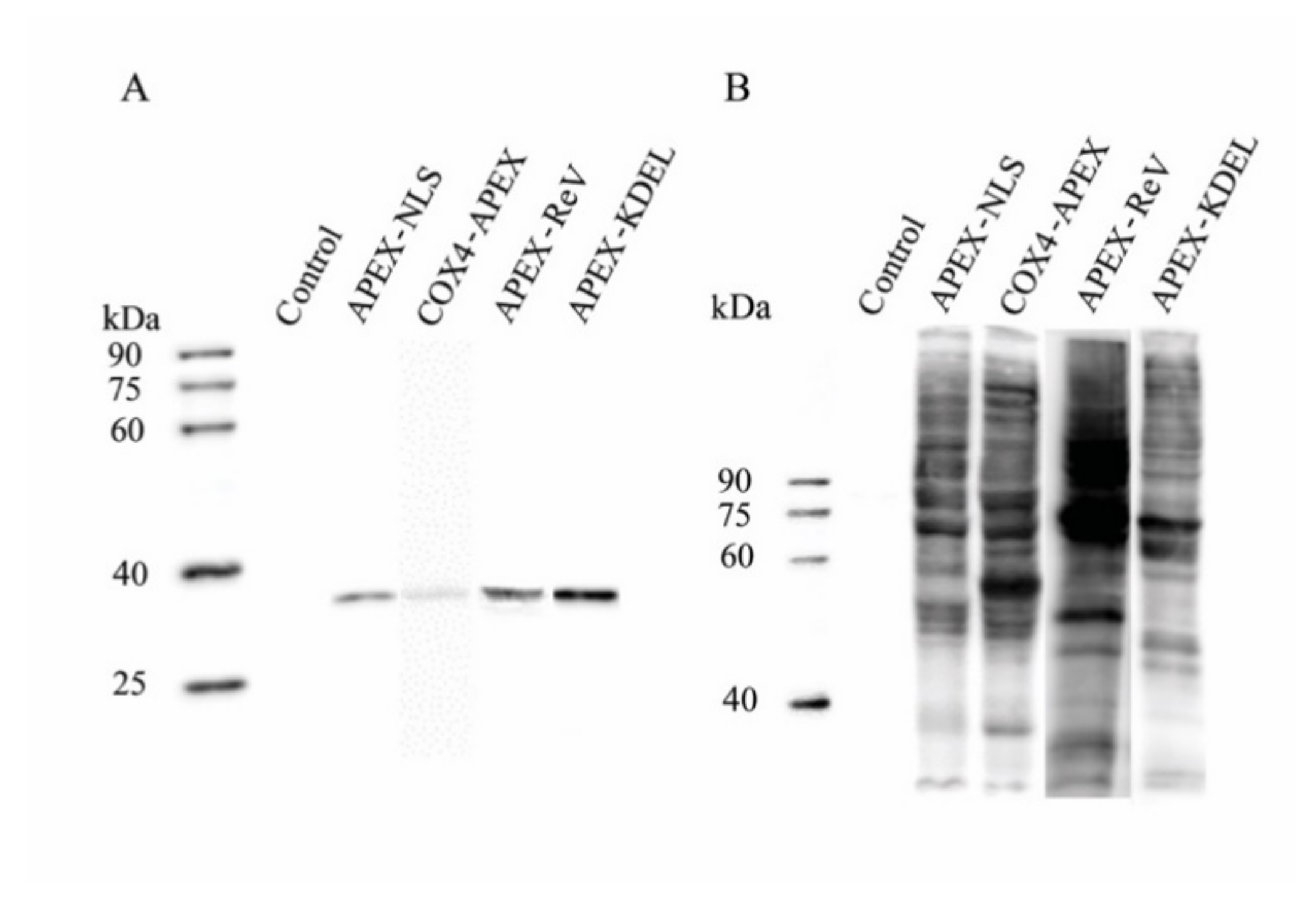
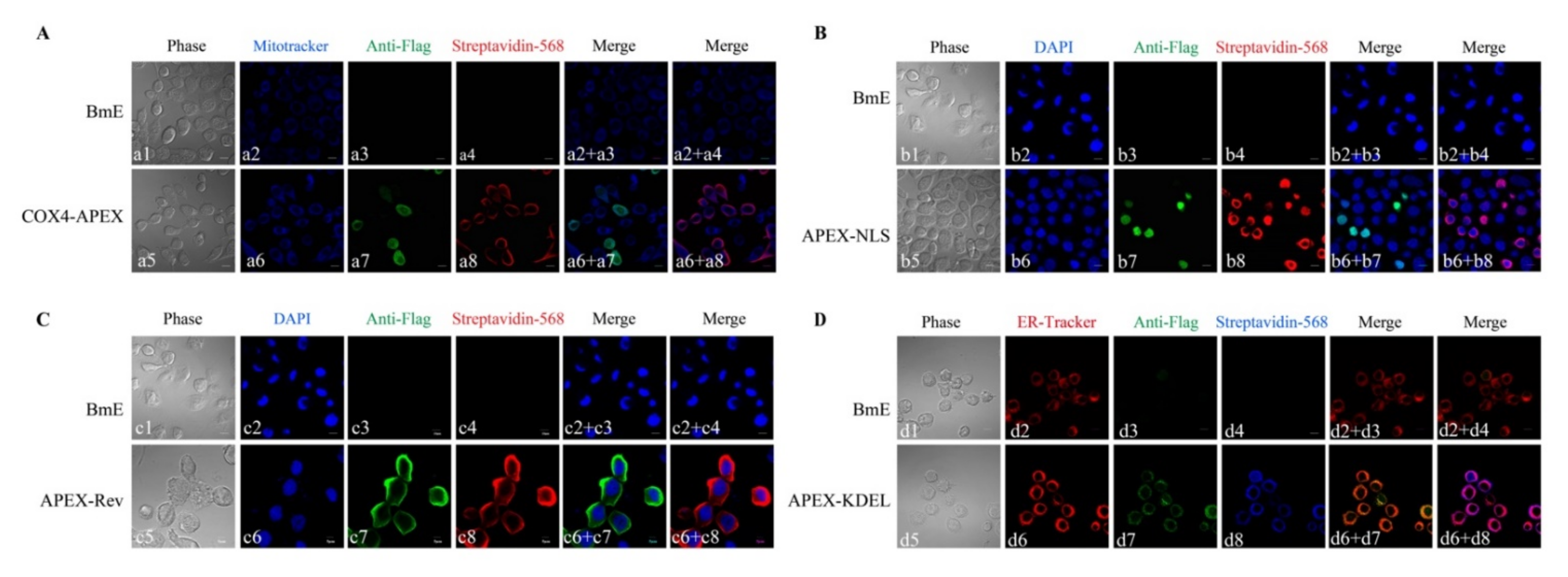
2.1. Expression of APEX in Transgenic BmE cells
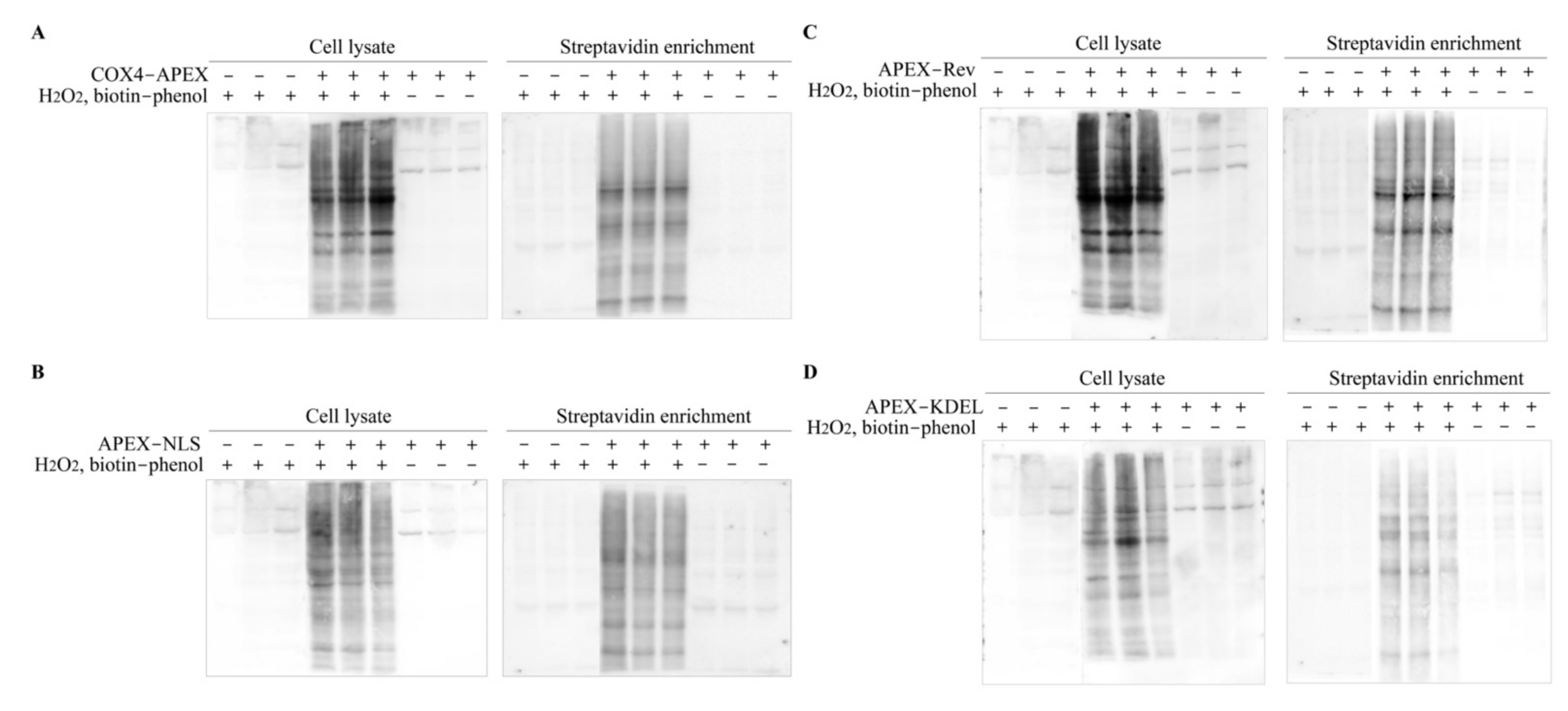
2.2. Verification of APEX-Mediated Protein Biotinylation
2.3. Enrichment of the Biotinylated Proteins
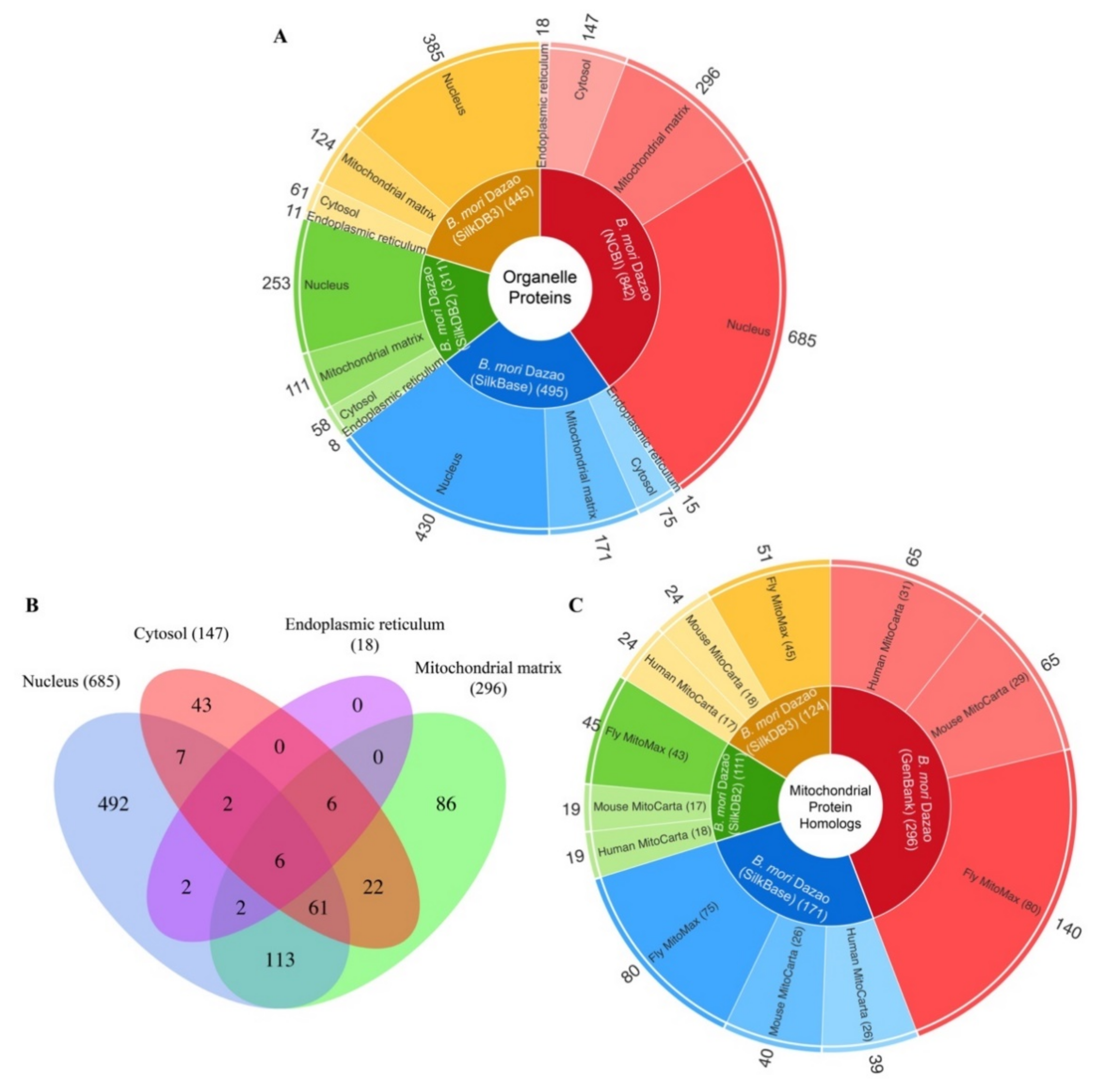
2.4. Identification and Annotation of the Biotinylated Proteins
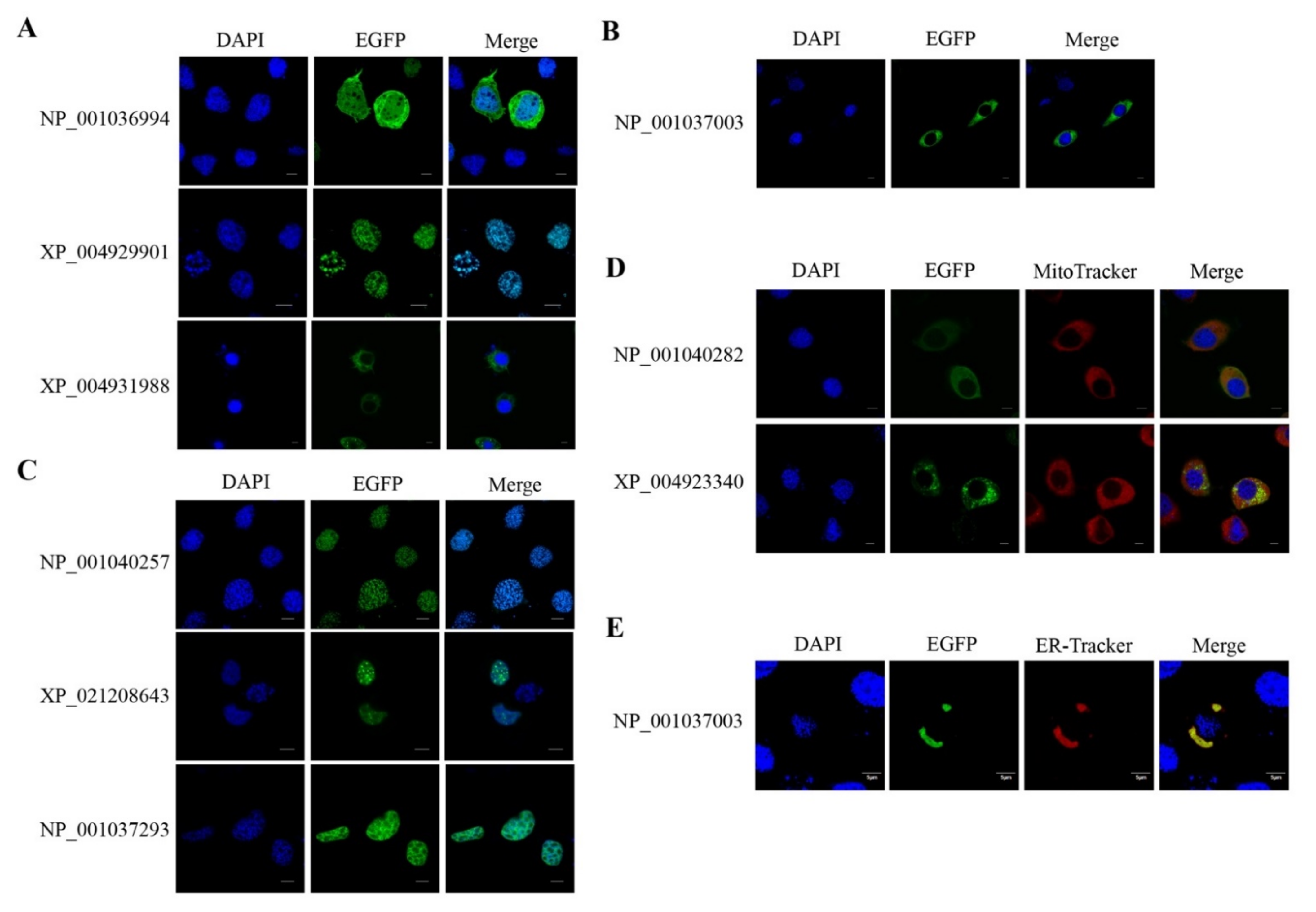
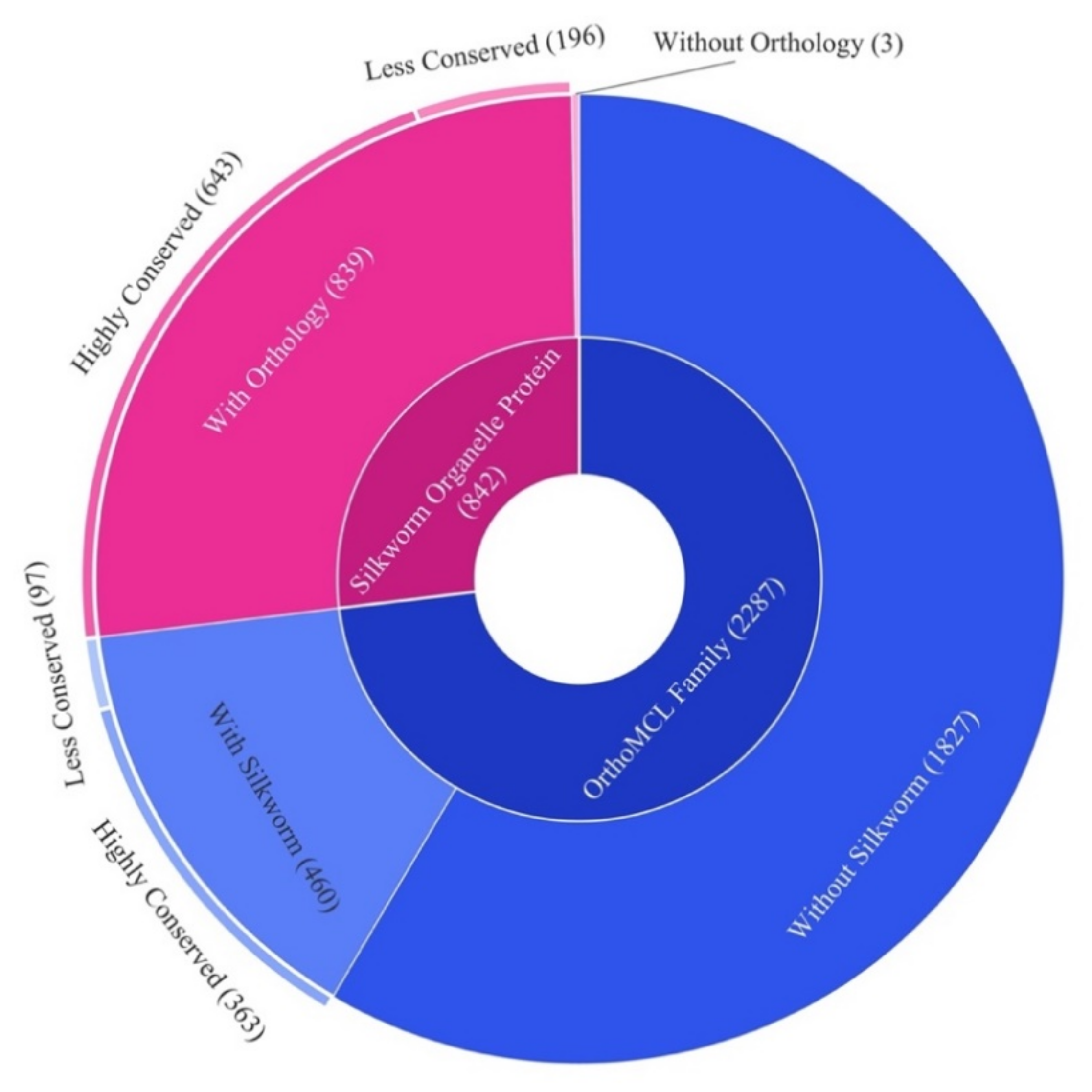
2.5. Orthology of the Organelle Proteins
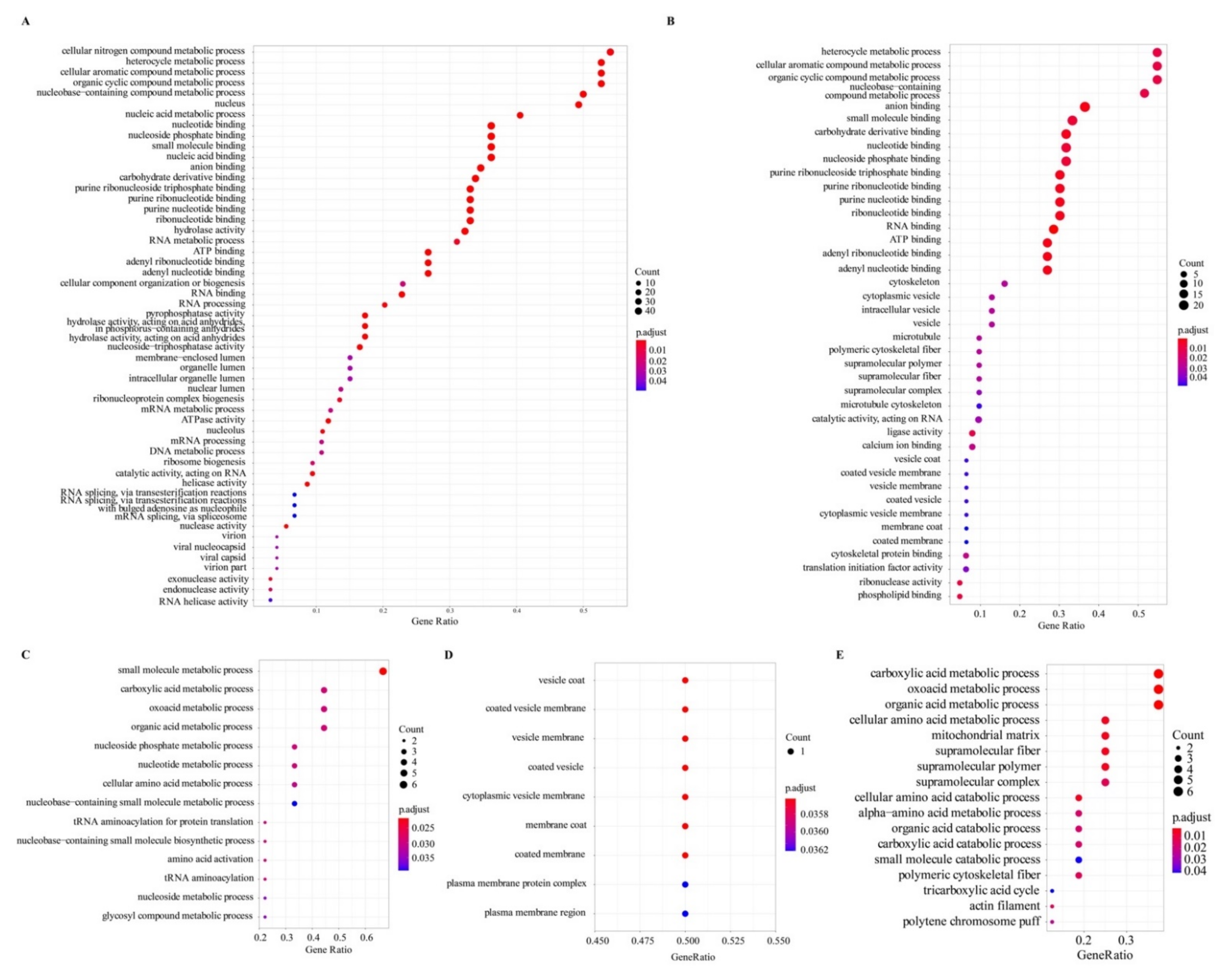
2.6. Gene Ontology Enrichment of the Identified Proteins
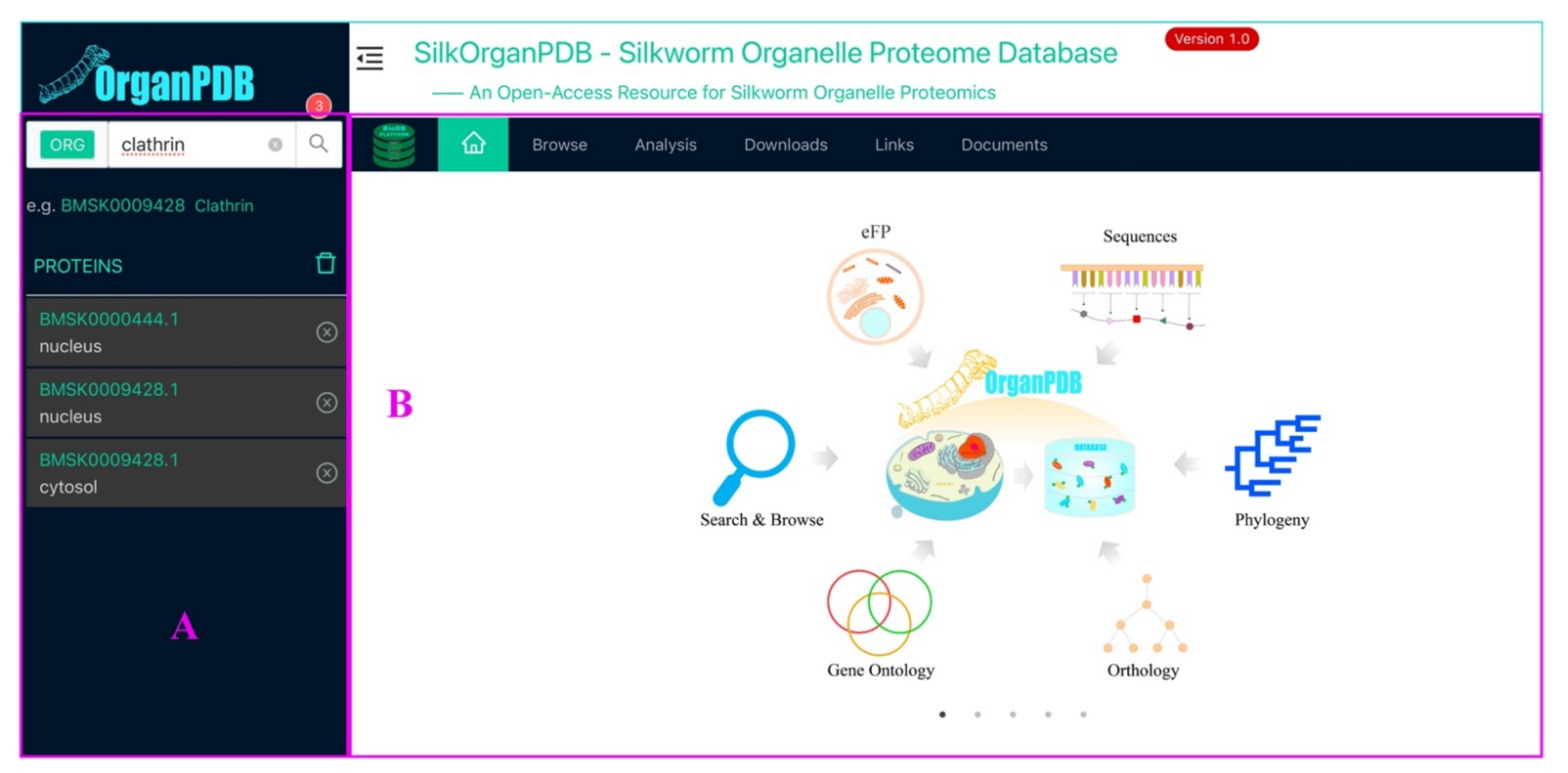
2.7. SilkOrganPDB Architecture and Implementation
2.7.1. Search and Browse Modules
2.7.2. eFP Module
2.7.3. Sequence and GO Module
2.7.4. Homologs and Phylogeny Module
2.7.5. BLAST Search and HMMER Search Modules
3. Discussion
4. Materials and Methods
4.1. Construction of APEX-Transgenic BmE Cell
4.2. Biotinylation of Organelle Proteins
4.3. Western Blotting and Streptavidin Blotting
4.4. Immunofluorescence Assay (IFA) of APEX Location and Biotinylated Proteins
4.5. Isolation of Biotinylated Proteins
4.6. Identification of Biotinylated Proteins
4.7. Homologous Search in Mitochondrial Protein Databases and Insect Proteomes
4.8. Gene Ontology Enrichment
4.9. Development of Silkworm Organelle Proteome Database (SilkOrganPDB)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xia, Q.; Zhou, Z.; Lu, C.; Cheng, D.; Dai, F.; Li, B.; Zhao, P.; Zha, X.; Cheng, T.; Chai, C.; et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 2004, 306, 1937–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Silkworm Genome, C. The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem. Mol. Biol. 2008, 38, 1036–1045. [Google Scholar] [CrossRef]
- Xia, Q.; Guo, Y.; Zhang, Z.; Li, D.; Xuan, Z.; Li, Z.; Dai, F.; Li, Y.; Cheng, D.; Li, R.; et al. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 2009, 326, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, M.; Jouraku, A.; Toyoda, A.; Yokoi, K.; Minakuchi, Y.; Katsuma, S.; Fujiyama, A.; Kiuchi, T.; Yamamoto, K.; Shimada, T. High-quality genome assembly of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2019, 107, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ye, L.; Lan, T.; Yu, M.; Liang, J.; Zhong, B. Comparative proteomic and phosphoproteomic analysis of the silkworm (Bombyx mori) posterior silk gland under high temperature treatment. Mol. Biol. Rep. 2012, 39, 8447–8456. [Google Scholar] [CrossRef]
- Xia, Q.; Cheng, D.; Duan, J.; Wang, G.; Cheng, T.; Zha, X.; Liu, C.; Zhao, P.; Dai, F.; Zhang, Z.; et al. Microarray-based gene expression profiles in multiple tissues of the domesticated silkworm, Bombyx mori. Genome Biol. 2007, 8, R162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Aso, Y.; Yamamoto, K.; Banno, Y.; Wang, Y.; Tsuchida, K.; Kawaguchi, Y.; Fujii, H. Proteome analysis of silk gland proteins from the silkworm, Bombyx mori. Proteomics 2006, 6, 2586–2599. [Google Scholar] [CrossRef]
- Li, J.; Hosseini Moghaddam, S.H.; Chen, X.; Chen, M.; Zhong, B. Shotgun strategy-based proteome profiling analysis on the head of silkworm Bombyx mori. Amino Acids 2010, 39, 751–761. [Google Scholar] [CrossRef]
- Li, J.Y.; Ye, L.P.; Che, J.Q.; Song, J.; You, Z.Y.; Wang, S.H.; Zhong, B.X. Proteome identification of the silkworm middle silk gland. Data Brief. 2016, 6, 903–907. [Google Scholar] [CrossRef]
- Dong, Z.; Zhao, P.; Zhang, Y.; Song, Q.; Zhang, X.; Guo, P.; Wang, D.; Xia, Q. Analysis of proteome dynamics inside the silk gland lumen of Bombyx mori. Sci. Rep. 2016, 6, 21158. [Google Scholar] [CrossRef] [Green Version]
- Bovilla, V.R.; Padwal, M.K.; Siripurapu, P.; Basu, B.; Mamillapalli, A. Developmental proteome dynamics of silk glands in the 5th instar larval stage of Bombyx mori L. (CSR2xCSR4). Biochim. Biophys. Acta 2016, 1864, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, Y.; Jiang, Y.; Xu, M. Proteome analysis of the silkworm (Bombyx mori. L.) colleterial gland during different development stages. Arch. Insect Biochem. Physiol. 2006, 61, 42–50. [Google Scholar] [CrossRef]
- Hou, Y.; Zou, Y.; Wang, F.; Gong, J.; Zhong, X.; Xia, Q.; Zhao, P. Comparative analysis of proteome maps of silkworm hemolymph during different developmental stages. Proteome Sci. 2010, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Chen, X.; Hosseini Moghaddam, S.H.; Chen, M.; Wei, H.; Zhong, B.X. Shotgun proteomics approach to characterizing the embryonic proteome of the silkworm, Bombyx mori, at labrum appearance stage. Insect Mol. Biol. 2009, 18, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Liu, Q.; Xia, Q.; Zhao, P. Proteome profile of spinneret from the silkworm, Bombyx mori. Proteomics 2017, 17. [Google Scholar] [CrossRef]
- Ye, X.; Tang, X.; Wang, X.; Che, J.; Wu, M.; Liang, J.; Ye, L.; Qian, Q.; Li, J.; You, Z.; et al. Improving Silkworm Genome Annotation Using a Proteogenomics Approach. J. Proteome Res. 2019, 18, 3009–3019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xia, D.; Zhao, Q.; Zhang, G.; Zhang, Y.; Qiu, Z.; Shen, D.; Lu, C. Label-free proteomic analysis of silkworm midgut infected by Bombyx mori nuclear polyhedrosis virus. J. Proteomics 2019, 200, 40–50. [Google Scholar] [CrossRef]
- Wu, P.; Shang, Q.; Huang, H.; Zhang, S.; Zhong, J.; Hou, Q.; Guo, X. Quantitative proteomics analysis provides insight into the biological role of Hsp90 in BmNPV infection in Bombyx mori. J. Proteomics 2019, 203, 103379. [Google Scholar] [CrossRef]
- Lu, D.; Xu, P.; Hou, C.; Gao, K.; Guo, X. Label-free LC-MS/MS proteomic analysis of the hemolymph of silkworm larvae infected with Beauveria bassiana. J. Invertebr. Pathol. 2019, 166, 107227. [Google Scholar] [CrossRef]
- Chen, S.; Dong, Z.; Ren, X.; Zhao, D.; Zhang, Y.; Tang, M.; Han, J.; Ye, L.; Zhao, P. Proteomic Identification of Immune-Related Silkworm Proteins Involved in the Response to Bacterial Infection. J. Insect Sci. 2019, 19. [Google Scholar] [CrossRef]
- Mao, F.; Lei, J.; Enoch, O.; Wei, M.; Zhao, C.; Quan, Y.; Yu, W. Quantitative proteomics of Bombyx mori after BmNPV challenge. J. Proteomics 2018, 181, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, X.; Xu, J.; Ma, Y.; Zhang, S.; Yu, D.; Fei, D.; Muhammad, A. iTRAQ-based quantitative proteomics analysis of molecular mechanisms associated with Bombyx mori (Lepidoptera) larval midgut response to BmNPV in susceptible and near-isogenic strains. J. Proteomics 2017, 165, 35–50. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yu, H.Z.; Xu, J.P.; Zhang, S.Z.; Yu, D.; Liu, M.H.; Wang, L.L. Comparative Subcellular Proteomics Analysis of Susceptible and Near-isogenic Resistant Bombyx mori (Lepidoptera) Larval Midgut Response to BmNPV infection. Sci. Rep. 2017, 7, 45690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, V.; Udeshi, N.D.; Lam, S.S.; Loh, K.H.; Cox, K.J.; Pedram, K.; Carr, S.A.; Ting, A.Y. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 2016, 11, 456–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Han, S.; Li, H.; Udeshi, N.D.; Svinkina, T.; Mani, D.R.; Xu, C.; Guajardo, R.; Xie, Q.; Li, T.; et al. Cell-Surface Proteomic Profiling in the Fly Brain Uncovers Wiring Regulators. Cell 2020, 180, 373.e15–386.e15. [Google Scholar] [CrossRef]
- Droujinine, I.A.; Meyer, A.S.; Wang, D.; Udeshi, N.D.; Hu, Y.; Rocco, D.; McMahon, J.A.; Yang, R.; Guo, J.; Mu, L.; et al. Proteomics of protein trafficking by in vivo tissue-specific labeling. Nat. Commun. 2021, 12, 2382. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Espenshade, P.J. Proximity-dependent biotin labelling in yeast using the engineered ascorbate peroxidase APEX2. Biochem. J. 2016, 473, 2463–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Hu, Y.; Udeshi, N.D.; Lau, T.Y.; Wirtz-Peitz, F.; He, L.; Ting, A.Y.; Carr, S.A.; Perrimon, N. Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 12093–12098. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Olzmann, J.A. Identification of Lipid Droplet Proteomes by Proximity Labeling Proteomics Using APEX2. Methods Mol. Biol. 2019, 2008, 57–72. [Google Scholar] [CrossRef]
- Reinke, A.W.; Mak, R.; Troemel, E.R.; Bennett, E.J. In vivo mapping of tissue- and subcellular-specific proteomes in Caenorhabditis elegans. Sci. Adv. 2017, 3, e1602426. [Google Scholar] [CrossRef] [Green Version]
- Hung, V.; Lam, S.S.; Udeshi, N.D.; Svinkina, T.; Guzman, G.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 2017, 6, e24463. [Google Scholar] [CrossRef] [Green Version]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Ami, G.O.H.; Web Presence Working, G. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- O’Brate, A.; Giannakakou, P. The importance of p53 location: Nuclear or cytoplasmic zip code? Drug Resist. Updat. 2003, 6, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Gonja, H.; Koike, R.; Fukuchi, S. Multiple-Localization and Hub Proteins. PLoS ONE 2016, 11, e0156455. [Google Scholar] [CrossRef] [PubMed]
- Veres, D.V.; Gyurko, D.M.; Thaler, B.; Szalay, K.Z.; Fazekas, D.; Korcsmaros, T.; Csermely, P. ComPPI: A cellular compartment-specific database for protein-protein interaction network analysis. Nucleic Acids Res. 2015, 43, D485–d493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Wei, Z.; Luo, Y.; Guo, H.; Zhang, G.; Xia, Q.; Wang, Y. SilkDB 3.0: Visualizing and exploring multiple levels of data for silkworm. Nucleic Acids Res. 2020, 48, D749–D755. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Duan, J.; Li, R.; Cheng, D.; Fan, W.; Zha, X.; Cheng, T.; Wu, Y.; Wang, J.; Mita, K.; Xiang, Z.; et al. SilkDB v2.0: A platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res. 2010, 38, D453–D456. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Kim, S.; Fermin, D.; Tsou, C.C.; Nesvizhskii, A.I. QPROT: Statistical method for testing differential expression using protein-level intensity data in label-free quantitative proteomics. J. Proteomics 2015, 129, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Xiang, H.; Liu, X.; Li, M.; Zhu, Y.; Wang, L.; Cui, Y.; Liu, L.; Fang, G.; Qian, H.; Xu, A.; et al. The evolutionary road from wild moth to domestic silkworm. Nat. Ecol. Evol. 2018, 2, 1268–1279. [Google Scholar] [CrossRef]
- Gershman, A.; Romer, T.G.; Fan, Y.; Razaghi, R.; Smith, W.A.; Timp, W. De novo genome assembly of the tobacco hornworm moth (Manduca sexta). G3 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Li, X.; Fan, D.; Zhang, W.; Liu, G.; Zhang, L.; Zhao, L.; Fang, X.; Chen, L.; Dong, Y.; Chen, Y.; et al. Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat. Commun. 2015, 6, 8212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Wu, J.; Wu, Y.; Chilukuri, R.V.; Huang, L.; Yamamoto, K.; Feng, L.; Li, W.; Chen, Z.; Guo, H.; et al. Genomic adaptation to polyphagy and insecticides in a major East Asian noctuid pest. Nat. Ecol. Evol. 2017, 1, 1747–1756. [Google Scholar] [CrossRef]
- Chen, W.; Yang, X.; Tetreau, G.; Song, X.; Coutu, C.; Hegedus, D.; Blissard, G.; Fei, Z.; Wang, P. A high-quality chromosome-level genome assembly of a generalist herbivore, Trichoplusia ni. Mol. Ecol. Resour. 2019, 19, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Tribolium Genome Sequencing, C.; Richards, S.; Gibbs, R.A.; Weinstock, G.M.; Brown, S.J.; Denell, R.; Beeman, R.W.; Gibbs, R.; Beeman, R.W.; Brown, S.J.; et al. The genome of the model beetle and pest Tribolium castaneum. Nature 2008, 452, 949–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nene, V.; Wortman, J.R.; Lawson, D.; Haas, B.; Kodira, C.; Tu, Z.J.; Loftus, B.; Xi, Z.; Megy, K.; Grabherr, M.; et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science 2007, 316, 1718–1723. [Google Scholar] [CrossRef] [Green Version]
- Hoskins, R.A.; Carlson, J.W.; Wan, K.H.; Park, S.; Mendez, I.; Galle, S.E.; Booth, B.W.; Pfeiffer, B.D.; George, R.A.; Svirskas, R.; et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 2015, 25, 445–458. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Jung, J.W.; Choi, B.S.; Jayakodi, M.; Lee, J.; Lim, J.; Yu, Y.; Choi, Y.S.; Lee, M.L.; Park, Y.; et al. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genomics 2015, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genomics 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Eilbeck, K.; Lewis, S.E.; Mungall, C.J.; Yandell, M.; Stein, L.; Durbin, R.; Ashburner, M. The Sequence Ontology: A tool for the unification of genome annotations. Genome Biol. 2005, 6, R44. [Google Scholar] [CrossRef] [Green Version]
- Mungall, C.J.; Emmert, D.B.; FlyBase, C. A Chado case study: An ontology-based modular schema for representing genome-associated biological information. Bioinformatics 2007, 23, i337–i346. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23, 205–211. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Xu, C.; Xu, J.; Luo, J.; Yu, B.; Meng, X.; Li, C.; Pan, G.; Zhou, Z. Proteomic Identification of Bombyx mori Organelles Using the Engineered Ascorbate Peroxidase APEX and Development of Silkworm Organelle Proteome Database (SilkOrganPDB). Int. J. Mol. Sci. 2021, 22, 5051. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095051
Li T, Xu C, Xu J, Luo J, Yu B, Meng X, Li C, Pan G, Zhou Z. Proteomic Identification of Bombyx mori Organelles Using the Engineered Ascorbate Peroxidase APEX and Development of Silkworm Organelle Proteome Database (SilkOrganPDB). International Journal of Molecular Sciences. 2021; 22(9):5051. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095051
Chicago/Turabian StyleLi, Tian, Chen Xu, Jinzhi Xu, Jian Luo, Bin Yu, Xianzhi Meng, Chunfeng Li, Guoqing Pan, and Zeyang Zhou. 2021. "Proteomic Identification of Bombyx mori Organelles Using the Engineered Ascorbate Peroxidase APEX and Development of Silkworm Organelle Proteome Database (SilkOrganPDB)" International Journal of Molecular Sciences 22, no. 9: 5051. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095051