
Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes

 ,
,  , , , and
, , , and
Abstract
:1. Adipose Tissue/β-Cell Cross-Talk: A Bidirectional Communication
2. Dysfunctional Adipose Tissue in Obesity: Alteration of the Adipocyte Secretome
3. Dysfunctional Adipose Tissue Secretome Affects β-Cell Functional Mass: The Natural History of an Announced Failure
3.1. How Adipose Tissue Secretome Influences β-Cell Compensatory Hyperplasia
3.2. How the Adipose Tissue Secretome Influences β-Cell Compensatory Insulin Hypersecretion
3.3. How the Adipose Tissue Secretome Influences β-Cell Insulin Secretory Dysfunction
3.4. How the Adipose Tissue Secretome Influences Loss of β-Cell Mass (Apoptosis/Dedifferentiation)
4. Future Perspectives and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gil, A.; Olza, J.; Gil-Campos, M.; Gomez-Llorente, C.; Aguilera, C. Is adipose tissue metabolically different at different sites? Int. J. Pediatr. Obes. 2011, 6 (Suppl. S1), 13–20. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, M.; Wolfrum, C. The origin and definition of brite versus white and classical brown adipocytes. Adipocyte 2014, 3, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Smorlesi, A.; Frontini, A.; Barbatelli, G.; Cint, S. White, brown and pink adipocytes: The extraordinary plasticity of the adipose organ. Eur. J. Endocrinol. 2014, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, S. Pink Adipocytes. Trends Endocrinol. Metab. 2018, 29, 651–666. [Google Scholar] [CrossRef]
- Wajchenberg, B.; Giannella-Neto, D.; da Silva, M.; Santos, R. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm. Metab. Res. 2002, 34, 616–621. [Google Scholar] [CrossRef]
- Ibrahim, M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Cignarelli, A.; Genchi, V.A.; Perrini, S.; Natalicchio, A.; Laviola, L.; Giorgino, F. Insulin and Insulin receptors in adipose tissue development. Int. J. Mol. Sci. 2019, 20, 759. [Google Scholar] [CrossRef] [Green Version]
- Meijssen, S.; Cabezas, M.; Ballieux, C.; Derksen, R.; Bilecen, S.; Erkelens, D. Insulin mediated inhibition of hormone sensitive lipase activity in vivo in relation to endogenous catecholamines in healthy subjects. J. Clin. Endocrinol. Metab. 2001, 86, 4193–4197. [Google Scholar] [CrossRef]
- Sadur, C.; Eckel, R. Insulin stimulation of adipose tissue lipoprotein lipase. Use of the euglycemic clamp technique. J. Clin. Investig. 1982, 69, 1119–1125. [Google Scholar] [CrossRef] [Green Version]
- Czech, M.P.; Tencerova, M.; Pedersen, D.J.; Aouadi, M. Insulin signalling mechanisms for triacylglycerol storage. Diabetologia 2013, 56, 949. [Google Scholar] [CrossRef] [Green Version]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.F.; Feng, D.D.; Chen, C. Contribution of adipocyte-derived factors to beta-cell dysfunction in diabetes. Int. J. Biochem. Cell Biol. 2006, 38, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M.T. Adipose tissue: An endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 2016, 26, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, V.; Shyng, S. Leptin-induced trafficking of K ATP channels: A mechanism to regulate pancreatic β-cell excitability and insulin secretion. Int. J. Mol. Sci. 2019, 20, 2660. [Google Scholar] [CrossRef] [Green Version]
- Morioka, T.; Asilmaz, E.; Hu, J.; Dishinger, J.; Kurpad, A.; Elias, C.; Li, H.; Elmquist, J.; Kennedy, R.; Kulkarni, R. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J. Clin. Investig. 2007, 117, 2860–2868. [Google Scholar] [CrossRef] [Green Version]
- Wijesekara, N.; Krishnamurthy, M.; Bhattacharjee, A.; Suhail, A.; Sweeney, G.; Wheeler, M.B. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J. Biol. Chem. 2010, 285, 33623–33631. [Google Scholar] [CrossRef] [Green Version]
- Cantley, J. The control of insulin secretion by adipokines: Current evidence for adipocyte-beta cell endocrine signalling in metabolic homeostasis. Mamm. Genome 2014, 25, 442–454. [Google Scholar] [CrossRef]
- Shirakawa, J.; De Jesus, D.F.; Kulkarni, R.N. Exploring inter-organ crosstalk to uncover mechanisms that regulate β-cell function and mass. Eur. J. Clin. Nutr. 2017, 71, 896–903. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Available online: https://www.who.int/health-topics/obesity#tab=tab_1 (accessed on 21 April 2022).
- Petrov, M.S.; Taylor, R. Intra-pancreatic fat deposition: Bringing hidden fat to the fore. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 153–168. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Honka, H.; Koffert, J.; Hannukainen, J.C.; Tuulari, J.J.; Karlsson, H.K.; Immonen, H.; Oikonen, V.; Tolvanen, T.; Soinio, M.; Salminen, P.; et al. The effects of bariatric surgery on pancreatic lipid metabolism and blood flow. J. Clin. Endocrinol. Metab. 2015, 100, 2015–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.; Cushman, S.; Periwal, V. Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PLoS Comput. Biol. 2009, 5, e324. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Jelenčić, V.; Valentić, S.; Šestan, M.; Wensveen, T.T.; Theurich, S.; Glasner, A.; Mendrila, D.; Štimac, D.; Wunderlich, F.T.; et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol. 2015, 16, 376–385. [Google Scholar] [CrossRef]
- Cifarelli, V.; Beeman, S.; Smith, G.; Yoshino, J.; Morozov, D.; Beals, J.; Kayser, B.; Watrous, J.; Jain, M.; Patterson, B.; et al. Decreased adipose tissue oxygenation associates with insulin resistance in individuals with obesity. J. Clin. Investig. 2020, 130, 6688–6699. [Google Scholar] [CrossRef] [PubMed]
- Maury, E.; Ehala-Aleksejev, K.; Guiot, Y.; Detry, R.; Vandenhooft, A.; Brichard, S.M. Adipokines oversecreted by omental adipose tissue in human obesity. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Maury, E.; Brichard, S.M.; Pataky, Z.; Carpentier, A.; Golay, A.; Bobbioni-Harsch, E. Effect of obesity on growth-related oncogene factor-alpha, thrombopoietin, and tissue inhibitor metalloproteinase-1 serum levels. Obesity 2010, 18, 1503–1509. [Google Scholar] [CrossRef]
- Jialal, I.; Adams-Huet, B.; Duong, F.; Smith, G. Relationship between retinol-binding protein-4/adiponectin and leptin/adiponectin ratios with insulin resistance and inflammation. Metab. Syndr. Relat. Disord. 2014, 12, 227–230. [Google Scholar] [CrossRef]
- Boucher, J.; Masri, B.; Daviaud, D.; Gesta, S.; Guigné, C.; Mazzucotelli, A.; Castan-Laurell, I.; Tack, I.; Knibiehler, B.; Carpéné, C.; et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology 2005, 146, 1764–1771. [Google Scholar] [CrossRef]
- Su, K.Z.; Li, Y.R.; Zhang, D.; Yuan, J.H.; Zhang, C.S.; Liu, Y.; Song, L.M.; Lin, Q.; Li, M.W.; Dong, J. Relation of circulating resistin to insulin resistance in type 2 diabetes and obesity: A systematic review and meta-analysis. Front. Physiol. 2019, 10, 1399. [Google Scholar] [CrossRef] [Green Version]
- Pieńkowska, J.; Brzeska, B.; Kaszubowski, M.; Kozak, O.; Jankowska, A.; Szurowska, E. The correlation between the MRI-evaluated ectopic fat accumulation and the incidence of diabetes mellitus and hypertension depends on body mass index and waist circumference ratio. PLoS ONE 2020, 15, e0226889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, R.; Lang, W.; Wadden, T.; Safford, M.; Knowler, W.; Bertoni, A.; Hill, J.; Brancati, F.; Peters, A.; Wagenknecht, L. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care 2011, 34, 1481–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porro, S.; Genchi, V.A.; Cignarelli, A.; Natalicchio, A.; Laviola, L.; Giorgino, F.; Perrini, S. Dysmetabolic adipose tissue in obesity: Morphological and functional characteristics of adipose stem cells and mature adipocytes in healthy and unhealthy obese subjects. J. Endocrinol. Investig. 2021, 44, 921–941. [Google Scholar] [CrossRef] [PubMed]
- Popko, K.; Gorska, E.; Stelmaszczyk-Emmel, A.; Plywaczewski, R.; Stoklosa, A.; Gorecka, D.; Pyrzak, B.; Demkow, U. Proinflammatory cytokines Il-6 and TNF-α and the development of inflammation in obese subjects. Eur. J. Med. Res. 2010, 15 (Suppl. S2), 120–122. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Monocyte Chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): Implication of macrophages resident in the AT. J. Clin. Endocrinol. Metab. 2005, 90, 2282–2289. [Google Scholar] [CrossRef]
- Charles, B.A.; Doumatey, A.; Huang, H.; Zhou, J.; Chen, G.; Shriner, D.; Adeyemo, A.; Rotimi, C.N. The roles of IL-6, IL-10, and IL-1RA in obesity and insulin resistance in african-americans. J. Clin. Endocrinol. Metab. 2011, 96, E2018–E2022. [Google Scholar] [CrossRef]
- Mishima, Y.; Kuyama, A.; Tada, A.; Takahashi, K.; Ishioka, T.; Kibata, M. Relationship between serum tumor necrosis factor-α and insulin resistance in obese men with Type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2001, 52, 119–123. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265. [Google Scholar] [CrossRef] [Green Version]
- Bahceci, M.; Gokalp, D.; Bahceci, S.; Tuzcu, A.; Atmaca, S.; Arikan, S. The correlation between adiposity and adiponectin, tumor necrosis factor alpha, interleukin-6 and high sensitivity C-reactive protein levels. Is adipocyte size associated with inflammation in adults? J. Endocrinol. Investig. 2007, 30, 210–214. [Google Scholar] [CrossRef]
- Alzamil, H. Elevated serum TNF-α is related to obesity in type 2 diabetes mellitus and is associated with glycemic control and insulin resistance. J. Obes. 2020, 2020, 5076858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaroop, J.; Rajarajeswari, D.; Naidu, J. Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J. Med. Res. 2012, 135, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Curat, C.A.; Wegner, V.; Sengenès, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumié, A. Macrophages in human visceral adipose tissue: Increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Baturcam, E.; Abubaker, J.; Tiss, A.; Abu-Farha, M.; Khadir, A.; Al-Ghimlas, F.; Al-Khairi, I.; Cherian, P.; Elkum, N.; Hammad, M.; et al. Physical exercise reduces the expression of RANTES and its CCR5 receptor in the adipose tissue of obese humans. Mediat. Inflamm. 2014, 2014, 627150. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, G.; Ben-Aicha, S.; Badimon, L. New insights into the role of adipose tissue in thrombosis. Cardiovasc. Res. 2017, 113, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Loukinova, E.; Dong, G.; Enamorado-Ayalya, I.; Thomas, G.R.; Chen, Z.; Schreiber, H.; Van Waes, C. Growth regulated oncogene-α expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene 2000, 19, 3477–3486. [Google Scholar] [CrossRef] [Green Version]
- Meissburger, B.; Stachorski, L.; Röder, E.; Rudofsky, G.; Wolfrum, C. Tissue inhibitor of matrix metalloproteinase 1 (TIMP1) controls adipogenesis in obesity in mice and in humans. Diabetologia 2011, 54, 1468–1479. [Google Scholar] [CrossRef] [Green Version]
- Alessi, M.C.; Poggi, M.; Juhan-Vague, I. Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Curr. Opin. Lipidol. 2007, 18, 240–245. [Google Scholar] [CrossRef]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef]
- Noh, H.J.; Kim, C.S.; Kang, J.H.; Park, J.Y.; Choe, S.Y.; Hong, S.M.; Yoo, H.; Park, T.; Yu, R. Quercetin suppresses MIP-1α-induced adipose inflammation by downregulating its receptors CCR1/CCR5 and inhibiting inflammatory signaling. J. Med. Food 2014, 17, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J. Clin. Endocrinol. Metab. 2008, 93, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Barouch, L. Leptin signaling and obesity: Cardiovascular consequences. Circ. Res. 2007, 101, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Hussain, Z.; Khan, J.A. Food intake regulation by leptin: Mechanisms mediating gluconeogenesis and energy expenditure. Asian Pac. J. Trop. Med. 2017, 10, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Genchi, V.A.; D’oria, R.; Palma, G.; Caccioppoli, C.; Cignarelli, A.; Natalicchio, A.; Laviola, L.; Giorgino, F.; Perrini, S. Impaired leptin signalling in obesity: Is leptin a new thermolipokine? Int. J. Mol. Sci. 2021, 22, 6445. [Google Scholar] [CrossRef] [PubMed]
- Dubuc, P. The development of obesity, hyperinsulinemia, and hyperglycemia in ob/ob mice. Metabolism 1976, 25, 1567–1574. [Google Scholar] [CrossRef]
- Considine, R.; Sinha, M.; Heiman, M.; Kriauciunas, A.; Stephens, T.; Nyce, M.; Ohannesian, J.; Marco, C.; McKee, L.; Bauer, T. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Zuo, H.; Shi, Z.; Yuan, B.; Dai, Y.; Wu, G.; Hussain, A. Association between serum leptin concentrations and insulin resistance: A population-based study from china. PLoS ONE 2013, 8, e54615. [Google Scholar] [CrossRef] [Green Version]
- Al Maskari, M.Y.; Alnaqdy, A.A. Correlation between serum leptin levels, body mass index and obesity in omanis. Sultan Qaboos Univ. Med. J. 2006, 6, 27–31. [Google Scholar]
- Hamed, E.A.; Zakary, M.M.; Ahmed, N.S.; Gamal, R.M. Circulating leptin and insulin in obese patients with and without type 2 diabetes mellitus: Relation to ghrelin and oxidative stress. Diabetes Red. Clin. Pract. 2011, 94, 434–441. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovren, F.; Pan, Y.; Quan, A.; Szmitko, P.E.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Chan, L.; Al-Omran, M.; Teoh, H.; et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. The pleiotropic actions of adiponectin are initiated via receptor-mediated activation of ceramidase activity. Nat. Med. 2011, 17, 55. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.; Tian, X.; Xu, A.; Yu, J.; Lau, C.; Hoo, R.; Wang, Y.; Lee, V.; Lam, K.; Vanhoutte, P.; et al. Adiponectin is required for PPARγ-mediated improvement of endothelial function in diabetic mice. Cell Metab. 2011, 14, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Mojiminiyi, O.A.; Abdella, N.A.; Al Arouj, M.; Ben Nakhi, A. Adiponectin, insulin resistance and clinical expression of the metabolic syndrome in patients with Type 2 diabetes. Int. J. Obes. 2007, 31, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Putz, D.M.; Goldner, W.S.; Bar, R.S.; Haynes, W.G.; Sivitz, W.I. Adiponectin and C-reactive protein in obesity, type 2 diabetes, and monodrug therapy. Metabolism 2004, 53, 1454–1461. [Google Scholar] [CrossRef]
- Kim, C.; Park, J.; Park, J.; Kang, E.; Ahn, C.; Cha, B.; Lim, S.; Kim, K.; Lee, H. Comparison of body fat composition and serum adiponectin levels in diabetic obesity and non-diabetic obesity. Obesity 2006, 14, 1164–1171. [Google Scholar] [CrossRef]
- Derosa, G.; Catena, G.; Gaudio, G.; D’Angelo, A.; Maffioli, P. Adipose tissue dysfunction and metabolic disorders: Is it possible to predict who will develop type 2 diabetes mellitus? Role of markers in the progression of diabetes in obese patients (The RESISTIN trial). Cytokine 2020, 127, 154947. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Benomar, Y.; Gertler, A.; De Lacy, P.; Crépin, D.; Hamouda, H.O.; Riffault, L.; Taouis, M. Central resistin overexposure induces insulin resistance through Toll-like receptor 4. Diabetes 2013, 62, 102–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, D.G.; Schindler, K.; Schaller, G.; Prager, G.; Wolzt, M.; Ludvik, B. Increased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric banding. J. Clin. Endocrinol. Metab. 2006, 91, 1578–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandeep, S.; Velmurugan, K.; Deepa, R.; Mohan, V. Serum visfatin in relation to visceral fat, obesity, and type 2 diabetes mellitus in Asian Indians. Metabolism 2007, 56, 565–570. [Google Scholar] [CrossRef]
- Castan-Laurell, I.; Vítkova, M.; Daviaud, D.; Dray, C.; Kováčiková, M.; Kovacova, Z.; Hejnova, J.; Stich, V.; Valet, P. Effect of hypocaloric diet-induced weight loss in obese women on plasma apelin and adipose tissue expression of apelin and APJ. Eur. J. Endocrinol. 2008, 158, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.V.; Laaksonen, D.E.; Karhu, T.; Karhunen, L.; Laitinen, T.; Kainulainen, S.; Rissanen, A.; Niskanen, L.; Herzig, K.H. Effect of diet-induced weight loss on plasma apelin and cytokine levels in individuals with the metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Xu, J.Y.; Stejskal, D.; Tam, S.; Zhang, J.; Wat, N.M.S.; Wong, W.K.; Lam, K.S.L. Adipocyte fatty acid–binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin. Chem. 2006, 52, 405–413. [Google Scholar] [CrossRef]
- Shan, T.; Liu, W.; Kuang, S. Fatty acid binding protein 4 expression marks a population of adipocyte progenitors in white and brown adipose tissues. FASEB J. 2013, 27, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Vasilenko, M.A.; Kirienkova, E.V.; Skuratovskaia, D.A.; Zatolokin, P.A.; Mironyuk, N.I.; Litvinova, L.S. The role of production of adipsin and leptin in the development of insulin resistance in patients with abdominal obesity. Dokl. Biochem. Biophys. 2017, 475, 271–276. [Google Scholar] [CrossRef]
- Guo, D.; Liu, J.; Zhang, P.; Yang, X.; Liu, D.; Lin, J.; Wei, X.; Xu, B.; Huang, C.; Zhou, X.; et al. Adiposity measurements and metabolic syndrome are linked through circulating neuregulin 4 and adipsin levels in obese adults. Front. Physiol. 2021, 12, 667330. [Google Scholar] [CrossRef]
- Cao, R.Y.; Zheng, H.; Redfearn, D.; Yang, J. FNDC5: A novel player in metabolism and metabolic syndrome. Biochimie 2019, 158, 111–116. [Google Scholar] [CrossRef]
- Shoukry, A.; Shalaby, S.M.; El-Arabi Bdeer, S.; Mahmoud, A.A.; Mousa, M.M.; Khalifa, A. Circulating serum irisin levels in obesity and type 2 diabetes mellitus. IUBMB Life 2016, 68, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Ahima, R.S. Resistin in rodents and humans. Diabetes Metab. J. 2013, 37, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieva-Vazquez, A.; Pérez-Fuentes, R.; Torres-Rasgado, E.; López-López, J.G.; Romero, J.R. Serum resistin levels are associated with adiposity and insulin sensitivity in obese hispanic subjects. Metab. Syndr. Relat. Disord. 2014, 12, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janowska, J.; Zahorska-Markiewicz, B.; Olszanecka-Glinianowicz, M. Relationship between serum resistin concentration and proinflammatory cytokines in obese women with impaired and normal glucose tolerance. Metabolism 2006, 55, 1495–1499. [Google Scholar] [CrossRef]
- Terra, X.; Auguet, T.; Quesada, I.; Aguilar, C.; Luna, A.M.; Hernández, M.; Sabench, F.; Porras, J.A.; Martínez, S.; Lucas, A.; et al. Increased levels and adipose tissue expression of visfatin in morbidly obese women: The relationship with pro-inflammatory cytokines. Clin. Endocrinol. 2012, 77, 691–698. [Google Scholar] [CrossRef]
- Chang, Y.H.; Chang, D.M.; Lin, K.C.; Shin, S.J.; Lee, Y.J. Visfatin in overweight/obesity, type 2 diabetes mellitus, insulin resistance, metabolic syndrome and cardiovascular diseases: A meta-analysis and systemic review. Diabetes Metab. Res. Rev. 2011, 27, 515–527. [Google Scholar] [CrossRef]
- Abd Rabo, S.A.; Mohammed, N.A.; Eissa, S.S.; Ali, A.A.; Ismail, S.M.; Gad, R.S. Serum visfatin in type 2 diabetes mellitus. Egypt. J. Intern. Med. 2013, 25, 27–32. [Google Scholar] [CrossRef]
- Hetta, H.; Ez-Eldeen, M.; Mohamed, G.; Gaber, M.; ElBadre, H.; Ahmed, E.; Abdellatief, R.; Abd-ElBaky, R.; Elkady, A.; Nafee, A.; et al. Visfatin serum levels in obese type 2 diabetic patients: Relation to proinflammatory cytokines and insulin resistance. Egypt J. Immunol. 2018, 25, 141–151. [Google Scholar]
- Kamińska, A.; Kopczyńska, E.; Bieliński, M.; Borkowska, A.; Junik, R. Visfatin concentrations in obese patients in relation to the presence of newly diagnosed glucose metabolism disorders. Endokrynol. Pol. 2015, 66, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Klöting, N.; Klöting, I. Visfatin: Gene expression in isolated adipocytes and sequence analysis in obese WOKW rats compared with lean control rats. Biochem. Biophys. Res. Commun. 2005, 332, 1070–1072. [Google Scholar] [CrossRef]
- De Luis, D.A.; Aller, R.; Gonzalez Sagrado, M.; Conde, R.; Izaola, O.; De la Fuente, B. Serum visfatin levels and metabolic syndrome criteria in obese female subjects. Diabetes Metab. Res. Rev. 2013, 29, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Vidal-Puig, A. Visfatin: The missing link between intra-abdominal obesity and diabetes? Trends Mol. Med. 2005, 11, 344–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.P.; Chung, F.M.; Chang, D.M.; Tsai, J.C.R.; Huang, H.F.; Shin, S.J.; Lee, Y.J. Elevated plasma level of visfatin/pre-B cell colony-enhancing factor in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2006, 91, 295–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaker, O.; El-Shehaby, A.; Zakaria, A.; Mostafa, N.; Talaat, S.; Katsiki, N.; Mikhailidis, D.P. Plasma visfatin and retinol binding protein-4 levels in patients with type 2 diabetes mellitus and their relationship to adiposity and fatty liver. Clin. Biochem. 2011, 44, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Esteghamati, A.; Alamdari, A.; Zandieh, A.; Elahi, S.; Khalilzadeh, O.; Nakhjavani, M.; Meysamie, A. Serum visfatin is associated with type 2 diabetes mellitus independent of insulin resistance and obesity. Diabetes Res. Clin. Pract. 2011, 91, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, I.; Karczewska-Kupczewska, M.; Adamska, A.; Nikolajuk, A.; Otziomek, E.; Straczkowski, M. Serum visfatin is differentially regulated by insulin and free Fatty acids in healthy men. J. Clin. Endocrinol. Metab. 2013, 98, 293–297. [Google Scholar] [CrossRef] [Green Version]
- Haider, D.G.; Schaller, G.; Kapiotis, S.; Maier, C.; Luger, A.; Wolzt, M. The release of the adipocytokine visfatin is regulated by glucose and insulin. Diabetologia 2006, 49, 1909–1914. [Google Scholar] [CrossRef]
- López-Bermejo, A.; Chico-Julià, B.; Fernàndez-Balsells, M.; Recasens, M.; Esteve, E.; Casamitjana, R.; Ricart, W.; Fernández-Real, J.M. Serum visfatin increases with progressive beta-cell deterioration. Diabetes 2006, 55, 2871–2875. [Google Scholar] [CrossRef] [Green Version]
- Dogru, T.; Sonmez, A.; Tasci, I.; Bozoglu, E.; Yilmaz, M.I.; Genc, H.; Erdem, G.; Gok, M.; Bingol, N.; Kilic, S.; et al. Plasma visfatin levels in patients with newly diagnosed and untreated type 2 diabetes mellitus and impaired glucose tolerance. Diabetes Res. Clin. Pract. 2007, 76, 24–29. [Google Scholar] [CrossRef]
- Li, L.; Yang, G.; Li, Q.; Tang, Y.; Yang, M.; Yang, H.; Li, K. Changes and relations of circulating visfatin, apelin, and resistin levels in normal, impaired glucose tolerance, and type 2 diabetic subjects. Exp. Clin. Endocrinol. Diabetes 2006, 114, 544–548. [Google Scholar] [CrossRef]
- Soriguer, F.; Garrido-Sanchez, L.; Garcia-Serrano, S.; Garcia-Almeida, J.M.; Garcia-Arnes, J.; Tinahones, F.J.; Garcia-Fuentes, E. Apelin levels are increased in morbidly obese subjects with type 2 diabetes mellitus. Obes. Surg. 2009, 19, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Dray, C.; Knauf, C.; Daviaud, D.; Waget, A.; Boucher, J.; Buléon, M.; Cani, P.D.; Attané, C.; Guigné, C.; Carpéné, C.; et al. Apelin stimulates glucose utilization in normal and obese insulin-resistant mice. Cell Metab. 2008, 8, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Than, A.; He, H.L.; Chua, S.H.; Xu, D.; Sun, L.; Leow, M.K.S.; Chen, P. Apelin enhances brown adipogenesis and browning of white adipocytes. J. Biol. Chem. 2015, 290, 14679–14691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trojnar, M.; Patro-Małysza, J.; Kimber-Trojnar, Ż.; Leszczyńska-Gorzelak, B.; Mosiewicz, J. Associations between fatty acid-binding protein 4–A proinflammatory adipokine and insulin resistance, gestational and type 2 diabetes mellitus. Cells 2019, 8, 227. [Google Scholar] [CrossRef] [Green Version]
- Uysal, K.T.; Scheja, L.; Wiesbrock, S.M.; Bonner-Weir, S.; Hotamisligil, G.S. Improved glucose and lipid metabolism in genetically obese mice lacking aP2. Endocrinology 2000, 141, 3388–3396. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Cao, H.; Kono, K.; Gorgun, C.Z.; Furuhashi, M.; Uysal, K.T.; Cao, Q.; Atsumi, G.; Malone, H.; Krishnan, B.; et al. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005, 1, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Steen, K.A.; Xu, H.; Bernlohr, D.A. FABP4/aP2 Regulates macrophage redox signaling and inflammasome activation via control of UCP2. Mol. Cell. Biol. 2017, 37, e00282-16. [Google Scholar] [CrossRef] [Green Version]
- Tuncman, G.; Erbay, E.; Hom, X.; De Vivo, I.; Campos, H.; Rimm, E.B.; Hotamisligil, G.S. A genetic variant at the fatty acid-binding protein aP2 locus reduces the risk for hypertriglyceridemia, type 2 diabetes, and cardiovascular disease. Proc. Natl. Acad. Sci. USA 2006, 103, 6970–6975. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Li, J.; Wang, H.; Ren, Y.; Bai, J. Associations of A-FABP with anthropometric and metabolic indices and inflammatory cytokines in obese patients with newly diagnosed type 2 diabetes. BioMed Res. Int. 2016, 2016, 9382092. [Google Scholar] [CrossRef]
- Cook, K.S.; Min, H.Y.; Johnson, D.; Chaplinsky, R.J.; Flier, J.S.; Hunt, C.R.; Spiegelman, B.M. Adipsin: A circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science 1987, 237, 402–405. [Google Scholar] [CrossRef]
- Zhou, Q.; Ge, Q.; Ding, Y.; Qu, H.; Wei, H.; Wu, R.; Yao, L.; Wei, Q.; Feng, Z.; Long, J.; et al. Relationship between serum adipsin and the first phase of glucose-stimulated insulin secretion in individuals with different glucose tolerance. J. Diabetes Investig. 2018, 9, 1128–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafere, G.G.; Wondafrash, D.Z.; Zewdie, K.A.; Assefa, B.T.; Ayza, M.A. Plasma adipsin as a biomarker and its implication in type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Marrano, N.; Biondi, G.; Borrelli, A.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Irisin and incretin hormones: Similarities, differences, and implications in type 2 diabetes and obesity. Biomolecules 2021, 11, 286. [Google Scholar] [CrossRef]
- Roca-Rivada, A.; Castelao, C.; Senin, L.L.; Landrove, M.O.; Baltar, J.; Crujeiras, A.B.; Seoane, L.M.; Casanueva, F.F.; Pardo, M. FNDC5/Irisin is not only a myokine but also an adipokine. PLoS ONE 2013, 8, e60563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Navarrete, J.M.; Ortega, F.; Serrano, M.; Guerra, E.; Pardo, G.; Tinahones, F.; Ricart, W.; Fernández-Real, J.M. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2013, 98, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Zaichenko, L.; Brinkoetter, M.; Thakkar, B.; Sahin-Efe, A.; Joung, K.E.; Tsoukas, M.A.; Geladari, E.V.; Huh, J.Y.; Dincer, F.; et al. Circulating irisin in relation to insulin resistance and the metabolic syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 4899–4907. [Google Scholar] [CrossRef]
- Stengel, A.; Hofmann, T.; Goebel-Stengel, M.; Elbelt, U.; Kobelt, P.; Klapp, B.F. Circulating levels of irisin in patients with anorexia nervosa and different stages of obesity—Correlation with body mass index. Peptides 2013, 39, 125–130. [Google Scholar] [CrossRef]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Spagnuolo, R.; Labarbuta, R.; Porreca, I.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; Perrini, S.; et al. The myokine irisin is released in response to saturated fatty acids and promotes pancreatic β-Cell survival and insulin secretion. Diabetes 2017, 66, 2849–2856. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Wong, M.D.S.; Toy, W.C.; Tan, C.S.H.; Liu, S.; Ng, X.W.; Tavintharan, S.; Sum, C.F.; Lim, S.C. Lower circulating irisin is associated with type 2 diabetes mellitus. J. Diabetes Complicat. 2013, 27, 365–369. [Google Scholar] [CrossRef]
- Du, X.L.; Jiang, W.X.; Lv, Z.T. Lower circulating irisin level in patients with diabetes mellitus: A systematic review and meta-analysis. Horm. Metab. Res. 2016, 48, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ding, Z.; Lv, G.; Li, J.; Zhou, P.; Zhang, J. Lower irisin level in patients with type 2 diabetes mellitus: A case-control study and meta-analysis. J. Diabetes 2016, 8, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Zhao, X.; Zhang, D.; Wang, R.; Feng, Y. Lower levels of irisin in patients with type 2 diabetes mellitus: A meta-analysis. Diabetes Res. Clin. Pract. 2021, 175, 108788. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Q.; Chen, D.; Sun, H.J.; Ding, L.; Wang, J.J.; Chen, Q.; Li, Y.H.; Zhou, Y.B.; Han, Y.; Zhang, F.; et al. FNDC5 overexpression and irisin ameliorate glucose/lipid metabolic derangements and enhance lipolysis in obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1867–1875. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; Ma, B.; Ma, X.; Wang, H.; Ni, Z.; Wang, B.; Li, X.; Jiang, P.; Umar, M.; Li, M. Anti-diabetic activity of recombinant irisin in STZ-induced insulin-deficient diabetic mice. Int. J. Biol. Macromol. 2016, 84, 457–463. [Google Scholar] [CrossRef]
- Xin, C.; Liu, J.; Zhang, J.; Zhu, D.; Wang, H.; Xiong, L.; Lee, Y.; Ye, J.; Lian, K.; Xu, C.; et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int. J. Obes. 2016, 40, 443–451. [Google Scholar] [CrossRef]
- Yasue, S.; Masuzaki, H.; Okada, S.; Ishii, T.; Kozuka, C.; Tanaka, T.; Fujikura, J.; Ebihara, K.; Hosoda, K.; Katsurada, A.; et al. Adipose tissue-specific regulation of angiotensinogen in obese humans and mice: Impact of nutritional status and adipocyte hypertrophy. Am. J. Hypertens. 2010, 23, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Cooper, R.; McFarlane-Anderson, N.; Bennett, F.I.; Wilks, R.; Puras, A.; Tewksbury, D.; Ward, R.; Forrester, T. ACE, angiotensinogen and obesity: A potential pathway leading to hypertension. J. Hum. Hypertens. 1997, 11, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Somers, K.R.; Becari, C.; Polonis, K.; Pfeifer, M.A.; Allen, A.M.; Kellogg, T.A.; Covassin, N.; Singh, P. Comparative expression of renin-angiotensin pathway proteins in visceral versus subcutaneous fat. Front. Physiol. 2018, 9, 1370. [Google Scholar] [CrossRef] [Green Version]
- de Farias Lelis, D.; de Freitas, D.F.; Machado, A.S.; Crespo, T.S.; Santos, S.H.S. Angiotensin-(1-7), adipokines and inflammation. Metabolism 2019, 95, 36–45. [Google Scholar] [CrossRef]
- Al-Daghri, N.M.; Bindahman, L.S.; Al-Attas, O.S.; Saleem, T.H.; Alokail, M.S.; Alkharfy, K.M.; Draz, H.M.; Yakout, S.; Mohamed, A.O.; Harte, A.L.; et al. Increased circulating ANG II and TNF-α represents important risk factors in obese saudi adults with hypertension irrespective of diabetic status and BMI. PLoS ONE 2012, 7, e51255. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-e-Silva, A.C. The anti-inflammatory potential of ACE2/Angiotensin-(1-7)/Mas receptor axis: Evidence from basic and clinical research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Mori, J.; Nakajima, H.; Kawabe, Y.; Tsuma, Y.; Fukuhara, S.; Kodo, K.; Ikoma, K.; Matoba, S.; Oudit, G.Y.; et al. Angiotensin 1-7 stimulates brown adipose tissue and reduces diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E131–E138. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.B.; Fernandes, A.B.; Febba, A.C.S.; Leite, A.P.O.; Leite, C.A.; Vitalle, M.S.S.; Jung, F.F.; Casarini, D.E. Association of Ang-(1-7) and des-Arg 9 BK as new biomarkers of obesity and cardiometabolic risk factors in adolescents. Hypertens. Res. 2021, 44, 969–977. [Google Scholar] [CrossRef]
- Kazafeos, K. Incretin effect: GLP-1, GIP, DPP4. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. S1), S32–S36. [Google Scholar] [CrossRef]
- Bosi, E.; Lucotti, P.; Setola, E.; Monti, L.; Piatti, P.M. Incretin-based therapies in type 2 diabetes: A review of clinical results. Diabetes Res. Clin. Pract. 2008, 82 (Suppl. S1), S102–S107. [Google Scholar] [CrossRef]
- Lambeir, A.M.; Durinx, C.; Scharpé, S.; De Meester, I. Dipeptidyl-peptidase IV from bench to bedside: An update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef]
- Lamers, D.; Famulla, S.; Wronkowitz, N.; Hartwig, S.; Lehr, S.; Ouwens, D.M.; Eckardt, K.; Kaufman, J.M.; Ryden, M.; Müller, S.; et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 2011, 60, 1917–1925. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.H.; Huri, H.Z.; Muniandy, S.; Al-Hamodi, Z.; Al-Absi, B.; Alsalahi, A.; Razif, M.F. Altered circulating concentrations of active glucagon-like peptide (GLP-1) and dipeptidyl peptidase 4 (DPP4) in obese subjects and their association with insulin resistance. Clin. Biochem. 2017, 50, 746–749. [Google Scholar] [CrossRef]
- Sell, H.; Blüher, M.; Klöting, N.; Schlich, R.; Willems, M.; Ruppe, F.; Knoefel, W.T.; Dietrich, A.; Fielding, B.A.; Arner, P.; et al. Adipose dipeptidyl peptidase-4 and obesity: Correlation with insulin resistance and depot-specific release from adipose tissue in vivo and in vitro. Diabetes Care 2013, 36, 4083–4090. [Google Scholar] [CrossRef] [Green Version]
- Rohmann, N.; Schlicht, K.; Geisler, C.; Hollstein, T.; Knappe, C.; Krause, L.; Hagen, S.; Beckmann, A.; Seoudy, A.K.; Wietzke-Braun, P.; et al. Circulating sDPP-4 is increased in obesity and insulin resistance but is not related to systemic metabolic inflammation. J. Clin. Endocrinol. Metab. 2021, 106, E592–E601. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, J.; Nargis, T.; Tantia, O.; Ghosh, S.; Chakrabarti, P. Increased plasma dipeptidyl peptidase-4 (DPP4) activity is an obesity-independent parameter for glycemic deregulation in type 2 diabetes patients. Front. Endocrinol. 2019, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Investig. 2002, 32 (Suppl. S3), 14–23. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Guerendiain, M.; Castellote, A.I.; Estruch, R.; Covas, M.I.; Fitó, M.; Salas-Salvadó, J.; Martínez-González, M.A.; Aros, F.; Lamuela-Raventós, R.M.; et al. Plasma fatty acid composition, estimated desaturase activities, and their relation with the metabolic syndrome in a population at high risk of cardiovascular disease. Clin. Nutr. 2014, 33, 90–97. [Google Scholar] [CrossRef]
- Ma, Y.; Xiong, J.; Zhang, X.; Qiu, T.; Pang, H.; Li, X.; Zhu, J.; Wang, J.; Pan, C.; Yang, X.; et al. Potential biomarker in serum for predicting susceptibility to type 2 diabetes mellitus: Free fatty acid 22:6. J. Diabetes Investig. 2021, 12, 950–962. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.Y.; Kim, O.Y.; Ham, B.M.; Kim, H.J.; Kwon, D.Y.; Jang, Y.; Lee, J.H. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC−Q-TOF MS). J. Proteome Res. 2010, 9, 4368–4375. [Google Scholar] [CrossRef]
- Ni, Y.; Zhao, L.; Yu, H.; Ma, X.; Bao, Y.; Rajani, C.; Loo, L.W.M.; Shvetsov, Y.B.; Yu, H.; Chen, T.; et al. Circulating unsaturated fatty acids delineate the metabolic status of obese individuals. EBioMedicine 2015, 2, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Hauner, H.; Bender, M.; Haastert, B.; Hube, F. Plasma concentrations of soluble TNF-alpha receptors in obese subjects. Int. J. Obes. Relat. Metab. Disord. 1998, 22, 1239–1243. [Google Scholar] [CrossRef] [Green Version]
- Marrano, N.; Biondi, G.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Functional loss of pancreatic islets in type 2 diabetes: How can we halt it? Metabolism 2020, 110, 154304. [Google Scholar] [CrossRef]
- Ye, R.; Onodera, T.; Scherer, P.E. Lipotoxicity and β cell maintenance in obesity and type 2 diabetes. J. Endocr. Soc. 2019, 3, 617–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-Cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saisho, Y. β-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. S4), 32–42. [Google Scholar] [CrossRef]
- Saisho, Y.; Butler, A.; Manesso, E.; Elashoff, D.; Rizza, R.; Butler, P. β-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.A.; Gannon, M. The beta cell in type 2 diabetes. Curr. Diabetes Rep. 2019, 19, 81. [Google Scholar] [CrossRef]
- Mezza, T.; Muscogiuri, G.; Sorice, G.P.; Clemente, G.; Hu, J.; Pontecorvi, A.; Holst, J.J.; Giaccari, A.; Kulkarni, R.N. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 2014, 63, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, S.; Uno, S.; Iwahashi, H.; Fujita, Y.; Yoshikawa, A.; Kozawa, J.; Okita, K.; Takiuchi, D.; Eguchi, H.; Nagano, H.; et al. Predominance of β-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 2053–2061. [Google Scholar] [CrossRef] [Green Version]
- UK Prospective Diabetes Study Group. UK prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: A progressive disease. Diabetes 1995, 44, 1249–1258. [Google Scholar] [CrossRef]
- Rhodes, C. Type 2 diabetes-a matter of beta-cell life and death? Science 2005, 307, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.; Ohmura, Y.; Sandoval, P.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.; Marchetti, P.; et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Mezza, T.; Cinti, F.; Cefalo, C.; Pontecorvi, A.; Kulkarni, R.; Giaccari, A. β-Cell fate in human insulin resistance and type 2 diabetes: A perspective on isletpPlasticity. Diabetes 2019, 68, 1121–1129. [Google Scholar] [CrossRef]
- Tanabe, K.; Okuya, S.; Tanizawa, Y.; Matsutani, A.; Oka, Y. Leptin induces proliferation of pancreatic beta cell line MIN6 through activation of mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 1997, 241, 765–768. [Google Scholar] [CrossRef]
- Islam, M.; Morton, N.; Hansson, A.; Emilsson, V. Rat insulinoma-derived pancreatic beta-cells express a functional leptin receptor that mediates a proliferative response. Biochem. Biophys. Res. Commun. 1997, 238, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.; Sjöholm, A.; Emilsson, V. Fetal pancreatic islets express functional leptin receptors and leptin stimulates proliferation of fetal islet cells. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 1246–1253. [Google Scholar] [CrossRef] [Green Version]
- Finegood, D.T.; McArthur, M.D.; Kojwang, D.; Thomas, M.J.; Topp, B.G.; Leonard, T.; Buckingham, R.E. Beta-cell mass dynamics in Zucker diabetic fatty rats. Rosiglitazone prevents the rise in net cell death. Diabetes 2001, 50, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, Y.; Sturis, J.; DePaoli, A.M.; Takeda, J.; Stoffel, M.; Tang, J.; Sun, X.; Polonsky, K.S.; Bell, G.I. Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes 1995, 44, 1447–1457. [Google Scholar] [CrossRef]
- Unger, R.; Zhou, Y. Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes 2001, 50 (Suppl. S1), S118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, R.; Zhou, Y.; Orci, L. Regulation of fatty acid homeostasis in cells: Novel role of leptin. Proc. Natl. Acad. Sci. USA 1999, 96, 2327–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.H.; McGarry, J.D.; Unger, R.H. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: Impairment in adipocyte-beta-cell relationships. Proc. Natl. Acad. Sci. USA 1994, 91, 10878–10882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.; Onyango, D.; Dunmore, S. Resistin down-regulates insulin receptor expression, and modulates cell viability in rodent pancreatic beta-cells. FEBS Lett. 2007, 581, 3273–3276. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Du, F.; Li, X.; Wang, M.; Duan, R.; Zhang, J.; Wu, Y.; Zhang, Q. Effects and underlying mechanisms of irisin on the proliferation and apoptosis of pancreatic β cells. PLoS ONE 2017, 12, e0175498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Dong, W.; Qian, L.; Wu, J.; Peng, Y. Visfatin inhibits apoptosis of pancreatic β-cell line, MIN6, via the mitogen-activated protein kinase/phosphoinositide 3-kinase pathway. J. Mol. Endocrinol. 2011, 47, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Zhang, N.; Zhang, Y.; Chen, Y.; Wang, L.; Zhu, Y.; Tang, H. Overexpression of apelin in Wharton’ jelly mesenchymal stem cell reverses insulin resistance and promotes pancreatic β cell proliferation in type 2 diabetic rats. Stem Cell Res. Ther. 2018, 9, 339. [Google Scholar] [CrossRef] [Green Version]
- Dirice, E.; Kahraman, S.; Jiang, W.; El Ouaamari, A.; De Jesus, D.; Teo, A.; Hu, J.; Kawamori, D.; Gaglia, J.; Mathis, D.; et al. Soluble factors secreted by T cells promote β-cell proliferation. Diabetes 2014, 63, 188–202. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhu, H.; Chen, X.; Peng, Y.; Wang, J.; Liu, F.; Shi, S.; Fu, B.; Lu, Y.; Hong, Q.; et al. TIMP-1 transgenic mice recover from diabetes induced by multiple low-dose streptozotocin. Diabetes 2007, 56, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Kono, T.; Sims, E.; Moss, D.; Yamamoto, W.; Ahn, G.; Diamond, J.; Tong, X.; Day, K.; Territo, P.; Hanenberg, H.; et al. Human adipose-derived stromal/stem cells protect against STZ-induced hyperglycemia: Analysis of hASC-derived paracrine effectors. Stem Cells 2014, 32, 1831–1842. [Google Scholar] [CrossRef] [Green Version]
- Tanday, N.; Irwin, N.; Moffett, R.; Flatt, P.; O’Harte, F. Beneficial actions of a long-acting apelin analogue in diabetes are related to positive effects on islet cell turnover and transdifferentiation. Diabetes Obes. Metab. 2020, 22, 2468–2478. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Englander, E.; Gomez, G.; Rastellini, C.; Quertermous, T.; Kundu, R.; Greeley, G. Pancreatic islet APJ deletion reduces islet density and glucose tolerance in mice. Endocrinology 2015, 156, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhao, H.; Du, M.; Wu, X. The effect of apelin-13 on pancreatic islet beta cell mass and myocardial fatty acid and glucose metabolism of experimental type 2 diabetic rats. Peptides 2019, 114, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.; Hobbs, M.; Dockter, J.; Oldstone, M.; Allison, J. Islet inflammation and hyperplasia induced by the pancreatic islet-specific overexpression of interleukin-6 in transgenic mice. Am. J. Pathol. 1994, 145, 157–166. [Google Scholar] [PubMed]
- Petropavlovskaia, M.; Makhlin, J.; Sampalis, J.; Rosenberg, L. Development of an in vitro pancreatic tissue model to study regulation of islet neogenesis associated protein expression. J. Endocrinol. 2006, 191, 65–81. [Google Scholar] [CrossRef]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Srivastava, S.; Pandey, H.; Tripathi, Y. Expression kinetics reveal the self-adaptive role of β cells during the progression of diabetes. Biomed. Pharmacother. 2018, 106, 472–482. [Google Scholar] [CrossRef]
- Bouzakri, K.; Plomgaard, P.; Berney, T.; Donath, M.; Pedersen, B.; Halban, P. Bimodal effect on pancreatic β-cells of secretory products from normal or insulin-resistant human skeletal muscle. Diabetes 2011, 60, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Yang, Z.; Yang, H.; Wang, L.; Wu, H.; Fan, Y.; Wang, W.; Fan, X.; Li, X. Regulation of insulin sensitivity, insulin production, and pancreatic β cell survival by angiotensin-(1-7) in a rat model of streptozotocin-induced diabetes mellitus. Peptides 2015, 64, 49–54. [Google Scholar] [CrossRef]
- Yuan, L.; Li, Y.; Li, G.; Song, Y.; Gong, X. Ang(1-7) treatment attenuates β-cell dysfunction by improving pancreatic microcirculation in a rat model of Type 2 diabetes. J. Endocrinol. Investig. 2013, 36, 931–937. [Google Scholar] [CrossRef]
- Lu, C.-L.; Wang, Y.; Yuan, L.; Li, Y.; Li, X.-Y. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects the function of pancreatic β cells by improving the function of islet microvascular endothelial cells. Int. J. Mol. Med. 2014, 34, 1293–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, X.; Gao, F.; Ma, X.; Huang, C.; Wang, Y.; Deng, H.; Wang, S.; Li, W.; Yuan, L. Activation of ACE2/angiotensin (1–7) attenuates pancreatic β cell dedifferentiation in a high-fat-diet mouse model. Metabolism 2018, 81, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Jiang, D.; Guo, W.; Guo, L.; Gao, M.; Bai, Y.; Wang, X.; Zhang, L. FABP4 inhibitor attenuates inflammation and endoplasmic reticulum stress of islet in leptin receptor knockout rats. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12808–12820. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Dunmore, S. Leptin decreases apoptosis and alters BCL-2: Bax ratio in clonal rodent pancreatic beta-cells. Diabetes Metab. Res. Rev. 2007, 23, 497–502. [Google Scholar] [CrossRef]
- Okuya, S.; Tanabe, K.; Tanizawa, Y.; Oka, Y. Leptin increases the viability of isolated rat pancreatic islets by suppressing apoptosis. Endocrinology 2001, 142, 4827–4830. [Google Scholar] [CrossRef]
- Zhang, D.; Xie, T.; Leung, P.S. Irisin ameliorates glucolipotoxicity-associated β-cell dysfunction and apoptosis via AMPK signaling and anti-inflammatory actions. Cell. Physiol. Biochem. 2018, 51, 924–937. [Google Scholar] [CrossRef]
- Xiang, R.; Mei, M.; Su, Y.; Li, L.; Wang, J.; Wu, L. Visfatin protects rat pancreatic β-cells against IFN-γ-induced apoptosis through AMPK and ERK1/2 signaling pathways. Biomed. Environ. Sci. 2015, 28, 169–177. [Google Scholar] [CrossRef]
- Han, X.; Sun, Y.; Scott, S.; Bleich, D. Tissue inhibitor of metalloproteinase-1 prevents cytokine-mediated dysfunction and cytotoxicity in pancreatic islets and beta-cells. Diabetes 2001, 50, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhu, H.; Wang, J.; Fu, B.; Lü, Y.; Hong, Q.; Xie, Y.; Chen, X. Tissue inhibitor of metalloproteinase-1 counteracts glucolipotoxicity in the pancreatic β-cell line INS-1. Chin. Med. J. 2011, 124, 258–261. [Google Scholar]
- Choi, S.; Choi, K.; Yoon, I.; Shin, J.; Kim, J.; Park, W.; Han, D.; Kim, S.; Ahn, C.; Kim, J.; et al. IL-6 protects pancreatic islet beta cells from pro-inflammatory cytokines-induced cell death and functional impairment in vitro and in vivo. Transpl. Immunol. 2004, 13, 43–53. [Google Scholar] [CrossRef]
- Linnemann, A.; Blumer, J.; Marasco, M.; Battiola, T.; Umhoefer, H.; Han, J.; Lamming, D.; Davis, D. Interleukin 6 protects pancreatic β cells from apoptosis by stimulation of autophagy. FASEB J. 2017, 31, 4140–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marasco, M.; Conteh, A.; Reissaus, C.; Cupit, J.; Appleman, E.; Mirmira, R.; Linnemann, A. Interleukin-6 reduces β-cell oxidative stress by linking autophagy with the antioxidant response. Diabetes 2018, 67, 1576–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Ahn, Y.; Park, C.; Chung, H.; Park, Y. Interleukin-6 protects MIN6 beta cells from cytokine-induced apoptosis. Ann. N. Y. Acad. Sci. 2003, 1005, 242–249. [Google Scholar] [CrossRef]
- Gómez-Banoy, N.; Guseh, J.S.; Li, G.; Rubio-Navarro, A.; Chen, T.; Poirier, B.A.; Putzel, G.; Rosselot, C.; Pabón, M.A.; Camporez, J.P.; et al. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat. Med. 2019, 25, 1739–1747. [Google Scholar] [CrossRef]
- Sharma, R.; Alonso, L. Lipotoxicity in the pancreatic beta cell: Not just survival and function, but proliferation as well? Curr. Diabetes Rep. 2014, 14, 492. [Google Scholar] [CrossRef] [Green Version]
- Prentki, M.; Madiraju, S. Glycerolipid metabolism and signaling in health and disease. Endocr. Rev. 2008, 29, 647–676. [Google Scholar] [CrossRef] [Green Version]
- Brelje, T.; Bhagroo, N.; Stout, L.; Sorenson, R. Beneficial effects of lipids and prolactin on insulin secretion and beta-cell proliferation: A role for lipids in the adaptation of islets to pregnancy. J. Endocrinol. 2008, 197, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Vernier, S.; Chiu, A.; Schober, J.; Weber, T.; Nguyen, P.; Luer, M.; McPherson, T.; Wanda, P.; Marshall, C.; Rohatgi, N.; et al. β-cell metabolic alterations under chronic nutrient overload in rat and human islets. Islets 2012, 4, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Fontés, G.; Zarrouki, B.; Hagman, D.K.; Latour, M.G.; Semache, M.; Roskens, V.; Moore, P.C.; Prentki, M.; Rhodes, C.J.; Jetton, T.L.; et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia 2010, 53, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Steil, G.; Trivedi, N.; Jonas, J.; Hasenkamp, W.; Sharma, A.; Bonner-Weir, S.; Weir, G. Adaptation of beta-cell mass to substrate oversupply: Enhanced function with normal gene expression. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E788–E796. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y. Mechanistic insights into pancreatic beta-cell mass regulation by glucose and free fatty acids. Anat. Cell Biol. 2015, 48, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vliet, S.; Koh, H.C.E.; Patterson, B.W.; Yoshino, M.; LaForest, R.; Gropler, R.J.; Klein, S.; Mittendorfer, B. Obesity is associated with increased basal and postprandial β-cell insulin secretion even in the absence of insulin resistance. Diabetes 2020, 69, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa, Y.; Okuya, S.; Ishihara, H.; Asano, T.; Yada, T.; Oka, Y. Direct stimulation of basal insulin secretion by physiological concentrations of leptin in pancreatic beta cells. Endocrinology 1997, 138, 4513–4516. [Google Scholar] [CrossRef]
- Fehmann, H.C.; Peiser, C.; Bode, H.P.; Stamm, M.; Staats, P.; Hedetoft, C.; Lang, R.E.; Göke, B. Leptin: A potent inhibitor of insulin secretion. Peptides 1997, 18, 1267–1273. [Google Scholar] [CrossRef]
- Zhao, A.Z.; Bornfeldt, K.E.; Beavo, J.A. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J. Clin. Investig. 1998, 102, 869–873. [Google Scholar] [CrossRef] [Green Version]
- Lupi, R.; Marchetti, P.; Maffei, M.; Del Guerra, S.; Benzi, L.; Marselli, L.; Bertacca, A.; Navalesi, R. Effects of acute or prolonged exposure to human leptin on isolated human islet function. Biochem. Biophys. Res. Commun. 1999, 256, 637–641. [Google Scholar] [CrossRef]
- Chetboun, M.; Abitbol, G.; Rozenberg, K.; Rozenfeld, H.; Deutsch, A.; Sampson, S.R.; Rosenzweig, T. Maintenance of redox state and pancreatic beta-cell function: Role of leptin and adiponectin. J. Cell. Biochem. 2012, 113, 1966–1976. [Google Scholar] [CrossRef]
- Seufert, J.; Kieffer, T.J.; Leech, C.A.; Holz, G.G.; Moritz, W.; Ricordi, C.; Habener, J.F. Leptin suppression of insulin secretion and gene expression in human pancreatic islets: Implications for the development of adipogenic diabetes mellitus. J. Clin. Endocrinol. Metab. 1999, 84, 670–676. [Google Scholar] [CrossRef]
- Seufert, J.; Kieffer, T.J.; Habener, J.F. Leptin inhibits insulin gene transcription and reverses hyperinsulinemia in leptin-deficient ob/ob mice. Proc. Natl. Acad. Sci. USA 1999, 96, 674–679. [Google Scholar] [CrossRef] [Green Version]
- Roduit, R.; Thorens, B. Inhibition of glucose-induced insulin secretion by long-term preexposure of pancreatic islets to leptin. FEBS Lett. 1997, 415, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Cases, J.A.; Gabriely, I.; Ma, X.H.; Yang, X.M.; Michaeli, T.; Fleischer, N.; Rossetti, L.; Barzilai, N. Physiological increase in plasma leptin markedly inhibits insulin secretion in vivo. Diabetes 2001, 50, 348–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallett, A.L.; Morton, N.M.; Cawthorne, M.A.; Emilsson, V. Leptin inhibits insulin secretion and reduces insulin mRNA levels in rat isolated pancreatic islets. Biochem. Biophys. Res. Commun. 1997, 238, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Murakami, T.; Mizuno, A.; Iida, M.; Kuwajima, M.; Shima, K. Leptin suppresses basal insulin secretion from rat pancreatic islets. Regul. Pept. 1997, 70, 179–182. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Wang, Z.L.; Wang, R.M.; Hurley, J.D.; Smith, D.M.; Ghatei, M.A.; Withers, D.J.; Gardiner, J.V.; Bailey, C.J.; Bloom, S.R. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Investig. 1997, 100, 2729–2736. [Google Scholar] [CrossRef]
- Fehmann, H.C.; Berghöfer, P.; Brandhorst, D.; Brandhorst, H.; Hering, B.; Bretzel, R.G.; Göke, B. Leptin inhibition of insulin secretion from isolated human islets. Acta Diabetol. 1997, 34, 249–252. [Google Scholar] [CrossRef]
- Saladin, R.; De Vos, P.; Guerre-Millot, M.; Leturque, A.; Girard, J.; Staels, B.; Auwerx, J. Transient increase in obese gene expression after food intake or insulin administration. Nature 1995, 377, 527–528. [Google Scholar] [CrossRef]
- Malmström, R.; Taskinen, M.R.; Karonen, S.L.; Yki-Järvinen, H. Insulin increases plasma leptin concentrations in normal subjects and patients with NIDDM. Diabetologia 1996, 39, 993–996. [Google Scholar] [CrossRef]
- Suzuki, T.; Imai, J.; Yamada, T.; Ishigaki, Y.; Kaneko, K.; Uno, K.; Hasegawa, Y.; Ishihara, H.; Oka, Y.; Katagiri, H. Interleukin-6 enhances glucose-stimulated insulin secretion from pancreatic beta-cells: Potential involvement of the PLC-IP3-dependent pathway. Diabetes 2011, 60, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Sato, N.; Tanaka, Y.; Ohtani, K.; Fukatsu, A.; Mori, M. Interleukin-6 stimulates insulin secretion in HIT-T 15 cells. Horm. Metab. Res. 1995, 27, 37–38. [Google Scholar] [CrossRef]
- Sandler, S.; Bendtzen, K.; Eizirik, D.L.; Welsh, M. Interleukin-6 affects insulin secretion and glucose metabolism of rat pancreatic islets in vitro. Endocrinology 1990, 126, 1288–1294. [Google Scholar] [CrossRef]
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Chen, P.; Jin, H.; Xie, X.; Gao, T.; Yang, L.; Yu, X. Circulating levels of irisin in middle-aged first-degree relatives of type 2 diabetes mellitus—Correlation with pancreatic β-cell function. Diabetol. Metab. Syndr. 2014, 6, 133. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilford, B.L.; Parson, J.C.; Grote, C.W.; Vick, S.N.; Ryals, J.M.; Wright, D.E. Increased FNDC5 is associated with insulin resistance in high fat-fed mice. Physiol. Rep. 2017, 5, e13319. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.E.; Samocha-Bonet, D.; Whitworth, P.T.; Fazakerley, D.J.; Turner, N.; Biden, T.J.; James, D.E.; Cantley, J. Identification of fatty acid binding protein 4 as an adipokine that regulates insulin secretion during obesity. Mol. Metab. 2014, 3, 465–473. [Google Scholar] [CrossRef]
- Liu, B.; Hassan, Z.; Amisten, S.; King, A.J.; Bowe, J.E.; Huang, G.C.; Jones, P.M.; Persaud, S.J. The novel chemokine receptor, G-protein-coupled receptor 75, is expressed by islets and is coupled to stimulation of insulin secretion and improved glucose homeostasis. Diabetologia 2013, 56, 2467–2476. [Google Scholar] [CrossRef] [Green Version]
- Pais, R.; Zietek, T.; Hauner, H.; Daniel, H.; Skurk, T. RANTES (CCL5) reduces glucose-dependent secretion of glucagon-like peptides 1 and 2 and impairs glucose-induced insulin secretion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G330–G337. [Google Scholar] [CrossRef] [Green Version]
- Gençoğlu, H.; Şahin, K.; Jones, P.M. Determining the insulin secretion potential for certain specific G-protein coupled receptors in MIN6 pancreatic beta cells. Turk. J. Med. Sci. 2019, 49, 403–411. [Google Scholar] [CrossRef]
- Wu, H.; Ghosh, S.; Perrard, X.D.; Feng, L.; Garcia, G.E.; Perrard, J.L.; Sweeney, J.F.; Peterson, L.E.; Chan, L.; Smith, C.W.; et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007, 115, 1029–1038. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.C.; Ljubicic, S.; Leibiger, B.; Kern, M.; Leibiger, I.B.; Moede, T.; Kelly, M.E.; Chatterjee Bhowmick, D.; Murano, I.; Cohen, P.; et al. Adipsin is an adipokine that improves β cell function in diabetes. Cell 2014, 158, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Zhao, Z.; Gao, H. Effect of hTIMP-1 overexpression in human umbilical cord mesenchymal stem cells on the repair of pancreatic islets in type-1 diabetic mice. Cell Biol. Int. 2021, 45, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Fjære, E.; Andersen, C.; Myrmel, L.S.; Petersen, R.K.; Hansen, J.B.; Tastesen, H.S.; Mandrup-Poulsen, T.; Brünner, N.; Kristiansen, K.; Madsen, L.; et al. Tissue inhibitor of matrix metalloproteinase-1 is required for high-fat diet-induced glucose intolerance and hepatic steatosis in mice. PLoS ONE 2015, 10, e0132910. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.E.P.; Onyango, D.J.; Ramanjaneya, M.; Conner, A.C.; Patel, S.T.; Dunmore, S.J.; Randeva, H.S. Visfatin regulates insulin secretion, insulin receptor signalling and mRNA expression of diabetes-related genes in mouse pancreatic beta-cells. J. Mol. Endocrinol. 2010, 44, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Caton, P.W.; Kieswich, J.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia 2011, 54, 3083–3092. [Google Scholar] [CrossRef] [Green Version]
- Sheng, F.; Ren, X.; Dai, X.; Xu, X.; Dong, M.; Pei, Q.; Qu, J.; Zhou, Z.; Zhou, H.; Liu, Z. Effect of nicotinamide mononucleotide on insulin secretion and gene expressions of PDX-1 and FoxO1 in RIN-m5f cells. J. Cent. South Univ. (Med. Sci.) 2011, 36, 958–963. [Google Scholar] [CrossRef]
- Sayers, S.R.; Beavil, R.L.; Fine, N.H.F.; Huang, G.C.; Choudhary, P.; Pacholarz, K.J.; Barran, P.E.; Butterworth, S.; Mills, C.E.; Cruickshank, J.K.; et al. Structure-functional changes in eNAMPT at high concentrations mediate mouse and human beta cell dysfunction in type 2 diabetes. Diabetologia 2020, 63, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Spinnler, R.; Gorski, T.; Stolz, K.; Schuster, S.; Garten, A.; Beck-Sickinger, A.G.; Engelse, M.A.; de Koning, E.J.P.; Körner, A.; Kiess, W.; et al. The adipocytokine Nampt and its product NMN have no effect on beta-cell survival but potentiate glucose stimulated insulin secretion. PLoS ONE 2013, 8, e054106. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Li, Q.; Wang, W.; Yu, P.; Pan, H.; Li, P.; Sun, Y.; Zhang, J. Apelin inhibits insulin secretion in pancreatic beta-cells by activation of PI3-kinase-phosphodiesterase 3B. Endocr. Res. 2009, 34, 142–154. [Google Scholar] [CrossRef]
- Ringström, C.; Nitert, M.D.; Bennet, H.; Fex, M.; Valet, P.; Rehfeld, J.F.; Friis-Hansen, L.; Wierup, N. Apelin is a novel islet peptide. Regul. Pept. 2010, 162, 44–51. [Google Scholar] [CrossRef] [Green Version]
- O’Harte, F.P.M.; Parthsarathy, V.; Hogg, C.; Flatt, P.R. Apelin-13 analogues show potent in vitro and in vivo insulinotropic and glucose lowering actions. Peptides 2018, 100, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Magnusson, C.; Ahrén, B. The apj receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul. Pept. 2005, 131, 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Harte, F.P.M.; Parthsarathy, V.; Hogg, C.; Flatt, P.R. Acylated apelin-13 amide analogues exhibit enzyme resistance and prolonged insulin releasing, glucose lowering and anorexic properties. Biochem. Pharmacol. 2017, 146, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, R.; Liu, Y.; Yang, J.; Wang, X.; Geng, L.; Xu, T.; He, J. Angiotensin-(1-7) improves islet function in a rat model of streptozotocin-induced diabetes mellitus by up-regulating the expression of Pdx1/Glut2. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, C.; Wang, L.; Cao, X.; Wang, Y.Y.; Yang, J.K. Antioxidant effect of angiotensin (1-7) in the protection of pancreatic β cell function. Mol. Med. Rep. 2016, 14, 1963–1969. [Google Scholar] [CrossRef] [Green Version]
- Brar, G.S.; Barrow, B.M.; Watson, M.; Griesbach, R.; Choung, E.; Welch, A.; Ruzsicska, B.; Raleigh, D.P.; Zraika, S. Neprilysin is required for angiotensin-(1-7)’s ability to enhance insulin secretion via its proteolytic activity to generate angiotensin-(1-2). Diabetes 2017, 66, 2201–2212. [Google Scholar] [CrossRef] [Green Version]
- Sahr, A.; Wolke, C.; MacZewsky, J.; Krippeit-Drews, P.; Tetzner, A.; Drews, G.; Venz, S.; Gürtler, S.; Van Den Brandt, J.; Berg, S.; et al. The angiotensin-(1-7)/Mas axis improves pancreatic β-cell function in vitro and in vivo. Endocrinology 2016, 157, 4677–4690. [Google Scholar] [CrossRef]
- Barbosa, M.A.; Barbosa, C.M.; Lima, T.C.; Dos Santos, R.A.S.; Alzamora, A.C. The novel angiotensin-(1-7) analog, A-1317, improves insulin resistance by restoring pancreatic β-cell functionality in rats with metabolic syndrome. Front. Pharmacol. 2020, 11, 1263. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R.M. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef] [Green Version]
- Ježek, J.; Dlasková, A.; Zelenka, J.; Jabůrek, M.; Ježek, P. H₂O₂-activated mitochondrial phospholipase iPLA₂γ prevents lipotoxic oxidative stress in synergy with UCP2, amplifies signaling via G-Protein-Coupled Receptor GPR40, and regulates insulin secretion in pancreatic β-cells. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Montaño, P.; García-González, V. Effects of dietary fatty acids in pancreatic beta cell metabolism, implications in homeostasis. Nutrients 2018, 10, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudez, B.; Ortega-Gomez, A.; Varela, L.M.; Villar, J.; Abia, R.; Muriana, F.J.G.; Lopez, S. Clustering effects on postprandial insulin secretion and sensitivity in response to meals with different fatty acid compositions. Food Funct. 2014, 5, 1374–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-Montaño, P.; Rodríguez-Velázquez, E.; Ibarra-López, E.; Frayde-Gómez, H.; Mas-Oliva, J.; Delgado-Coello, B.; Rivero, I.A.; Alatorre-Meda, M.; Aguilera, J.; Guevara-Olaya, L.; et al. Fatty acid and lipopolysaccharide effect on beta cells proteostasis and its impact on insulin secretion. Cells 2019, 8, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghislain, J.; Poitout, V. Targeting lipid GPCRs to treat type 2 diabetes mellitus—Progress and challenges. Nat. Rev. Endocrinol. 2021, 17, 162–175. [Google Scholar] [CrossRef]
- Ježek, P.; Jabůrek, M.; Holendová, B.; Plecitá-Hlavatá, L. Fatty acid-stimulated insulin secretion vs. lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef] [Green Version]
- Neuman, J.C.; Schaid, M.D.; Brill, A.L.; Fenske, R.J.; Kibbe, C.R.; Fontaine, D.A.; Sdao, S.M.; Brar, H.K.; Connors, K.M.; Wienkes, H.N.; et al. Enriching islet phospholipids with eicosapentaenoic acid reduces prostaglandin E 2 signaling and enhances diabetic β-cell function. Diabetes 2017, 66, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Graciano, M.F.; Leonelli, M.; Curi, R.; Carpinelli, A.R. Omega-3 fatty acids control productions of superoxide and nitrogen oxide and insulin content in INS-1E cells. J. Physiol. Biochem. 2016, 72, 699–710. [Google Scholar] [CrossRef]
- Lucena, C.F.; Roma, L.P.; Graciano, M.F.R.; Veras, K.; Simões, D.; Curi, R.; Carpinelli, A.R. Omega-3 supplementation improves pancreatic islet redox status: In vivo and in vitro studies. Pancreas 2015, 44, 287–295. [Google Scholar] [CrossRef]
- Kato, T.; Shimano, H.; Yamamoto, T.; Ishikawa, M.; Kumadaki, S.; Matsuzaka, T.; Nakagawa, Y.; Yahagi, N.; Nakakuki, M.; Hasty, A.H.; et al. Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of SREBP-1c in pancreatic islets. Diabetes 2008, 57, 2382–2392. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Ohara-Imaizumi, M.; Kubota, N.; Hashimoto, S.; Eto, K.; Kanno, T.; Kubota, T.; Wakui, M.; Nagai, R.; Noda, M.; et al. Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia 2008, 51, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, J.R.; Keating, D.J.; Chen, C.; Parkington, H.C. Adiponectin increases insulin content and cell proliferation in MIN6 cells via PPARγ-dependent and PPARγ-independent mechanisms. Diabetes Obes. Metab. 2012, 14, 983–989. [Google Scholar] [CrossRef]
- Lee, Y.H.; Magkos, F.; Mantzoros, C.S.; Kang, E.S. Effects of leptin and adiponectin on pancreatic β-cell function. Metabolism 2011, 60, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Nogueiras, R.; Dieguez, C.; Ahrén, B. Dual action of adiponectin on insulin secretion in insulin-resistant mice. Biochem. Biophys. Res. Commun. 2004, 321, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Rakatzi, I.; Mueller, H.; Ritzeler, O.; Tennagels, N.; Eckel, J. Adiponectin counteracts cytokine- and fatty acid-induced apoptosis in the pancreatic beta-cell line INS-1. Diabetologia 2004, 47, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staiger, K.; Stefan, N.; Staiger, H.; Brendel, M.D.; Brandhorst, D.; Bretzel, R.G.; Machicao, F.; Kellerer, M.; Stumvoll, M.; Fritsche, A.; et al. Adiponectin is functionally active in human islets but does not affect insulin secretory function or beta-cell lipoapoptosis. J. Clin. Endocrinol. Metab. 2005, 90, 6707–6713. [Google Scholar] [CrossRef] [Green Version]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.U.; Ha, K.H.; Han, S.J.; Kim, H.J.; Kim, D.J. The association of adiponectin and visceral fat with insulin resistance and β-cell dysfunction. J. Korean Med. Sci. 2018, 34, e7. [Google Scholar] [CrossRef]
- Nakamura, A.; Miyoshi, H.; Ukawa, S.; Nakamura, K.; Nakagawa, T.; Terauchi, Y.; Tamakoshi, A.; Atsumi, T. Serum adiponectin and insulin secretion: A direct or inverse association? J. Diabetes Investig. 2018, 9, 1106–1109. [Google Scholar] [CrossRef] [Green Version]
- Fasshauer, M.; Kralisch, S.; Klier, M.; Lossner, U.; Bluher, M.; Klein, J.; Paschke, R. Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2003, 301, 1045–1050. [Google Scholar] [CrossRef]
- Zhang, S.; Kim, K.H. TNF-alpha inhibits glucose-induced insulin secretion in a pancreatic beta-cell line (INS-1). FEBS Lett. 1995, 377, 237–239. [Google Scholar] [CrossRef] [Green Version]
- Hostens, K.; Pavlovic, D.; Zambre, Y.; Ling, Z.; Van Schravendijk, C.; Eizirik, D.L.; Pipeleers, D.G. Exposure of human islets to cytokines can result in disproportionately elevated proinsulin release. J. Clin. Investig. 1999, 104, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiotra, P.C.; Tsigos, C.; Raptis, S.A. TNFalpha and leptin inhibit basal and glucose-stimulated insulin secretion and gene transcription in the HIT-T15 pancreatic cells. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1018–1026. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, V.; Halban, P.A.; Rouiller, D.G. Tumor necrosis factor-alpha modifies adhesion properties of rat islet B cells. J. Clin. Investig. 1993, 91, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Shinjo, T.; Iwashita, M.; Yamashita, A.; Sano, T.; Tsuruta, M.; Matsunaga, H.; Sanui, T.; Asano, T.; Nishimura, F. IL-17A synergistically enhances TNFα-induced IL-6 and CCL20 production in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2016, 477, 241–246. [Google Scholar] [CrossRef]
- Bugliani, M.; Syed, F.; Paula, F.M.M.; Omar, B.A.; Suleiman, M.; Mossuto, S.; Grano, F.; Cardarelli, F.; Boggi, U.; Vistoli, F.; et al. DPP-4 is expressed in human pancreatic beta cells and its direct inhibition improves beta cell function and survival in type 2 diabetes. Mol. Cell. Endocrinol. 2018, 473, 186–193. [Google Scholar] [CrossRef]
- Morita, A.; Mukai, E.; Hiratsuka, A.; Takatani, T.; Iwanaga, T.; Lee, E.Y.; Miki, T. Distinct effects of dipeptidyl peptidase-4 inhibitor and glucagon-like peptide-1 receptor agonist on islet morphology and function. Endocrine 2016, 51, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Ardestani, A.; Dharmadhikari, G.; Laue, S.; Schumann, D.M.; Kerr-Conte, J.; Pattou, F.; Klein, T.; Maedler, K. The DPP-4 inhibitor linagliptin restores β-cell function and survival in human isolated islets through GLP-1 stabilization. J. Clin. Endocrinol. Metab. 2013, 98, E1163–E1172. [Google Scholar] [CrossRef] [Green Version]
- Akarte, A.S.; Srinivasan, B.P.; Gandhi, S.; Sole, S. Chronic DPP-IV inhibition with PKF-275-055 attenuates inflammation and improves gene expressions responsible for insulin secretion in streptozotocin induced diabetic rats. Eur. J. Pharm. Sci. 2012, 47, 456–463. [Google Scholar] [CrossRef]
- Mu, J.; Petrov, A.; Eiermann, G.J.; Woods, J.; Zhou, Y.P.; Li, Z.; Zycband, E.; Feng, Y.; Zhu, L.; Roy, R.S.; et al. Inhibition of DPP-4 with sitagliptin improves glycemic control and restores islet cell mass and function in a rodent model of type 2 diabetes. Eur. J. Pharmacol. 2009, 623, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Nakata, M.; Okada, T.; Ozawa, K.; Yada, T. Resistin induces insulin resistance in pancreatic islets to impair glucose-induced insulin release. Biochem. Biophys. Res. Commun. 2007, 353, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Yang, Y.; Sun, C.; Fang, H.; Nie, L.; Li, L.; Liu, Y.; Yang, Z. Resistin inhibits glucose-stimulated insulin secretion through Mir-494 by target on STXBP5. Acta Endocrinol. 2017, 13, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Sassek, M.; Pruszynska-Oszmalek, E.; Kołodziejski, P.A.; Szczepankiewicz, D.; Kaczmarek, P.; Wieloch, M.; Kurto, K.; Nogowski, L.; Nowak, K.W.; Strowski, M.Z.; et al. Resistin is produced by rat pancreatic islets and regulates insulin and glucagon in vitro secretion. Islets 2016, 8, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, J.; Choudhury, A.I.; Asare-Anane, H.; Selman, C.; Lingard, S.; Heffron, H.; Herrera, P.; Persaud, S.J.; Withers, D.J. Pancreatic deletion of insulin receptor substrate 2 reduces beta and alpha cell mass and impairs glucose homeostasis in mice. Diabetologia 2007, 50, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Ronald Kahn, C. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Bulum, T.; Tomić, M.; Vučković-Rebrina, S.; Roso, V.; Vučić Lovrenčić, M.; Duvnjak, L. Preserved C-peptide secretion in patients with type 1 diabetes and incipient chronic complications is associated with lower serum resistin and higher uric acid levels. J. Diabetes Metab. Disord. 2020, 19, 1185–1189. [Google Scholar] [CrossRef]
- Pham, M.N.; Kolb, H.; Mandrup-Poulsen, T.; Battelino, T.; Ludvigsson, J.; Pozzilli, P.; Roden, M.; Schloot, N.C. Serum adipokines as biomarkers of beta-cell function in patients with type 1 diabetes: Positive association with leptin and resistin and negative association with adiponectin. Diabetes Metab. Res. Rev. 2013, 29, 166–170. [Google Scholar] [CrossRef]
- Curran, A.M.; Ryan, M.F.; Drummond, E.; Gibney, E.R.; Gibney, M.J.; Roche, H.M.; Brennan, L. Uncovering factors related to pancreatic beta-cell function. PLoS ONE 2016, 11, e0161350. [Google Scholar] [CrossRef] [Green Version]
- Ramracheya, R.D.; Muller, D.S.; Wu, Y.; Whitehouse, B.J.; Huang, G.C.; Amiel, S.A.; Karalliedde, J.; Viberti, G.; Jones, P.M.; Persaud, S.J. Direct regulation of insulin secretion by angiotensin II in human islets of Langerhans. Diabetologia 2006, 49, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Yu, L.; Gao, L. Activation of angiotensin type 2 receptors partially ameliorates streptozotocin-induced diabetes in male rats by islet protection. Endocrinology 2014, 155, 790–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebelmann, M.; Wensing, J.; Verspohl, E.J. The impact of ANG II and IV on INS-1 cells and on blood glucose and plasma insulin. J. Recept. Signal Transduct. Res. 2010, 30, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.; AlSiraj, Y.; Chen, J.; Cassis, L.A. Pancreatic AT1aR deficiency decreases insulin secretion in obese C57BL/6 mice. Am. J. Hypertens. 2019, 32, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.S.; Thienel, C.; Plutino, Y.; Kampe, K.; Dror, E.; Traub, S.; Timper, K.; Bédat, B.; Pattou, F.; Kerr-Conte, J.; et al. Angiotensin II induces interleukin-1β-mediated islet inflammation and β-cell dysfunction independently of vasoconstrictive effects. Diabetes 2015, 64, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, L.; Sopontammarak, B.; Menikdiwela, K.R.; Moustaid-Moussa, N. Endoplasmic reticulum (ER) stress in part mediates effects of angiotensin II in pancreatic beta cells. Diabetes Metab. Syndr. Obes. 2020, 13, 2843–2853. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Lau, Y.S.; Miller, A.A.; Ku, J.M.; Potocnik, S.; Ye, J.M.; Woodman, O.L.; Herbert, T.P. Angiotensin II causes β-cell dysfunction through an ER stress-induced proinflammatory response. Endocrinology 2017, 158, 3162–3173. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Ibata, T.; Kobayashi, T.; Dong, T.; Yoshimoto, T.; Yonezaki, K.; Nagata, H.; et al. Angiotensin II induces cholesterol accumulation and impairs insulin secretion by regulating ABCA1 in beta cells. J. Lipid Res. 2018, 59, 1906–1915. [Google Scholar] [CrossRef] [Green Version]
- Ihoriya, C.; Satoh, M.; Kuwabara, A.; Sasaki, T.; Kashihara, N. Angiotensin II regulates islet microcirculation and insulin secretion in mice. Microcirculation 2014, 21, 112–123. [Google Scholar] [CrossRef]
- Carlsson, P.O.; Berne, C.; Jansson, L. Angiotensin II and the endocrine pancreas: Effects on islet blood flow and insulin secretion in rats. Diabetologia 1998, 41, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Fliser, D.; Schaefer, F.; Schmid, D.; Veldhuis, J.D.; Ritz, E. Angiotensin II affects basal, pulsatile, and glucose-stimulated insulin secretion in humans. Hypertension 1997, 30, 1156–1161. [Google Scholar] [CrossRef]
- Lau, T.; Carlsson, P.O.; Leung, P.S. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 2004, 47, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikellis, C.; Wookey, P.J.; Candido, R.; Andrikopoulos, S.; Thomas, M.C.; Cooper, M.E. Improved islet morphology after blockade of the renin- angiotensin system in the ZDF rat. Diabetes 2004, 53, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, J.; Li, Y.; Zhou, X.; Qiao, S.; Han, D. Telmisartan protects against high glucose/high lipid-induced apoptosis and insulin secretion by reducing the oxidative and ER stress. Cell Biochem. Funct. 2019, 37, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Kwan, Y.C.; Lau, T.; Carlsson, P.O.; Po, S.L. Angiotensin II type 1 receptor blockade improves beta-cell function and glucose tolerance in a mouse model of type 2 diabetes. Diabetes 2006, 55, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, C.; Gan, Z.; Wang, X.; Yi, Q.; Liu, Y.; Wang, Y.; Lu, B.; Du, H.; Shao, J.; et al. Improved glucose-stimulated insulin secretion by selective intraislet inhibition of angiotensin II type 1 receptor expression in isolated islets of db/db mice. Int. J. Endocrinol. 2013, 2013, 319586. [Google Scholar] [CrossRef] [PubMed]
- Iwai, M.; Kanno, H.; Tomono, Y.; Inaba, S.; Senba, I.; Furuno, M.; Mogi, M.; Horiuchi, M. Direct renin inhibition improved insulin resistance and adipose tissue dysfunction in type 2 diabetic KK-A(y) mice. J. Hypertens. 2010, 28, 1471–1481. [Google Scholar] [CrossRef]
- Shao, J.; Iwashita, N.; Ikeda, F.; Ogihara, T.; Uchida, T.; Shimizu, T.; Uchino, H.; Hirose, T.; Kawamori, R.; Watada, H. Beneficial effects of candesartan, an angiotensin II type 1 receptor blocker, on beta-cell function and morphology in db/db mice. Biochem. Biophys. Res. Commun. 2006, 344, 1224–1233. [Google Scholar] [CrossRef]
- Li, X.; Yuan, L.; Xu, G.; Qi, C.; Li, J.; Li, H.; Cheng, S. Effect of renin angiotensin system blockade on the islet microvessel density of diabetic rats and its relationship with islet function. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 684–688. [Google Scholar] [CrossRef]
- Lupi, R.; Del Guerra, S.; Bugliani, M.; Boggi, U.; Mosca, F.; Torri, S.; Del Prato, S.; Marchetti, P. The direct effects of the angiotensin-converting enzyme inhibitors, zofenoprilat and enalaprilat, on isolated human pancreatic islets. Eur. J. Endocrinol. 2006, 154, 355–361. [Google Scholar] [CrossRef]
- Yuan, L.; Li, X.; Xui, G.L.; Qi, C.J. Effects of renin-angiotensin system blockade on islet function in diabetic rats. J. Endocrinol. Investig. 2010, 33, 13–19. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Chueire, V.B.; Muscelli, E. Effect of free fatty acids on insulin secretion, insulin sensitivity and incretin effect—A narrative review. Arch. Endocrinol. Metab. 2021, 65, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.F.; Deng, Y.; Zhou, Y.; Fan, R.R.; Chan, J.C.N.; Laybutt, D.R.; Luzuriaga, J.; Xu, G. Pharmacological reduction of NEFA restores the efficacy of incretin-based therapies through GLP-1 receptor signalling in the beta cell in mouse models of diabetes. Diabetologia 2013, 56, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natalicchio, A.; Biondi, G.; Marrano, N.; Labarbuta, R.; Tortosa, F.; Spagnuolo, R.; D’Oria, R.; Carchia, E.; Leonardini, A.; Cignarelli, A.; et al. Long-term exposure of pancreatic β-cells to palmitate results in SREBP-1C-dependent decreases in GLP-1 receptor signaling via CREB and AKT and insulin secretory response. Endocrinology 2016, 157, 2243–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Hao, L.; Li, S.; Lin, S.; Lv, L.; Chen, Y.; Cui, H.; Zi, T.; Chu, X.; Na, L.; et al. Elevated circulating stearic acid leads to a major lipotoxic effect on mouse pancreatic beta cells in hyperlipidaemia via a miR-34a-5p-mediated PERK/p53-dependent pathway. Diabetologia 2016, 59, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Gesmundo, I.; Pardini, B.; Gargantini, E.; Gamba, G.; Birolo, G.; Fanciulli, A.; Banfi, D.; Congiusta, N.; Favaro, E.; Deregibus, M.C.; et al. Adipocyte-derived extracellular vesicles regulate survival and function of pancreatic β cells. JCI Insight 2021, 6, e141962. [Google Scholar] [CrossRef]
- Maedler, K.; Sergeev, P.; Ehses, J.; Mathe, Z.; Bosco, D.; Berney, T.; Dayer, J.; Reinecke, M.; Halban, P.; Donath, M.Y. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc. Natl. Acad. Sci. USA 2004, 101, 8138–8143. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Schulthess, F.; Bielman, C.; Berney, T.; Bonny, C.; Prentki, M.; Donath, M.Y.; Roduit, R. Glucose and leptin induce apoptosis in human beta-cells and impair glucose-stimulated insulin secretion through activation of c-Jun N-terminal kinases. FASEB J. 2008, 22, 1905–1913. [Google Scholar] [CrossRef] [Green Version]
- Hekerman, P.; Zeidler, J.; Korfmacher, S.; Bamberg-Lemper, S.; Knobelspies, H.; Zabeau, L.; Tavernier, J.; Becker, W. Leptin induces inflammation-related genes in RINm5F insulinoma cells. BMC Mol. Biol. 2007, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; López, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338–351. [Google Scholar] [CrossRef]
- Oh, Y.; Bae, G.; Park, E.; Jun, H. MicroRNA-181c inhibits interleukin-6-mediated beta cell apoptosis by targeting TNF-α expression. Molecules 2019, 24, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.; Lee, Y.; Park, E.; Jun, H. Interleukin-6 treatment induces beta-cell apoptosis via STAT-3-mediated nitric oxide production. Diabetes Metab. Res. Rev. 2011, 27, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, T.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The role of inflammation in β-cell dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Zhao, D.; Qiu, J.; Zhang, C.; Ji, C.; Chen, X.; Liu, F.; Guo, X. Resistin induces rat insulinoma cell RINm5F apoptosis. Mol. Biol. Rep. 2009, 36, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Fukaya, M.; Nogueira, T.; Delaroche, D.; Welsh, N.; Marselli, L.; Marchetti, P.; Haefliger, J.; Eizirik, D.; Cardozo, A. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in β-cells. Cell Death Differ. 2012, 19, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Pinto, C.; García, M.; Gómez, L.; Ballesteros, A.; Zaballos, A.; Flores, J.; Mellado, M.; Rodríguez-Frade, J.; Balomenos, D.; Martínez-A, C. Leukocyte attraction through the CCR5 receptor controls progress from insulitis to diabetes in non-obese diabetic mice. Eur. J. Immunol. 2004, 34, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Dunmore, S.; Brown, J. The role of adipokines in β-cell failure of type 2 diabetes. J. Endocrinol. 2013, 216, T37–T45. [Google Scholar] [CrossRef] [Green Version]
- Kamper, M.; Tsimpoukidi, O.; Chatzigeorgiou, A.; Lymberi, M.; Kamper, E.F. The antioxidant effect of angiotensin II receptor blocker, losartan, in streptozotocin-induced diabetic rats. Transl. Res. 2010, 156, 26–36. [Google Scholar] [CrossRef]
- Frantz, E.D.C.; Crespo-Mascarenhas, C.; Barreto-Vianna, A.R.C.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Renin-angiotensin system blockers protect pancreatic islets against diet-induced obesity and insulin resistance in mice. PLoS ONE 2013, 8, e67192. [Google Scholar] [CrossRef]
- Graus-Nunes, F.; de Souza Marinho, T.; Barbosa-da-Silva, S.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential effects of angiotensin receptor blockers on pancreatic islet remodelling and glucose homeostasis in diet-induced obese mice. Mol. Cell. Endocrinol. 2017, 439, 54–64. [Google Scholar] [CrossRef]
- Chu, K.Y.; Leung, P.S. Angiotensin II Type 1 receptor antagonism mediates uncoupling protein 2-driven oxidative stress and ameliorates pancreatic islet beta-cell function in young Type 2 diabetic mice. Antioxid. Redox Signal. 2007, 9, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Li, X.; Li, J.; Li, H.-L.; Cheng, S.-S. Effects of renin-angiotensin system blockade on the islet morphology and function in rats with long-term high-fat diet. Acta Diabetol. 2013, 50, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, W.; Ruan, Y.; Yang, L.; Xu, N.; Chen, R.; Yang, R.; Sun, J.; Zhang, Z. Reversal of angiotensin ll-induced β-cell dedifferentiation via inhibition of NF-κb signaling. Mol. Med. 2018, 24, 43. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Tong, A.; Xu, Y. Pancreatic β-cell function is inhibited by miR-3666 in type 2 diabetes mellitus by targeting adiponectin. Braz. J. Med. Biol. Res. 2019, 52, e8344. [Google Scholar] [CrossRef]
- Kim, J.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.; Hofmann, S.; Schraw, T.; Durand, J.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Wang, M.; Wang, Q.; Scherer, P. Adiponectin-mediated antilipotoxic effects in regenerating pancreatic islets. Endocrinology 2015, 156, 2019–2028. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Saget, S.; Lu, C.; Hay, W.; Karsenty, G.; Shao, J. Adiponectin promotes maternal β-cell expansion through placental lactogen expression. Diabetes 2021, 70, 132–142. [Google Scholar] [CrossRef]
- Xie, K.; Xu, B.; Zhang, Y.; Chen, M.; Ji, Y.; Wang, J.; Huang, Z.; Zhou, K.; Xia, Y.; Tang, W. A multi-method evaluation of the effects of Inflammatory cytokines (IL-1β, IFN-γ, TNF-α) on pancreatic β-cells. J. Cell. Physiol. 2018, 233, 9375–9382. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Baquerizo, H.; Sumoski, W. Cytotoxic effects of cytokines on islet beta-cells: Evidence for involvement of eicosanoids. Endocrinology 1990, 126, 67–71. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Sumoski, W.; Rajotte, R.V.; Warnock, G.L. Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture. J. Clin. Endocrinol. Metab. 1990, 71, 152–156. [Google Scholar] [CrossRef]
- Stephens, L.A.; Thomas, H.E.; Ming, L.; Grell, M.; Darwiche, R.; Volodin, L.; Kay, T.W. Tumor necrosis factor-alpha-activated cell death pathways in NIT-1 insulinoma cells and primary pancreatic beta cells. Endocrinology 1999, 140, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, N.; Yagui, K.; Tokuyama, Y.; Yamada, K.; Suzuki, Y.; Miyazaki, J.; Hashimoto, N.; Makino, H.; Saito, Y.; Kanatsuka, A. Tumor necrosis factor alpha signaling pathway and apoptosis in pancreatic beta cells. Metabolism 1999, 48, 1485–1492. [Google Scholar] [CrossRef]
- Natalicchio, A.; De Stefano, F.; Orlando, M.R.; Melchiorre, M.; Leonardini, A.; Cignarelli, A.; Labarbuta, R.; Marchetti, P.; Perrini, S.; Laviola, L.; et al. Exendin-4 prevents c-Jun N-terminal protein kinase activation by Tumor Necrosis Factor-α (TNFα) and inhibits TNFα-induced apoptosis in insulin-secreting cells. Endocrinology 2010, 151, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Parkash, J.; Chaudhry, M.; Rhoten, W. Tumor necrosis factor-alpha-induced changes in insulin-producing beta-cells. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol. 2005, 286, 982–993. [Google Scholar] [CrossRef]
- Chang, I.; Kim, S.; Kim, J.; Cho, N.; Kim, Y.; Kim, H.; Lee, M.; Kim, K.; Lee, M. Nuclear factor kappaB protects pancreatic beta-cells from tumor necrosis factor-alpha-mediated apoptosis. Diabetes 2003, 52, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Cottet, S.; Dupraz, P.; Hamburger, F.; Dolci, W.; Jaquet, M.; Thorens, B. cFLIP protein prevents tumor necrosis factor-alpha-mediated induction of caspase-8-dependent apoptosis in insulin-secreting betaTc-Tet cells. Diabetes 2002, 51, 1805–1814. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, J.; Amo, K.; Ohminami, H.; Orime, K.; Togashi, Y.; Ito, Y.; Tajima, K.; Koganei, M.; Sasaki, H.; Takeda, E.; et al. Protective effects of dipeptidyl peptidase-4 (DPP-4) inhibitor against increased β cell apoptosis induced by dietary sucrose and linoleic acid in mice with diabetes. J. Biol. Chem. 2011, 286, 25467–25476. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and oleic acid: The yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [Green Version]
- Sargsyan, E.; Artemenko, K.; Manukyan, L.; Bergquist, J.; Bergsten, P. Oleate protects beta-cells from the toxic effect of palmitate by activating pro-survival pathways of the ER stress response. Biochim. Biophys. Acta 2016, 1861, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Geer, E.B.; Shen, W. Gender differences in insulin resistance, body composition, and energy balance. Gend. Med. 2009, 6 (Suppl. S1), 60–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaulet, M.; Perez-Llamas, F.; Fuente, T.; Zamora, S.; Tebar, F.J. Anthropometric, computed tomography and fat cell data in an obese population: Relationship with insulin, leptin, tumor necrosis factor-alpha, sex hormone-binding globulin and sex hormones. Eur. J. Endocrinol. 2000, 143, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, O.K.W.; Cheng, A.S.L. Gender differences in adipocyte metabolism and liver cancer progression. Front. Genet. 2016, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Shimomura, L.; Kishida, K.; Maeda, N.; Kuriyama, H.; Nagaretani, H.; Matsuda, M.; Kondo, H.; Furuyama, N.; Kihara, S.; et al. Androgens decrease plasma adiponectin, an insulin-sensitizing adipocyte-derived protein. Diabetes 2002, 51, 2734–2741. [Google Scholar] [CrossRef] [Green Version]


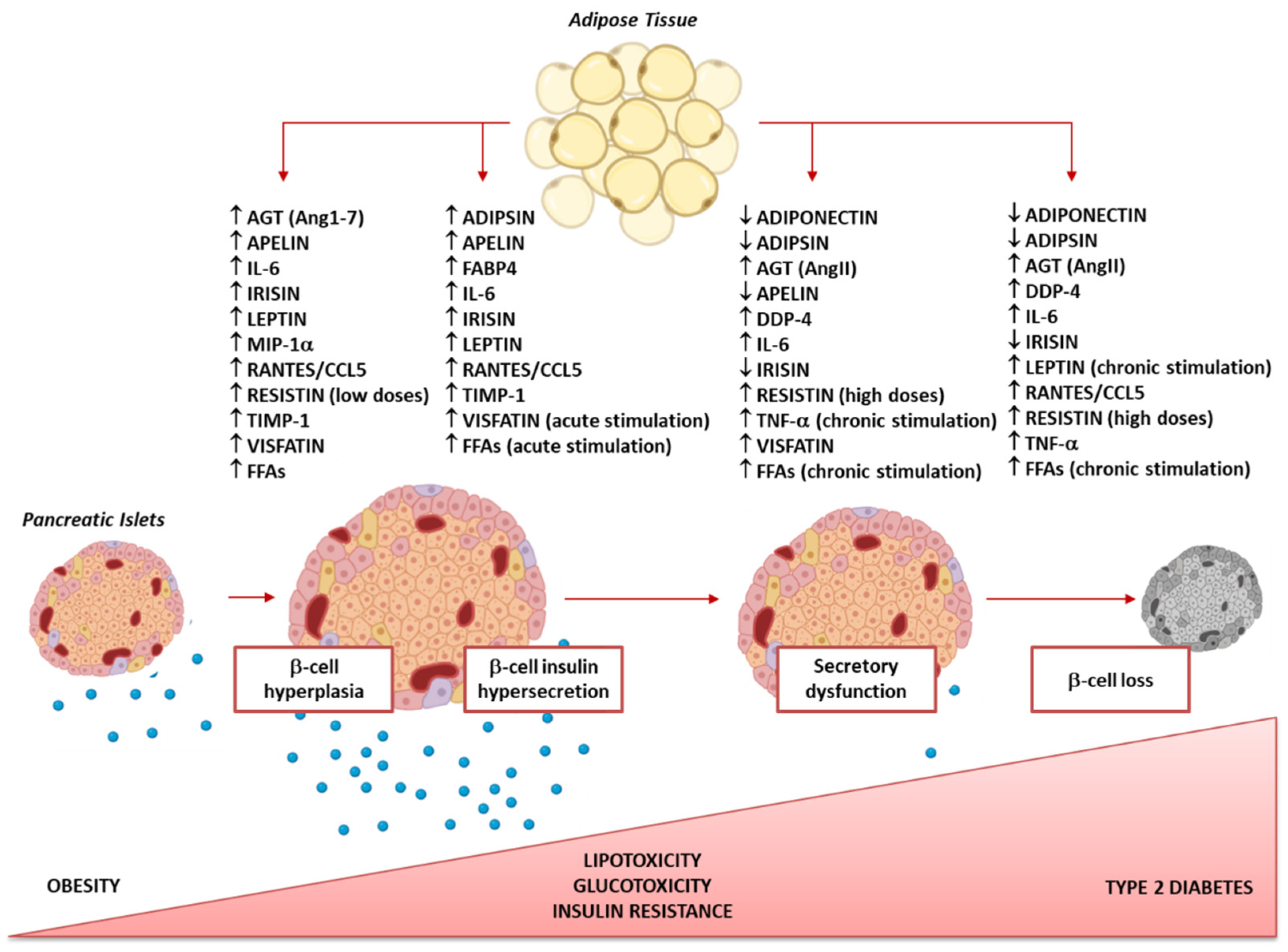{kind=link}
| Adipokines | Obese vs. Normal-Weight | Obese with T2D vs. Non-Diabetic Obese | Refs. |
|---|---|---|---|
| Adiponectin | ↓ | ↓ | [29,41,68,69] |
| Adipsin | ↑ | = | [70,79,80] |
| AGT (AngII, Ang1-7) | ↑ (↑, ↓) | N/A | [128,129,132,135] |
| Apelin | ↑ | ↓ | [30,75,76,101,102] |
| DPP-4 | ↑ | = | [140,141,142] |
| FABP4 | ↑ | ↑ | [77,78,110] |
| GRO-alpha | ↑ | N/A | [28,47] |
| IL-6 | ↑ | ↑ | [35,37,41] |
| Irisin | ↑ | ↓ | [81,82,121,122,123,124] |
| Leptin | ↑ | = | [57,60] |
| MCP-1 | ↑ | N/A | [36,40] |
| MIP-1α and MIP-1β | ↑ | N/A | [27,50,51,52] |
| PAI-1 | ↑ | N/A | [49] |
| RANTES/CCL5 | ↑ | N/A | [45] |
| Resistin | ↑ | ↑ | [71,72,84,85] |
| TIMP-1 | ↑ | N/A | [28,48] |
| TNF-α | ↑ | ↑ | [35,38,42,150] |
| TPO | ↑ | N/A | [28] |
| Visfatin | ↑ | ↑ | [73,74,86,87,88,89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biondi, G.; Marrano, N.; Borrelli, A.; Rella, M.; Palma, G.; Calderoni, I.; Siciliano, E.; Lops, P.; Giorgino, F.; Natalicchio, A. Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 5522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105522
Biondi G, Marrano N, Borrelli A, Rella M, Palma G, Calderoni I, Siciliano E, Lops P, Giorgino F, Natalicchio A. Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes. International Journal of Molecular Sciences. 2022; 23(10):5522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105522
Chicago/Turabian StyleBiondi, Giuseppina, Nicola Marrano, Anna Borrelli, Martina Rella, Giuseppe Palma, Isabella Calderoni, Edoardo Siciliano, Pasquale Lops, Francesco Giorgino, and Annalisa Natalicchio. 2022. "Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes" International Journal of Molecular Sciences 23, no. 10: 5522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105522