
Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases MUTYH and hOGG1 in Colorectal Cancer Patients

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Screening for Mutations and Gene Variants
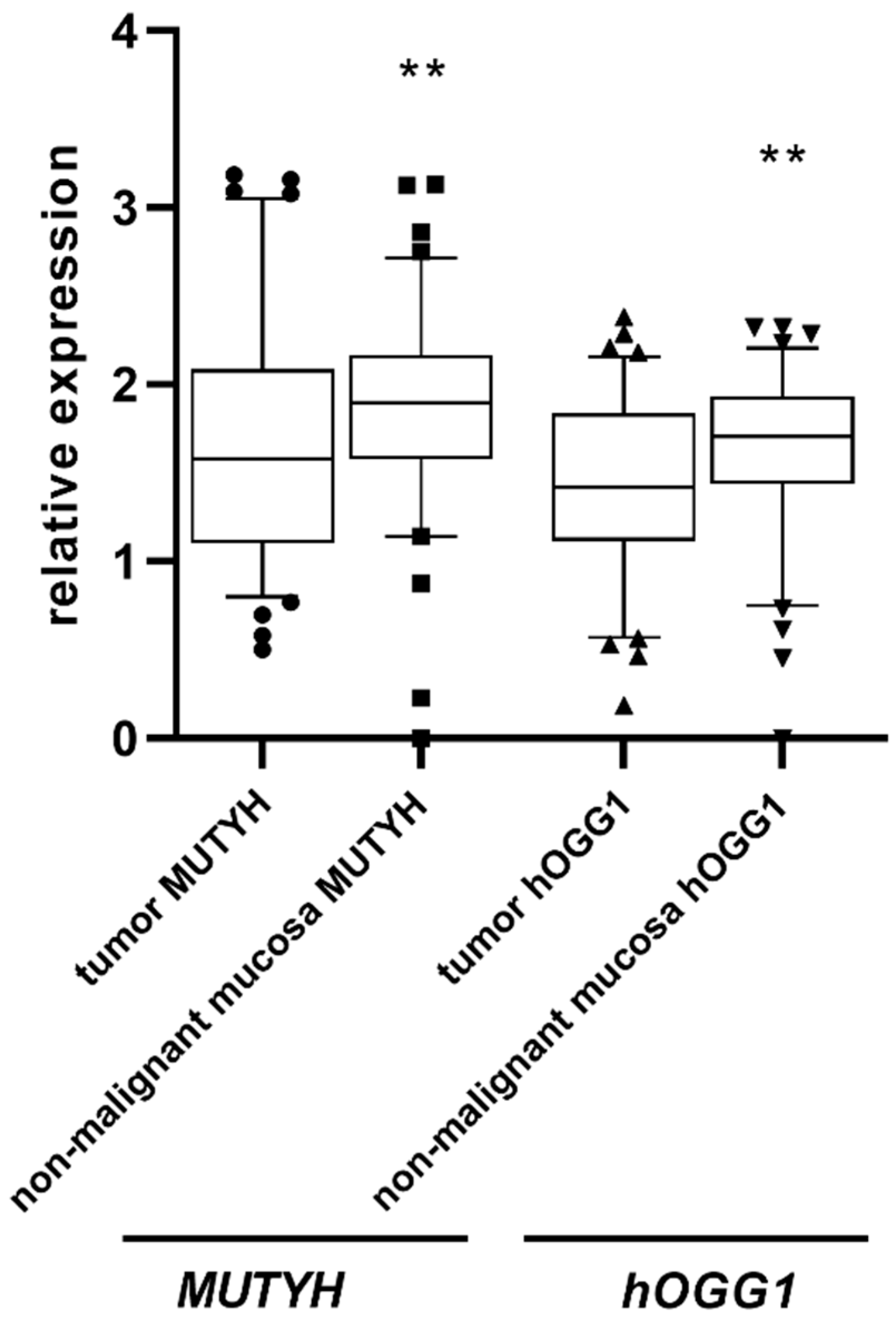
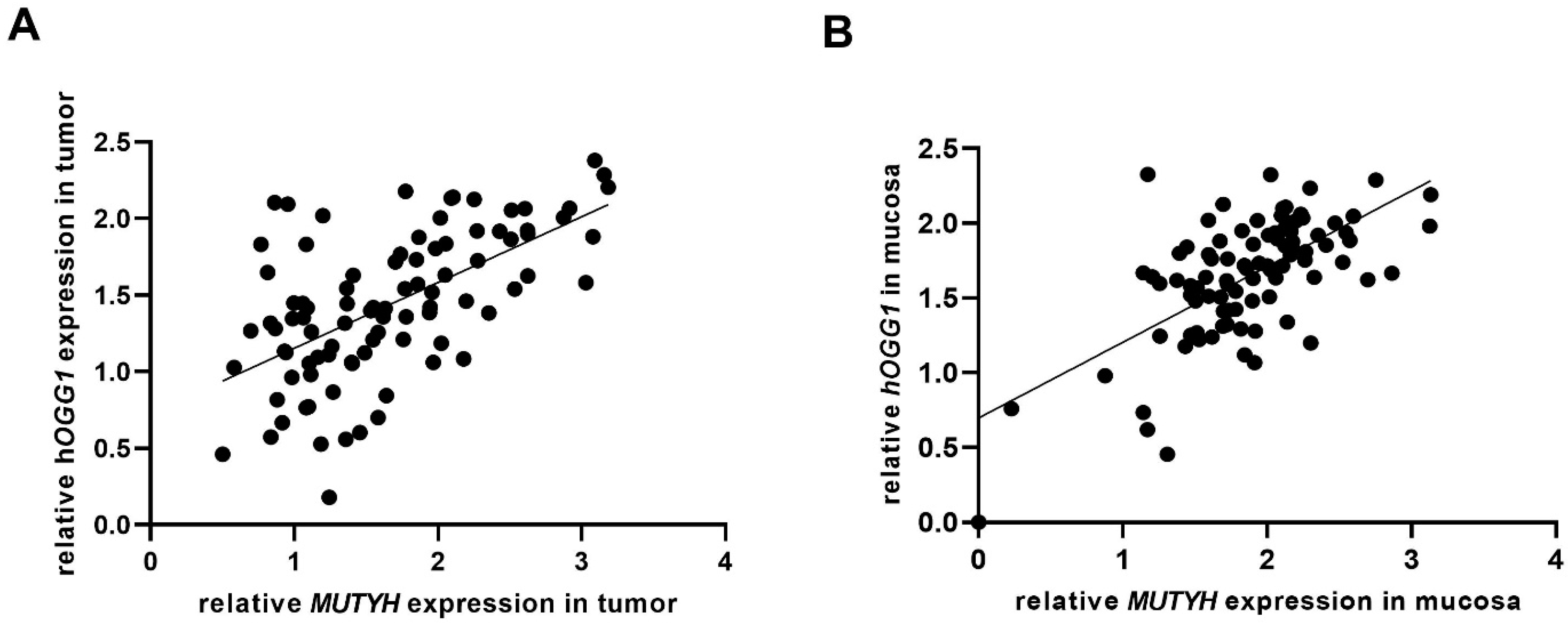
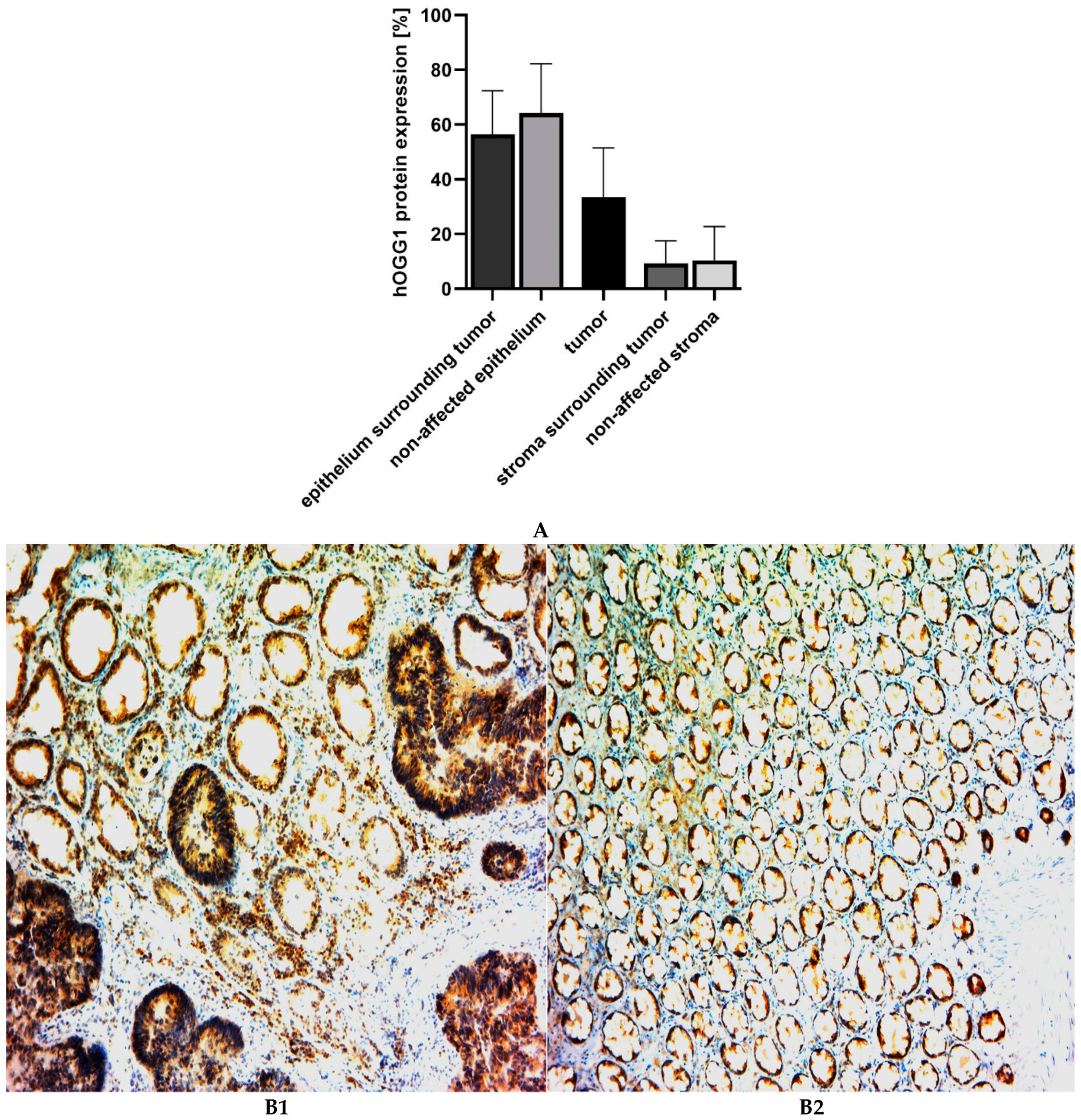
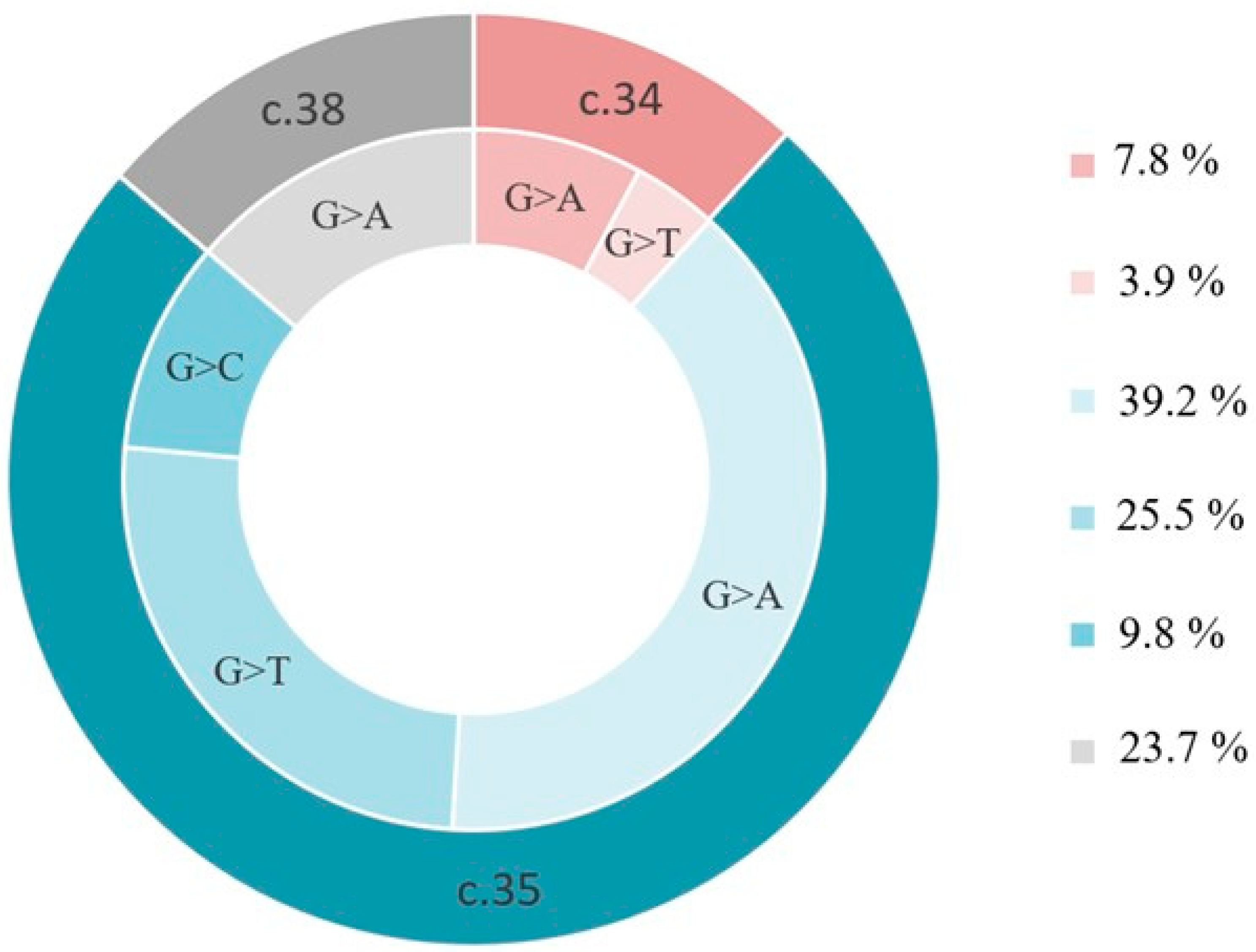
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Brenner, H.; Chen, C. The colorectal cancer epidemic: Challenges and opportunities for primary, secondary and tertiary prevention. Br. J. Cancer 2018, 119, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef]
- Collins, A.R.; Azqueta, A.; Langie, S.A. Effects of micronutrients on DNA repair. Eur. J. Nutr. 2012, 51, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Jenab, M.; Gunter, M.J. Adiposity and gastrointestinal cancers: Epidemiology, mechanisms and future directions. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 659–670. [Google Scholar] [CrossRef]
- Kompella, P.; Vasquez, K.M. Obesity and cancer: A mechanistic overview of metabolic changes in obesity that impact genetic instability. Mol. Carcinog. 2019, 58, 1531–1550. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, M.; Sakumi, K.; Fukumura, R.; Furuichi, M.; Iwasaki, Y.; Hokama, M.; Ikemura, T.; Tsuzuki, T.; Gondo, Y.; Nakabeppu, Y. 8-oxoguanine causes spontaneous de novo germline mutations in mice. Sci. Rep. 2014, 4, 4689. [Google Scholar] [CrossRef] [Green Version]
- Dizdaroglu, M. Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Mutat. Res. 2015, 763, 212–245. [Google Scholar] [CrossRef]
- Wallace, S.S. DNA glycosylases search for and remove oxidized DNA bases. Environ. Mol. Mutagen. 2013, 54, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Vodenkova, S.; Opattova, A.; Vodickova, L. DNA damage and repair measured by comet assay in cancer patients. Mutat. Res. Toxicol. Environ. Mutagen. 2019, 843, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Urbanova, M.; Makovicky, P.; Tomasova, K.; Kroupa, M.; Stetina, R.; Opattova, A.; Kostovcikova, K.; Siskova, A.; Schneiderova, M.; et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. Int. J. Mol. Sci. 2020, 21, 2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, E.F.R.; Ribeiro, M.L.; Magro, D.O.; Carvalho, J.; Kanno, D.T.; Martinez, C.A.R.; Coy, C.S.R. Tissue expresion of the genes mutyh and ogg1 in patients with sporadic colorectal cancer. ABCD Arq. Bras. Cir. Dig. 2017, 30, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular basis of colorectal cancer. New Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, S.A.; Morreau, H.; De Miranda, N.F.C.C.; Van Wezel, T. The missing heritability of familial colorectal cancer. Mutagenesis 2020, 35, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Aimé, A.; Coulet, F.; Lefevre, J.H.; Colas, C.; Cervera, P.; Flejou, J.-F.; Lascols, O.; Soubrier, F.; Parc, Y. Somatic c.34G > T KRAS mutation: A new prescreening test for MUTYH-associated polyposis? Cancer Genet. 2015, 208, 390–395. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2011, 121, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Zinsky, R.; Bölükbas, S.; Bartsch, H.; Schirren, J.; Fisseler-Eckhoff, A. Analysis of KRAS Mutations of Exon 2 Codons 12 and 13 by SNaPshot Analysis in Comparison to Common DNA Sequencing. Gastroenterol. Res. Pr. 2010, 2010, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Slyskova, J.; Cordero, F.; Pardini, B.; Korenkova, V.; Vymetalkova, V.; Bielik, L.; Vodickova, L.; Pitule, P.; Liska, V.; Matejka, V.M.; et al. Post-treatment recovery of suboptimal DNA repair capacity and gene expression levels in colorectal cancer patients. Mol. Carcinog. 2014, 54, 769–778. [Google Scholar] [CrossRef]
- Jiraskova, K.; Hughes, D.J.; Brezina, S.; Gumpenberger, T.; Veskrnova, V.; Buchler, T.; Schneiderova, M.; Levy, M.; Liska, V.; Vodenkova, S.; et al. Functional polymorphisms in DNA repair genes are associated with sporadic colorectal cancer susceptibility and clinical outcome. Int. J. Mol. Sci. 2018, 20, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodenkova, S.; Jiraskova, K.; Urbanova, M.; Kroupa, M.; Slyskova, J.; Schneiderova, M.; Levy, M.; Buchler, T.; Liska, V.; Vodickova, L.; et al. Base excision repair capacity as a determinant of prognosis and therapy response in colon cancer patients. DNA Repair 2018, 72, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Dolwani, S.; Williams, G.T.; West, K.P.; Newman, J.; Stock, D.; Griffiths, A.P.; Best, J.; Cheadle, J.P.; Sampson, J.R. Analysis of inherited MYH/(MutYH) mutations in British Asian patients with colorectal cancer. Gut 2007, 56, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görgens, H.; Krüger, S.; Kuhlisch, E.; Pagenstecher, C.; Höhl, R.; Schackert, H.K.; Müller, A. Microsatellite stable colorectal cancers in clinically suspected hereditary nonpolyposis colorectal cancer patients without vertical transmission of disease are unlikely to be caused by biallelic germline mutations in MYH. J. Mol. Diagn. 2006, 8, 178–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, R.P.; Hoogerbrugge, N. NTHL1 defines novel cancer syndrome. Oncotarget 2015, 6, 34069–34070. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Slyskova, J.; Korenkova, V.; Collins, A.; Prochazka, P.; Vodickova, L.; Svec, J.; Lipska, L.; Levy, M.; Schneiderová, M.; Liška, V.; et al. Functional, Genetic, and Epigenetic Aspects of Base and Nucleotide Excision Repair in Colorectal Carcinomas. Clin. Cancer Res. 2012, 18, 5878–5887. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Vodenkova, S.; Buchler, T.; Vodickova, L. DNA repair capacity and response to treatment of colon cancer. Pharmacogenomics 2019, 20, 1225–1233. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D. PD-L1, TMB, and other potential predictors of response to immunotherapy for hepatocellular carcinoma: How can they assist drug clinical trials? Expert Opin. Investig. Drugs 2022, 31, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dowling, E.K.; Yan, G.; Dereli, Z.; Bozorgui, B.; Imanirad, P.; Elnaggar, J.H.; Luna, A.; Menter, D.G.; Pilié, P.G.; et al. Precision Combination Therapies Based on Recurrent Oncogenic Coalterations. Cancer Discov. 2022, OF1–OF18. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Kaina, B. Impact of DNA repair on the dose-response of colorectal cancer formation induced by dietary carcinogens. Food Chem. Toxicol. 2017, 106, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Melis, J.P.; Van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [Green Version]
- Lerner, L.K.; Moreno, N.C.; Rocha, C.R.R.; Munford, V.; Santos, V.; Soltys, D.T.; Garcia, C.C.M.; Sarasin, A.; Menck, C.F. XPD/ERCC2 mutations interfere in cellular responses to oxidative stress. Mutagenesis 2019, 34, 341–354. [Google Scholar] [CrossRef]
- Lee, T.-H.; Kang, T.-H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Shi, T.-Y.; Zhu, M.-L.; Wang, M.-Y.; Li, Q.-X.; Wei, Q.-Y. Associations of Lys939Gln and Ala499Val polymorphisms of the XPC gene with cancer susceptibility: A meta-analysis. Int. J. Cancer 2013, 133, 1765–1775. [Google Scholar] [CrossRef]
- Slyskova, J.; Naccarati, A.; Pardini, B.; Polakova, V.; Vodickova, L.; Smerhovsky, Z.; Levy, M.; Lipska, L.; Liska, V.; Vodicka, P. Differences in nucleotide excision repair capacity between newly diagnosed colorectal cancer patients and healthy controls. Mutagenesis 2012, 27, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Stetina, R.; Polakova, V.; Tulupova, E.; Naccarati, A.; Vodickova, L.; Kumar, R.; Hanova, M.; Pardini, B.; Slyskova, J.; et al. Association of DNA repair polymorphisms with DNA repair functional outcomes in healthy human subjects. Carcinogenesis 2007, 28, 657–664. [Google Scholar] [CrossRef]
- Guo, C.-L.; Han, F.-F.; Wang, H.-Y.; Wang, L. Meta-analysis of the association between hOGG1 Ser326Cys polymorphism and risk of colorectal cancer based on case–control studies. J. Cancer Res. Clin. Oncol. 2012, 138, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Kinnersley, B.; Buch, S.; Castellví-Bel, S.; Farrington, S.M.; Forsti, A.; Hampe, J.; Hemminki, K.; Hofstra, R.M.W.; Northwood, E.; Palles, C.; et al. Re: Role of the oxidative DNA damage repair gene OGG1 in colorectal tumorigenesis. J. Natl. Cancer Inst. 2013, 106, dju086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuk, J.; Ozernov-Palchik, O.; Kim, H.; Lakshminarayanan, K.; Gabrieli, J.D.E.; Tallal, P.; Gaab, N. Enhanced Syllable Discrimination Thresholds in Musicians. PLoS ONE 2013, 8, e80546. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients (n = 193) | ||
|---|---|---|
| Gender | Male | 122 (63.2%) |
| Female | 71 (36.8%) | |
| Age of diagnosis | Median | 69.5 |
| Range | 38–91 | |
| Smoker | Smoker | 29 (15%) |
| Non-smoker | 85 (44%) | |
| Ex-smoker | 79 (41%) | |
| TNM stage | I | 45 (23%) |
| II | 57 (29.5%) | |
| II | 58 (30%) | |
| IV | 34 (17.5%) | |
| MSI phenotype | MSI-high | 16 (8.3%) |
| MSI-low (stable) | 177 (91.7%) | |
| Localization | colon | 131 (67.5%) |
| rectosigmoideum | 19 (9.8%) | |
| rectum | 44 (22.7%) | |
| Grading (n = 64) | NDA | 4 (6.25%) |
| 0 | 6 (9.38%) | |
| 1 | 3 (4.68%) | |
| 1–2 | 6 (9.38%) | |
| 2 | 36 (56.25%) | |
| 2–3 | 3 (4.68%) | |
| 3 | 5 (7.81%) | |
| 4 | 1 (1.56%) |
| Somatic Mutations | |||
|---|---|---|---|
| Carrier/Gender/Age | Gene Position | Protein Position | Reference, rs# |
| P137/F/60 | c.141 G>A | p.Lys47Lys | novel |
| P157/F/77 | c.695 C>T | p.Thr232Ile | novel |
| P257/M/70 | c.38 C>T | p.Ala13Val | rs587780747 |
| Germline Mutation | |||
| P145/M/85 | c.312 C>A | p.Tyr104X | [23] |
| P13/F/75 | c.1187 G>A | p.Gly396Asp | [24], rs36053993 |
| Benign Exonic SNPs | |||
| Frequency in our Sample Group | Gene Position | Protein Position | SNP ID |
| 16/193 | c.64 G>A | p.Val22Met | rs3219484 |
| 1/193 | c.312C>T | p.Tyr104Tyr | rs121908380 |
| 31/193 | c.1014G>C | p.Gln338His | rs3219489 |
| 2/193 | c.1276C>T | p.Arg426Cys | rs150792276 |
| 1/193 | c.1431G>C | p.Thr477Thr | novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavec, M.J.; Urbanova, M.; Makovicky, P.; Opattová, A.; Tomasova, K.; Kroupa, M.; Kostovcikova, K.; Siskova, A.; Navvabi, N.; Schneiderova, M.; et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases MUTYH and hOGG1 in Colorectal Cancer Patients. Int. J. Mol. Sci. 2022, 23, 5704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105704
Kavec MJ, Urbanova M, Makovicky P, Opattová A, Tomasova K, Kroupa M, Kostovcikova K, Siskova A, Navvabi N, Schneiderova M, et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases MUTYH and hOGG1 in Colorectal Cancer Patients. International Journal of Molecular Sciences. 2022; 23(10):5704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105704
Chicago/Turabian StyleKavec, Miriam J., Marketa Urbanova, Pavol Makovicky, Alena Opattová, Kristyna Tomasova, Michal Kroupa, Klara Kostovcikova, Anna Siskova, Nazila Navvabi, Michaela Schneiderova, and et al. 2022. "Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases MUTYH and hOGG1 in Colorectal Cancer Patients" International Journal of Molecular Sciences 23, no. 10: 5704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105704