
Integrated mRNA and miRNA Transcriptome Analysis Suggests a Regulatory Network for UV–B-Controlled Terpenoid Synthesis in Fragrant Woodfern (Dryopteris fragrans)



Abstract
:1. Introduction
2. Results
2.1. Cell Death, Photosynthetic Pigments Content, ROS and Peroxidase Activity in Fragrant Woodfern Leaves after UV–B Treatment
2.2. Terpenoid Content in Fragrant Woodfern Leaves under UV–B Treatment
2.3. Overview of RNA-Seq Dynamics and Small RNA Sequencing
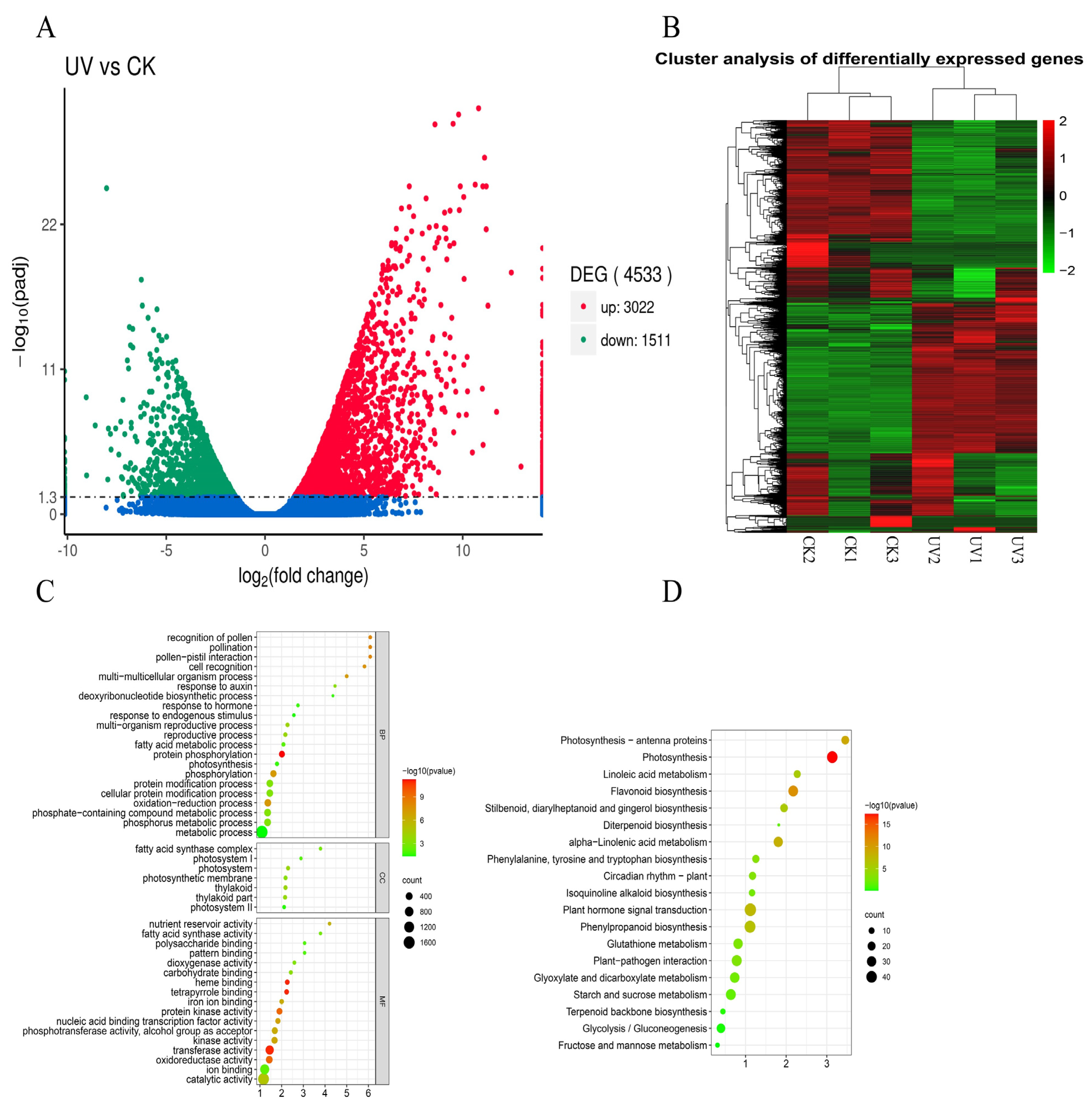
2.4. Differentially Expressed Genes Annotation by GO Term and KEGG Pathway
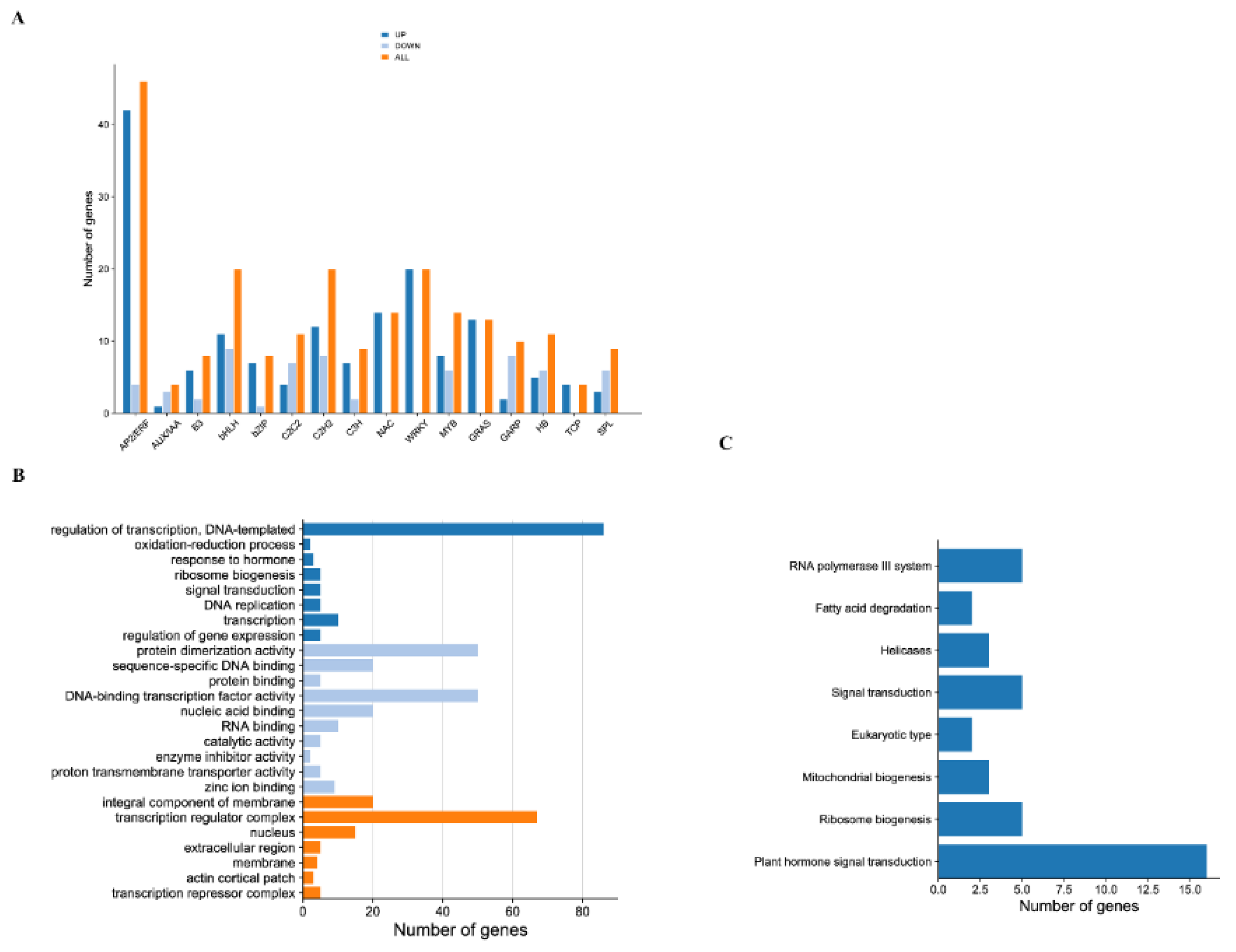
2.5. Transcription Factors
2.6. Identification of Target Genes of Differentially Expressed miRNA in Tuber Expansion Compared with Initiation Stage
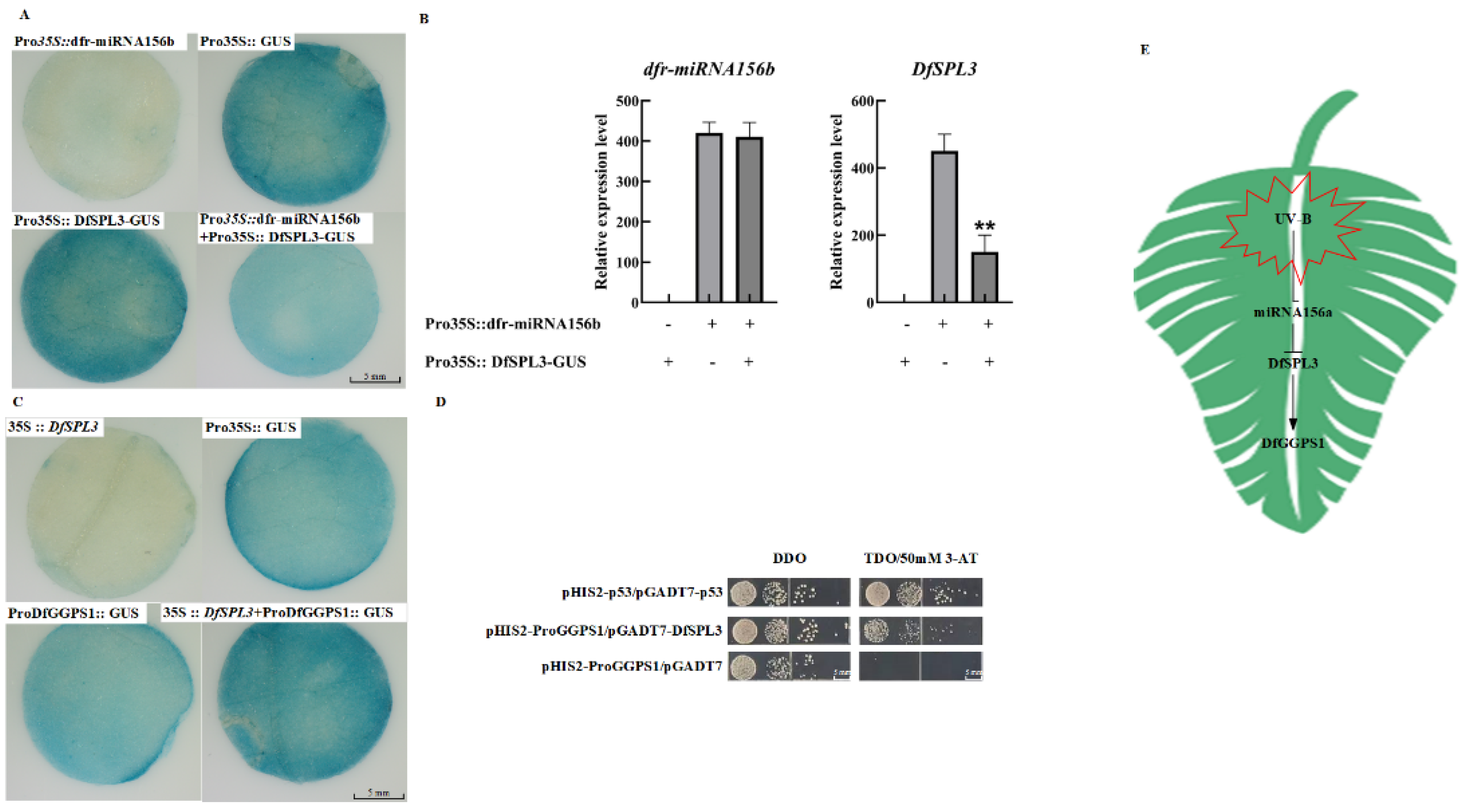
2.7. The dfr–miR156b–DfSPL3 Module Regulates the Expression of DfGGPS1
2.8. qRT-PCR Analysis of DEGs and DEMs Data
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Treatment
4.2. Enzyme-Labeled Instrument Analysis of Photosynthetic Pigments
4.3. Measurement of Cell Death
4.4. Assay on ROS and Peroxidase Activity in Fragrant Woodfern Plants
4.5. Determination of the Secondary Metabolite Content of Dryopteris Serrata by GC–MS
4.5.1. Sample Processing before Sample Loading in GC–MS Experiment
4.5.2. GC–MS Instrument Experimental Conditions
4.6. RNA Extraction, Library Construction and Sequencing
4.7. Identification and Functional Annotation of Differentially Expressed Genes and miRNAs
4.8. Validation of the DEGs and DEMs Data Using qRT-PCR
4.9. One-Hybrid Screening
4.10. Agrobacterium-Mediated Transient Transformation in Tobacco (Nicotiana Benthamiana)
4.11. Statistics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DEG | Differentially expressed genes |
| UV–B | Ultraviolet-B |
| GO | gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DEM | Differentially expressed miRNA |
| MVA | pathway Mevalonate pathway |
| MEP | pathway 2-C-methylerythritol 4-phosphate pathway |
References
- Chen, L.; Zhang, D.; Song, C.; Wang, H.; Tang, X.; Chang, Y. Transcriptomic Analysis and Specific Expression of Transcription Factor Genes in the Root and Sporophyll of Dryopteris fragrans (L.) Schott. Int. J. Mol. Sci. 2020, 21, 7296. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, X.; Shen, Z.; Cheng, W.; Zeng, Z.; Chen, Y.; Tang, C.; Jiang, T. The effect of isoflavaspidic acid PB extracted from Dryopteris fragrans (L.) Schott on planktonic and biofilm growth of dermatophytes and the possible mechanism of antibiofilm. J. Ethnopharmacol. 2019, 241, 111956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, L.; Duan, D.H.; Zhang, Y.H.; Huang, S.X.; Chang, Y. Cytotoxicity-Guided Isolation of Two New Phenolic Derivatives from Dryopteris fragrans (L.) Schott. Molecules 2018, 23, 1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.L.; Zhang, D.R.; Li, J.; Wang, H.M.; Song, C.H.; Tang, X.; Guan, Y.; Chang, Y.; Wang, W.F. Albicanol Alleviates D-Galactose-Induced Aging and Improves Behavioral Ability via by Alleviating Oxidative Stress-Induced Damage. Neurochem. Res. 2021, 46, 1058–1067. [Google Scholar] [CrossRef]
- Guan, Y.; Zhao, X.; Song, N.; Cui, Y.; Chang, Y. Albicanol antagonizes Cd-induced apoptosis through a NO/iNOS-regulated mitochondrial pathway in chicken liver cells. Food Funct. 2021, 12, 1757–1768. [Google Scholar] [CrossRef]
- Yadav, R.K.; Sangwan, R.S.; Sabir, F.; Srivastava, A.K.; Sangwan, N.S. Effect of prolonged water stress on specialized secondary metabolites, peltate glandular trichomes, and pathway gene expression in Artemisia annua L. Plant Physiol. Biochem. 2014, 74, 70–83. [Google Scholar] [CrossRef]
- Qureshi, M.I.; Israr, M.; Abdin, M.Z.; Iqbal, M. Responses of Artemisia annua L. to lead and salt-induced oxidative stress. Environ. Exp. Bot. 2005, 53, 185–193. [Google Scholar]
- Wang, H.; Han, J.; Kanagarajan, S.; Lundgren, A.; Brodelius, P.E. Studies on the expression of sesquiterpene synthases using promoter-β-glucuronidase fusions in transgenic Artemisia annua L. PLoS ONE 2013, 8, e80643. [Google Scholar] [CrossRef] [Green Version]
- Takshak, S.; Agrawal, S.B. Defense potential of secondary metabolites in medicinal plants under UV–B stress. J. Photochem. Photobiol. B 2019, 193, 51–88. [Google Scholar] [CrossRef]
- Nagegowda, D.A. Plant volatile terpenoid metabolism: Biosynthetic genes, transcriptional regulation and subcellular compartmentation. FEBS Lett. 2010, 584, 2965–2973. [Google Scholar] [CrossRef] [Green Version]
- Vranová, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Booth, J.K.; Page, J.E.; Bohlmann, J. Terpene synthases from Cannabis sativa. PLoS ONE 2017, 12, e0173911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Q.; Zhang, A.; Zhou, W.; Jiang, R.; Yang, Z.; Yang, H.; Qin, X.; Ding, S.; Lu, Q.; et al. The Phytol Phosphorylation Pathway Is Essential for the Biosynthesis of Phylloquinone, which Is Required for Photosystem I Stability in Arabidopsis. Mol. Plant 2017, 10, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Nagegowda, D.A.; Gupta, P. Advances in biosynthesis, regulation, and metabolic engineering of plant specialized terpenoids. Plant Sci. 2020, 294, 110457. [Google Scholar] [CrossRef]
- Li, S.; Liu, L.; Zhuang, X.; Yu, Y.; Liu, X.; Cui, X.; Ji, L.; Pan, Z.; Cao, X.; Mo, B.; et al. MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 2013, 153, 562–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, K.; Chen, L.; Zou, Y.; Liu, H.; Tian, Y.; Li, D.; Wang, R.; Zhao, F.; Ferguson, B.J.; et al. MicroRNA167-Directed Regulation of the Auxin Response Factors GmARF8a and GmARF8b Is Required for Soybean Nodulation and Lateral Root Development. Plant Physiol. 2015, 168, 984–999. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.N.; Rothstein, S.J.; Spangenberg, G.; Kant, S. Role of microRNAs involved in plant response to nitrogen and phosphorous limiting conditions. Front. Plant Sci. 2015, 6, 629. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, S.L.; Li, J.L.; Hu, X.H.; Wang, H.; Cao, X.L.; Xu, Y.J.; Zhao, Z.X.; Xiao, Z.Y.; Yang, N.; et al. Osa-miR169 Negatively Regulates Rice Immunity against the Blast Fungus Magnaporthe oryzae. Front. Plant Sci. 2017, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Mathiyalagan, R.; Subramaniyam, S.; Natarajan, S.; Kim, Y.J.; Sun, M.S.; Kim, S.Y.; Kim, Y.J.; Yang, D.C. Insilico profiling of microRNAs in Korean ginseng (Panax ginseng Meyer). J. Ginseng Res. 2013, 37, 227–247. [Google Scholar] [CrossRef] [Green Version]
- Vashisht, I.; Mishra, P.; Pal, T.; Chanumolu, S.; Singh, T.R.; Chauhan, R.S. Erratum to: Mining NGS transcriptomes for miRNAs and dissecting their role in regulating growth, development, and secondary metabolites production in different organs of a medicinal herb, Picrorhiza kurroa. Planta 2015, 241, 1269–1270. [Google Scholar] [CrossRef] [PubMed]
- Andrady, A.L.; Aucamp, P.J.; Austin, A.T.; Bais, A.F.; Ballaré, C.L.; Björn, L.O.; Bornman, J.F.; Caldwell, M.; Cullen, A.P.; Erickson, D.J.; et al. Environmental effects of ozone depletion and its interactions with climate change: Progress report, 2011. Photochem. Photobiol. Sci. 2012, 11, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballaré, C.L.; Rousseau, M.C.; Searles, P.S.; Zaller, J.G.; Giordano, C.V.; Robson, T.M.; Caldwell, M.M.; Sala, O.E.; Scopel, A.L. Impacts of solar ultraviolet-B radiation on terrestrial ecosystems of Tierra del Fuego (southern Argentina): An overview of recent progress. J. Photochem. Photobiol. B 2001, 62, 67–77. [Google Scholar] [CrossRef]
- Holzinger, A.; Lütz, C. Algae and UV irradiation: Effects on ultrastructure and related metabolic functions. Micron 2006, 37, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.A.; Hectors, K.; O’Brien, N.M.; Guisez, Y.; Potters, G. Plant stress and human health: Do human consumers benefit from UV–B acclimated crops? Plant Sci. 2008, 175, 449–458. [Google Scholar] [CrossRef]
- Salama, H.M.; Al Watban, A.A.; Al-Fughom, A.T. Effect of ultraviolet radiation on chlorophyll, carotenoid, protein and proline contents of some annual desert plants. Saudi J. Biol. Sci. 2011, 18, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Morales, L.O.; Tegelberg, R.; Brosché, M.; Keinänen, M.; Lindfors, A.; Aphalo, P.J. Effects of solar UV-A and UV–B radiation on gene expression and phenolic accumulation in Betula pendula leaves. Tree Physiol. 2010, 30, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Doma, M.; Abhayankar, G.; Reddy, V.D.; Kavi Kishor, P.B. Carbohydrate and elicitor enhanced withanolide (withaferin A and withanolide A) accumulation in hairy root cultures of Withania somnifera (L.). Indian J. Exp. Biol. 2012, 50, 484–490. [Google Scholar]
- Dolzhenko, Y.; Bertea, C.M.; Occhipinti, A.; Bossi, S.; Maffei, M.E. UV–B modulates the interplay between terpenoids and flavonoids in peppermint (Mentha x piperita L.). J. Photochem. Photobiol. B 2010, 100, 67–75. [Google Scholar] [CrossRef]
- Wei, N.; Xin, P.; Ma, L.; Lei, C.; Lu, X.; Jing, W.; Tian, J.; Li, X.; Wei, W.; Lin, Z. Enhanced secondary metabolites production and antioxidant activity in postharvest Lonicera japonica Thunb. in response to UV radiation. Innov. Food Sci. Emerg. 2012, 13, 231–243. [Google Scholar]
- Casati, P.; Walbot, V. Rapid transcriptome responses of maize (Zea mays) to UV–B in irradiated and shielded tissues. Genome Biol. 2004, 5, R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.P.; Guo, Y.T.; Wang, J.W.; Tan, R.X. Nitric oxide potentiates oligosaccharide-induced artemisinin production in Artemisia annua hairy roots. J. Integr. Plant Biol. 2008, 50, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Zheng, L.P.; Zhang, B.; Zou, T. Stimulation of artemisinin synthesis by combined cerebroside and nitric oxide elicitation in Artemisia annua hairy roots. Appl. Microbiol. Biotechnol. 2009, 85, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.W.; Wan, Y.L.; Li, P.Z.; Zhang, B. Stimulation of Artemisinin Production in Artemisia annua Hairy Roots by Ag-SiO2 Core-shell Nanoparticles. Curr. Nanosci. 2013, 9, 363–370. [Google Scholar]
- Yu, N.; Cai, W.J.; Wang, S.; Shan, C.M.; Wang, L.J.; Chen, X.Y. Temporal control of trichome distribution by microRNA156-targeted SPL genes in Arabidopsis thaliana. Plant Cell 2010, 22, 2322–2335. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Birkenbihl, R.P.; Jach, G.; Saedler, H.; Huijser, P. Functional dissection of the plant-specific SBP-domain: Overlap of the DNA-binding and nuclear localization domains. J. Mol. Biol. 2005, 352, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Nazarenus, T.J.; Stone, J.M. Identification of a consensus DNA-binding site for the Arabidopsis thaliana SBP domain transcription factor, AtSPL14, and binding kinetics by surface plasmon resonance. Biochemistry 2008, 47, 3645–3653. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.J.; Björn, L.O. The effect of ultraviolet radiation on the accumulation of medicinal compounds in plants. Fitoterapia 2009, 80, 207–218. [Google Scholar] [CrossRef]
- Takshak, S.; Agrawal, S.B. Secondary metabolites and phenylpropanoid pathway enzymes as influenced under supplemental ultraviolet-B radiation in Withania somnifera Dunal, an indigenous medicinal plant. J. Photochem. Photobiol. B 2014, 140, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Song, T.; Ma, Z.; Dong, L.; Zhan, L.; Xing, Y.; Liu, J.; Song, J.; Wang, S.; Cai, H. Overexpression of MsSiR enhances alkali tolerance in alfalfa (Medicago sativa L.) by increasing the glutathione content. Plant Physiol. Biochem. 2020, 154, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Yang, N.; Meng, J.; Wang, D.; Fu, L.; Wang, J.; Cang, J. Adaptability of winter wheat Dongnongdongmai 1 (Triticum aestivum L.) to overwintering in alpine regions. Plant Biol. 2021, 23, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Krupa, S.V. Ultraviolet-B radiation, ozone and plant biology. Environ. Pollut. 2000, 110, 193–194. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, C.; Xu, C.; Sun, J.; Grierson, D.; Zhang, B.; Chen, K. Integration of Metabolite Profiling and Transcriptome Analysis Reveals Genes Related to Volatile Terpenoid Metabolism in Finger Citron (C. medica var. sarcodactylis). Molecules 2019, 24, 2564. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, L.; Zheng, X.; Zhang, J.; Yang, L.; Tan, R.; Zhao, S. Overexpression of SmMYB9b enhances tanshinone concentration in Salvia miltiorrhiza hairy roots. Plant Cell Rep. 2017, 36, 1297–1309. [Google Scholar] [CrossRef]
- Tan, H.; Xiao, L.; Gao, S.; Li, Q.; Chen, J.; Xiao, Y.; Ji, Q.; Chen, R.; Chen, W.; Zhang, L. TRICHOME AND ARTEMISININ REGULATOR 1 Is Required for Trichome Development and Artemisinin Biosynthesis in Artemisia annua. Mol. Plant 2015, 8, 1396–1411. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, Y.; Shen, S.; Yin, X.; Klee, H.; Zhang, B.; Chen, K.; Hancock, R. Transcription factor CitERF71 activates the terpene synthase gene CitTPS16 involved in the synthesis of E-geraniol in sweet orange fruit. J. Exp. Bot. 2017, 68, 4929–4938. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.J.; Xue, X.Y.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 2012, 24, 2635–2648. [Google Scholar] [CrossRef] [Green Version]
- Chini, A.; Fonseca, S.; Fernández, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; García-Casado, G.; López-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef]
- Hou, X.; Lee, L.Y.; Xia, K.; Yan, Y.; Yu, H. DELLAs modulate jasmonate signaling via competitive binding to JAZs. Dev. Cell 2010, 19, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, X.; Zhong, L.; Zhou, X.; Tang, Y.; Liu, Y.; Li, J.; Zheng, H.; Zhan, R.; Chen, L. PatJAZ6 Acts as a Repressor Regulating JA-Induced Biosynthesis of Patchouli Alcohol in Pogostemon Cablin. Int. J. Mol. Sci. 2019, 20, 6038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Sun, L.; Qi, T.; Zhang, B.; Peng, W.; Liu, Y.; Xie, D. The bHLH transcription factor MYC3 interacts with the Jasmonate ZIM-domain proteins to mediate jasmonate response in Arabidopsis. Mol. Plant 2011, 4, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Saifi, M.; Yogindran, S.; Nasrullah, N.; Nissar, U.; Gul, I.; Abdin, M.Z. Co-expression of anti-miR319g and miRStv_11 lead to enhanced steviol glycosides content in Stevia rebaudiana. BMC Plant Biol. 2019, 19, 274. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.X.; Wang, L.J.; Zhao, B.; Shan, C.M.; Zhang, Y.H.; Chen, D.F.; Chen, X.Y. Progressive regulation of sesquiterpene biosynthesis in Arabidopsis and Patchouli (Pogostemon cablin) by the miR156-targeted SPL transcription factors. Mol. Plant 2015, 8, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Reeves, P.H.; Ellis, C.M.; Ploense, S.E.; Wu, M.F.; Yadav, V.; Tholl, D.; Chételat, A.; Haupt, I.; Kennerley, B.J.; Hodgens, C.; et al. A regulatory network for coordinated flower maturation. PLoS Genet. 2012, 8, e1002506. [Google Scholar] [CrossRef] [Green Version]
- Mandaokar, A.; Thines, B.; Shin, B.; Lange, B.M.; Choi, G.; Koo, Y.J.; Yoo, Y.J.; Choi, Y.D.; Choi, G.; Browse, J. Transcriptional regulators of stamen development in Arabidopsis identified by transcriptional profiling. Plant J. 2006, 46, 984–1008. [Google Scholar] [CrossRef]
- Wellburn, A.R. The Spectral Determination of Chlorophylls a and b, as well as Total Carotenoids, Using Various Solvents with Spectrophotometers of Different Resolution. J. Plant Physiol. 1994, 5, 28–29. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A systems biology approach for pathway level analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total | CK1 | CK2 | CK3 | UV1 | UV2 | UV3 |
|---|---|---|---|---|---|---|---|
| raw_reads | - | 24,839,653 | 20,892,938 | 22,535,960 | 20,967,576 | 22,164,559 | 23,328,773 |
| clean_reads | - | 23,961,150 | 20,220,033 | 21,734,750 | 20,252,088 | 21,327,355 | 22,519,655 |
| clean_bases | - | 7.19G | 6.07G | 6.52G | 6.08G | 6.40G | 6.76G |
| error_rate (%) | - | 0.02 | 0.02 | 0.02 | 0.03 | 0.03 | 0.03 |
| Q20 (%) | - | 98.12 | 98.11 | 98.12 | 97.88 | 97.85 | 97.66 |
| Q30 (%) | - | 94.42 | 94.36 | 94.39 | 94.01 | 93.81 | 93.44 |
| GC_pct (%) | - | 46.92 | 46.03 | 45.9 | 48.37 | 47.32 | 47.3 |
| N50 | 2035 | - | - | - | - | - | - |
| N90 | 526 | - | - | - | - | - | - |
| Number of transcripts | 141,963 | - | - | - | - | - | - |
| Number of Unigenes | 69,493 | - | - | - | - | - | - |
| Sample | Total Reads | Clean Reads | Mapped sRNA | Known miRNA | Novel miRNA | Total miRNA |
|---|---|---|---|---|---|---|
| CK1 | 15,490,143 (100.00%) | 14,585,008 (94.16%) | 6,307,041 (58.03%) | 4 | 72 | 76 |
| CK2 | 12,790,994 (100.00%) | 12,417,161 (97.08%) | 4,561,120 (51.72%) | 6 | 71 | 77 |
| CK3 | 15,716,150 (100.00%) | 15,055,676 (95.80%) | 6,417,540 (67.93%) | 5 | 72 | 77 |
| UV1 | 17,010,191 (100.00%) | 16,386,263 (96.33%) | 6,917,716 (61.08%) | 6 | 77 | 83 |
| UV2 | 13,075,551 (100.00%) | 12,297,545 (94.05%) | 4,580,695 (57.97%) | 7 | 79 | 86 |
| UV3 | 11,728,662 (100.00%) | 11,077,797 (94.45%) | 4,023,081 (54.21%) | 6 | 78 | 84 |
| Sum | 85,811,691 | 81,819,450 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Guan, Y.; Zhang, D.; Tang, X.; Chang, Y. Integrated mRNA and miRNA Transcriptome Analysis Suggests a Regulatory Network for UV–B-Controlled Terpenoid Synthesis in Fragrant Woodfern (Dryopteris fragrans). Int. J. Mol. Sci. 2022, 23, 5708. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105708
Song C, Guan Y, Zhang D, Tang X, Chang Y. Integrated mRNA and miRNA Transcriptome Analysis Suggests a Regulatory Network for UV–B-Controlled Terpenoid Synthesis in Fragrant Woodfern (Dryopteris fragrans). International Journal of Molecular Sciences. 2022; 23(10):5708. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105708
Chicago/Turabian StyleSong, Chunhua, Yalin Guan, Dongrui Zhang, Xun Tang, and Ying Chang. 2022. "Integrated mRNA and miRNA Transcriptome Analysis Suggests a Regulatory Network for UV–B-Controlled Terpenoid Synthesis in Fragrant Woodfern (Dryopteris fragrans)" International Journal of Molecular Sciences 23, no. 10: 5708. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105708