
UL34 Deletion Restricts Human Cytomegalovirus Capsid Formation and Maturation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
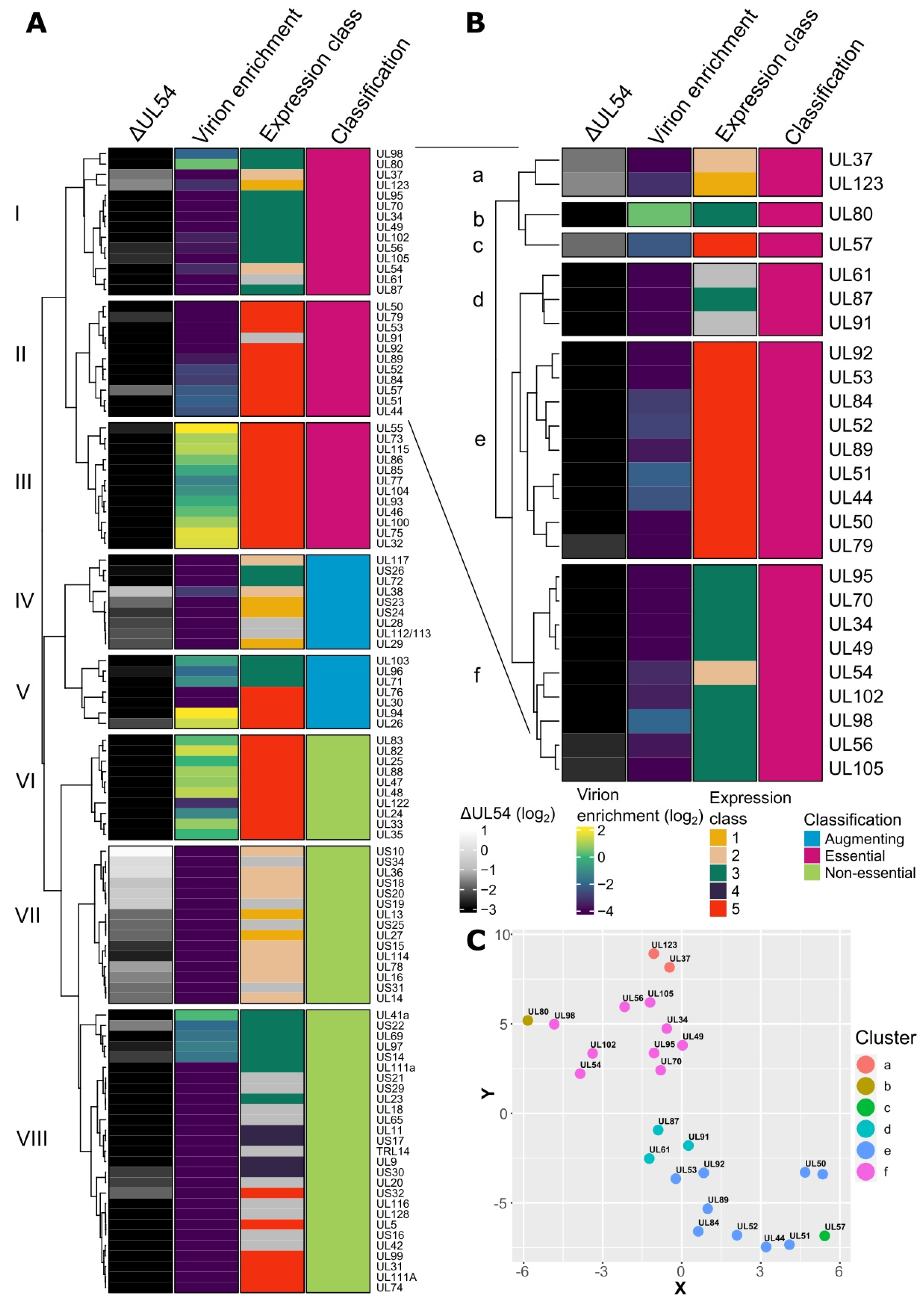
2.1. Characterisation of HCMV Genes by RNA Sequencing and Bioinformatic Analysis
2.2. UL34 Is an Augmenting HCMV Gene Not Required for Viral Genome Replication or vAC Formation
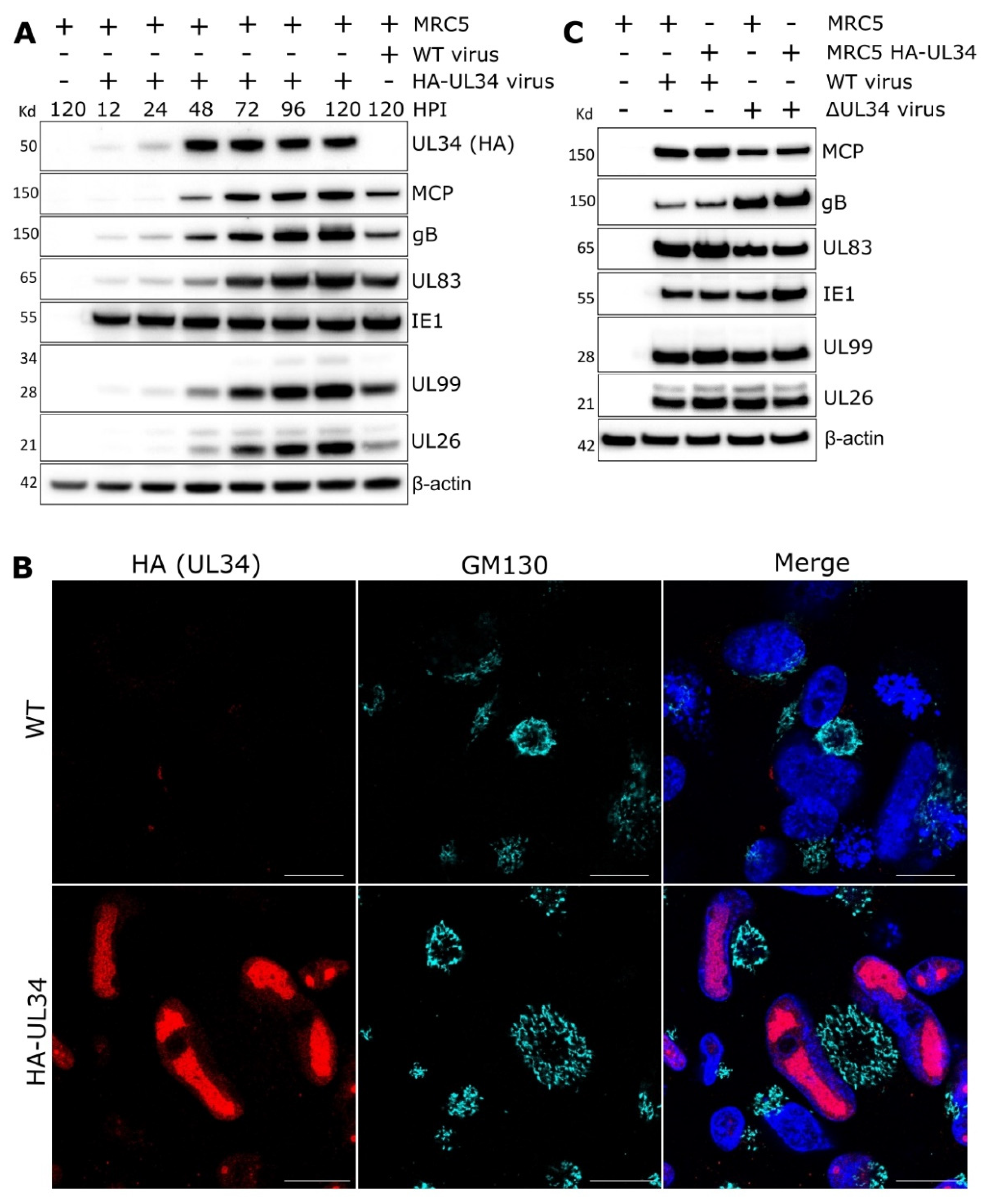
2.3. UL34 Is Expressed with Leaky Late Kinetics and Localises to the Nucleus
2.4. UL34 Interacts with Host Nuclear Regulators but Does Not Modulate Lamina Integrity
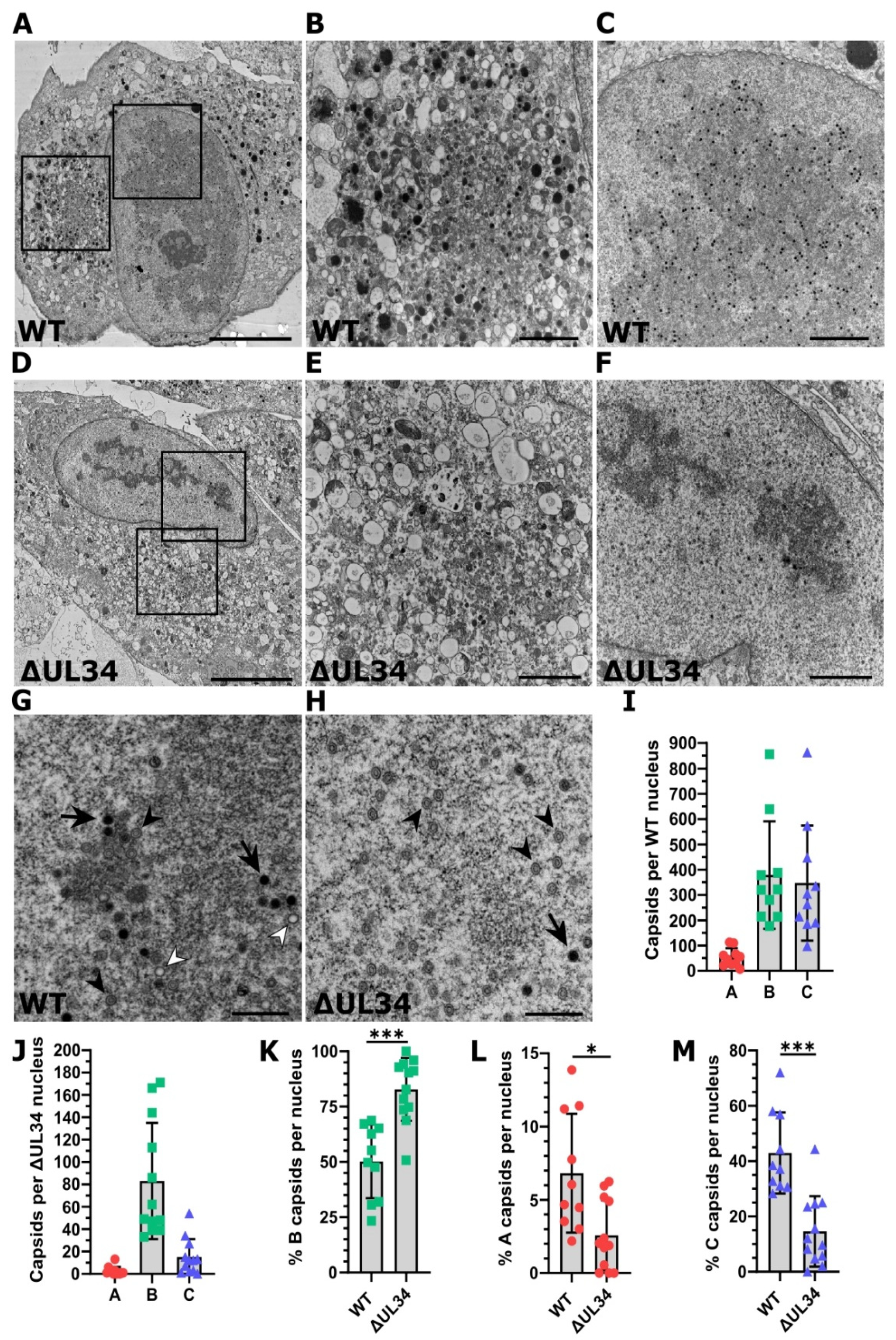
2.5. Cells Infected with HCMV Lacking UL34 Display Reduced Capsids That Fail to Mature
3. Discussion
4. Methods
4.1. Cells and Viruses
4.2. Cloning and Stable Cell Line Generation
4.3. IE1 Fluorescent Focus and Spread Assay
4.4. BAC Recombineering
4.5. Densitometry-Based Protein Quantification
4.6. Western Blotting
4.7. Intracellular Viral Genome Quantitation
4.8. Immunofluorescence Confocal Microscopy
4.9. RT-qPCR
4.10. High Pressure Freezing and Transmission Electron Microscopy
4.11. RNA-seq
4.12. Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Goodrum, F.; Caviness, K.; Zagallo, P. Human cytomegalovirus persistence. Cell. Microbiol. 2012, 14, 644–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanan, P.; Razonable, R.R. Cytomegalovirus infections in solid organ transplantation: A review. Infect. Chemother. 2013, 45, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Bonalumi, S.; Trapanese, A.; Santamaria, A.; D’Emidio, L.; Mobili, L. Cytomegalovirus infection in pregnancy: Review of the literature. J. Prenat. Med. 2011, 5, 1–8. [Google Scholar] [PubMed]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Shenk, T. Human cytomegalovirus genome. Curr. Top. Microbiol. Immunol. 2008, 325, 1–19. [Google Scholar] [CrossRef]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 2013, 4, e00332-13. [Google Scholar] [CrossRef] [Green Version]
- Kalejta, R.F. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 2008, 72, 249–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.S.; Hertel, L. Onset of human cytomegalovirus replication in fibroblasts requires the presence of an intact vimentin cytoskeleton. J. Virol. 2009, 83, 7015–7028. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Jeong, H.; Park, K.; Lee, S.; Shim, J.Y.; Kim, H.; Song, Y.; Park, S.; Park, H.Y.; Kim, V.N.; et al. STING facilitates nuclear import of herpesvirus genome during infection. Proc. Natl. Acad. Sci. USA 2021, 118, 33. [Google Scholar] [CrossRef]
- Paulus, C.; Nevels, M. The human cytomegalovirus major immediate-early proteins as antagonists of intrinsic and innate antiviral host responses. Viruses 2009, 1, 760–779. [Google Scholar] [CrossRef] [Green Version]
- Shenk, T.; Alwine, J.C. Human Cytomegalovirus: Coordinating Cellular Stress, Signaling, and Metabolic Pathways. Annu. Rev. Virol. 2014, 1, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Muylaert, I.; Tang, K.-W.; Elias, P. Replication and recombination of herpes simplex virus DNA. J. Biol. Chem. 2011, 286, 15619–15624. [Google Scholar] [CrossRef] [Green Version]
- Weekes, M.P.; Tomasec, P.; Huttlin, E.L.; Fielding, C.A.; Nusinow, D.; Stanton, R.J.; Wang, E.C.Y.; Aicheler, R.; Murrell, I.; Wilkinson, G.W.G.; et al. Quantitative temporal viromics: An approach to investigate host-pathogen interaction. Cell 2014, 157, 1460–1472. [Google Scholar] [CrossRef] [Green Version]
- Strang, B.L.; Boulant, S.; Chang, L.; Knipe, D.M.; Kirchhausen, T.; Coen, D.M. Human cytomegalovirus UL44 concentrates at the periphery of replication compartments, the site of viral DNA synthesis. J. Virol. 2012, 86, 2089–2095. [Google Scholar] [CrossRef] [Green Version]
- Caragliano, E.; Bonazza, S.; Frascaroli, G.; Tang, J.; Soh, T.K.; Grünewald, K.; Bosse, J.B.; Brune, W. Human cytomegalovirus forms phase-separated compartments at viral genomes to facilitate viral replication. Cell Rep. 2022, 38, 110469. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Trang, P.; Shah, S.; Atanasov, I.; Kim, Y.-H.; Bai, Y.; Zhou, Z.H.; Liu, F. Dissecting human cytomegalovirus gene function and capsid maturation by ribozyme targeting and electron cryomicroscopy. Proc. Natl. Acad. Sci. USA 2005, 102, 7103–7108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogner, E.; Radsak, K.; Stinski, M.F. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J. Virol. 1998, 72, 2259–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, E.M.; Kleine-Albers, J.; Gabaev, I.; Babic, M.; Wagner, K.; Binz, A.; Degenhardt, I.; Kalesse, M.; Jonjic, S.; Bauerfeind, R.; et al. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J. Virol. 2013, 87, 1720–1732. [Google Scholar] [CrossRef] [Green Version]
- Scheffczik, H.; Savva, C.G.; Holzenburg, A.; Kolesnikova, L.; Bogner, E. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res. 2002, 30, 1695–1703. [Google Scholar] [CrossRef] [Green Version]
- Borst, E.M.; Wagner, K.; Binz, A.; Sodeik, B.; Messerle, M. The essential human cytomegalovirus gene UL52 is required for cleavage-packaging of the viral genome. J. Virol. 2008, 82, 2065–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, J.D. Herpes simplex virus capsid assembly and DNA packaging: A present and future antiviral drug target. Trends Microbiol. 2011, 19, 606–613. [Google Scholar] [CrossRef]
- Sanchez, V.; Britt, W. Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions. Viruses 2021, 14, 15. [Google Scholar] [CrossRef]
- Alwine, J.C. The human cytomegalovirus assembly compartment: A masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Path. 2012, 8, e1002878. [Google Scholar] [CrossRef] [Green Version]
- Dietz, A.N.; Villinger, C.; Becker, S.; Frick, M.; von Einem, J. A Tyrosine-Based Trafficking Motif of the Tegument Protein pUL71 Is Crucial for Human Cytomegalovirus Secondary Envelopment. J. Virol. 2018, 92, e00907-17. [Google Scholar] [CrossRef] [Green Version]
- Schauflinger, M.; Fischer, D.; Schreiber, A.; Chevillotte, M.; Walther, P.; Mertens, T.; von Einem, J. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J. Virol. 2011, 85, 3821–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Womack, A.; Shenk, T. Human cytomegalovirus tegument protein pUL71 is required for efficient virion egress. mBio 2010, 1, e00282-10. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.L.; Bresnahan, W.A. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J. Virol. 2012, 86, 2523–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.C.; Yu, Q.C.; Enquist, L.; Shenk, T. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 2003, 77, 10594–10605. [Google Scholar] [CrossRef] [Green Version]
- Ahlqvist, J.; Mocarski, E. Cytomegalovirus UL103 controls virion and dense body egress. J. Virol. 2011, 85, 5125–5135. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.L.; Korneev, D.V.; Purdy, J.G.; de Marco, A.; Mathias, R.A. The host exosome pathway underpins biogenesis of the human cytomegalovirus virion. Elife 2020, 9, e58288. [Google Scholar] [CrossRef]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gower, J.C. A General Coefficient of Similarity and Some of Its Properties. Biometrics 1971, 27, 857. [Google Scholar] [CrossRef]
- Hummel, M.; Edelmann, D.; Kopp-Schneider, A. Clustering of samples and variables with mixed-type data. PLoS ONE 2017, 12, e0188274. [Google Scholar] [CrossRef] [Green Version]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Van Der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2625. [Google Scholar]
- Battista, M.C.; Bergamini, G.; Boccuni, M.C.; Campanini, F.; Ripalti, A.; Landini, M.P. Expression and characterization of a novel structural protein of human cytomegalovirus, pUL25. J. Virol. 1999, 73, 3800–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldick, C.J., Jr.; Marchini, A.; Patterson, C.E.; Shenk, T. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 1997, 71, 4400–4408. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Cruz, L.; Sandhu, P.K.; Buchkovich, N.J. UL88 Mediates the Incorporation of a Subset of Proteins into the Virion Tegument. J. Virol. 2020, 94, e00474-20. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Yu, D.; Shenk, T. UL26-deficient human cytomegalovirus produces virions with hypophosphorylated pp28 tegument protein that is unstable within newly infected cells. J. Virol. 2006, 80, 3541–3548. [Google Scholar] [CrossRef] [Green Version]
- Mathers, C.; Schafer, X.; Martínez-Sobrido, L.; Munger, J. The human cytomegalovirus UL26 protein antagonizes NF-κB activation. J. Virol. 2014, 88, 14289–14300. [Google Scholar] [CrossRef] [Green Version]
- Isomura, H.; Stinski, M.F.; Murata, T.; Yamashita, Y.; Kanda, T.; Toyokuni, S.; Tsurumi, T. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J. Virol. 2011, 85, 6629–6644. [Google Scholar] [CrossRef] [Green Version]
- Omoto, S.; Mocarski, E.S. Cytomegalovirus UL91 is essential for transcription of viral true late (γ2) genes. J. Virol. 2013, 87, 8651–8664. [Google Scholar] [CrossRef] [Green Version]
- Omoto, S.; Mocarski, E.S. Transcription of true late (γ2) cytomegalovirus genes requires UL92 function that is conserved among beta- and gammaherpesviruses. J. Virol. 2014, 88, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Kamil, J.P.; Coughlin, M.; Reim, N.I.; Coen, D.M. Human Cytomegalovirus UL50 and UL53 Recruit Viral Protein Kinase UL97, Not Protein Kinase C, for Disruption of Nuclear Lamina and Nuclear Egress in Infected Cells. J. Virol. 2014, 88, 249–262. [Google Scholar] [CrossRef] [Green Version]
- McMahon, T.P.; Anders, D.G. Interactions between human cytomegalovirus helicase-primase proteins. Virus Res. 2002, 86, 39–52. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penfold, M.E.T.; Mocarski, E.S. Formation of Cytomegalovirus DNA Replication Compartments Defined by Localization of Viral Proteins and DNA Synthesis. Virology 1997, 239, 46–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, R.; Mocarski, E.S. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 2012, 20, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Biegalke, B.J. Human cytomegalovirus UL34 binds to multiple sites within the viral genome. J. Virol. 2013, 87, 3587–3591. [Google Scholar] [CrossRef] [Green Version]
- Nobre, L.V.; Nightingale, K.; Ravenhill, B.J.; Antrobus, R.; Soday, L.; Nichols, J.; Davies, J.A.; Seirafian, S.; Wang, E.C.Y.; Davison, A.J.; et al. Human cytomegalovirus interactome analysis identifies degradation hubs, domain associations and viral protein functions. eLife 2019, 8, e49894. [Google Scholar] [CrossRef]
- Toyo-oka, K.; Mori, D.; Yano, Y.; Shiota, M.; Iwao, H.; Goto, H.; Inagaki, M.; Hiraiwa, N.; Muramatsu, M.; Wynshaw-Boris, A.; et al. Protein phosphatase 4 catalytic subunit regulates Cdk1 activity and microtubule organization via NDEL1 dephosphorylation. J. Cell Biol. 2008, 180, 1133–1147. [Google Scholar] [CrossRef] [Green Version]
- Hamirally, S.; Kamil, J.P.; Ndassa-Colday, Y.M.; Lin, A.J.; Jahng, W.J.; Baek, M.-C.; Noton, S.; Silva, L.A.; Simpson-Holley, M.; Knipe, D.M.; et al. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Path. 2009, 5, e1000275. [Google Scholar] [CrossRef] [Green Version]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef] [Green Version]
- Eilbrecht, M.; Le-Trilling, V.T.K.; Trilling, M. Mouse Cytomegalovirus M34 Encodes a Non-essential, Nuclear, Early-Late Expressed Protein Required for Efficient Viral Replication. Front. Cell. Infect. Microbiol. 2020, 10, 171. [Google Scholar] [CrossRef]
- Rana, R.; Biegalke, B.J. Human cytomegalovirus UL34 early and late proteins are essential for viral replication. Viruses 2014, 6, 476–488. [Google Scholar] [CrossRef] [Green Version]
- Slayton, M.; Hossain, T.; Biegalke, B.J. pUL34 binding near the human cytomegalovirus origin of lytic replication enhances DNA replication and viral growth. Virology 2018, 518, 414–422. [Google Scholar] [CrossRef]
- Biegalke, B.J.; Lester, E.; Branda, A.; Rana, R. Characterization of the human cytomegalovirus UL34 gene. J. Virol. 2004, 78, 9579–9583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPierre, L.A.; Biegalke, B.J. Identification of a novel transcriptional repressor encoded by human cytomegalovirus. J. Virol. 2001, 75, 6062–6069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariamé, B.; Kappler-Gratias, S.; Kappler, M.; Balor, S.; Gallardo, F.; Bystricky, K. Real-Time Visualization and Quantification of Human Cytomegalovirus Replication in Living Cells Using the ANCHOR DNA Labeling Technology. J. Virol. 2018, 92, e00571-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lawry, S.T.; Cohen, A.L.; Jia, S. Chromosome boundary elements and regulation of heterochromatin spreading. Cell. Mol. Life Sci. 2014, 71, 4841–4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevels, M.; Nitzsche, A.; Paulus, C. How to control an infectious bead string: Nucleosome-based regulation and targeting of herpesvirus chromatin. Rev. Med. Virol. 2011, 21, 154–180. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pang, J.; Dong, L.; Yu, X. Structural basis for genome packaging, retention, and ejection in human cytomegalovirus. Nat. Commun. 2021, 12, 4538. [Google Scholar] [CrossRef] [PubMed]
- Paulus, C.; Nitzsche, A.; Nevels, M. Chromatinisation of herpesvirus genomes. Rev. Med. Virol. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Richmond, T.J.; Finch, J.T.; Rushton, B.; Rhodes, D.; Klug, A. Structure of the nucleosome core particle at 7 A resolution. Nature 1984, 311, 532–537. [Google Scholar] [CrossRef]
- Zalckvar, E.; Paulus, C.; Tillo, D.; Asbach-Nitzsche, A.; Lubling, Y.; Winterling, C.; Strieder, N.; Mücke, K.; Goodrum, F.; Segal, E.; et al. Nucleosome maps of the human cytomegalovirus genome reveal a temporal switch in chromatin organization linked to a major IE protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13126–13131. [Google Scholar] [CrossRef] [Green Version]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, C.; Hausmann, G.; Basler, K. Beta-catenin hits chromatin: Regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 2009, 10, 276–286. [Google Scholar] [CrossRef]
- Barker, N.; Hurlstone, A.; Musisi, H.; Miles, A.; Bienz, M.; Clevers, H. The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 2001, 20, 4935–4943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ozawa, Y.; Lee, H.; Wen, Y.-D.; Tan, T.-H.; Wadzinski, B.E.; Seto, E. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 2005, 19, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, R.; Mocarski, E.S.; Conway, J.F. The A, B, Cs of herpesvirus capsids. Viruses 2015, 7, 899–914. [Google Scholar] [CrossRef]
- Borst, E.M.; Bauerfeind, R.; Binz, A.; Stephan, T.M.; Neuber, S.; Wagner, K.; Steinbrück, L.; Sodeik, B.; Lenac Roviš, T.; Jonjić, S.; et al. The Essential Human Cytomegalovirus Proteins pUL77 and pUL93 Are Structural Components Necessary for Viral Genome Encapsidation. J. Virol. 2016, 90, 5860–5875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Alain, S.; Baumert, T.F.; Ligat, G.; Hantz, S. Structures and Divergent Mechanisms in Capsid Maturation and Stabilization Following Genome Packaging of Human Cytomegalovirus and Herpesviruses. Life 2021, 11, 150. [Google Scholar] [CrossRef]
- Yu, D.; Smith, G.A.; Enquist, L.W.; Shenk, T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 2002, 76, 2316–2328. [Google Scholar] [CrossRef] [Green Version]
- Britt, W.J. Human cytomegalovirus: Propagation, quantification, and storage. Curr. Protoc. Microbiol. 2010, 18, 14E.3.1–14E.3.17. [Google Scholar] [CrossRef]
- Zhu, H.; Shen, Y.; Shenk, T. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 1995, 69, 7960–7970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, B.; Sullivan, C.; Sarnow, P.; Thomas, R.; Bricout, F.; Nicolas, J.C.; Fleckenstein, B.; Levine, A.J. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 1984, 132, 325–338. [Google Scholar] [CrossRef]
- Kirill Tsyganov, A.J.; Stuart, P.; Archer, K.; Powell, D. RNAsik: A Pipeline for complete and reproducible RNA-seq analysis that runs anywhere with speed and ease. J. Open Source Softw. 2018, 3, 583. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Kucukural, A.; Yukselen, O.; Ozata, D.M.; Moore, M.J.; Garber, M. DEBrowser: Interactive differential expression analysis and visualization tool for count data. BMC Genom. 2019, 20, 6. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turner, D.L.; Templin, R.M.; Barugahare, A.A.; Russ, B.E.; Turner, S.J.; Ramm, G.; Mathias, R.A. UL34 Deletion Restricts Human Cytomegalovirus Capsid Formation and Maturation. Int. J. Mol. Sci. 2022, 23, 5773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105773
Turner DL, Templin RM, Barugahare AA, Russ BE, Turner SJ, Ramm G, Mathias RA. UL34 Deletion Restricts Human Cytomegalovirus Capsid Formation and Maturation. International Journal of Molecular Sciences. 2022; 23(10):5773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105773
Chicago/Turabian StyleTurner, Declan L., Rachel M. Templin, Adele A. Barugahare, Brendan E. Russ, Stephen J. Turner, Georg Ramm, and Rommel A. Mathias. 2022. "UL34 Deletion Restricts Human Cytomegalovirus Capsid Formation and Maturation" International Journal of Molecular Sciences 23, no. 10: 5773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105773