
The Small Molecule GAL-201 Efficiently Detoxifies Soluble Amyloid β Oligomers: New Approach towards Oral Disease-Modifying Treatment of Alzheimer’s Disease

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Pharmacokinetics
2.2. Measuring Isolated Neurons Resting Potential
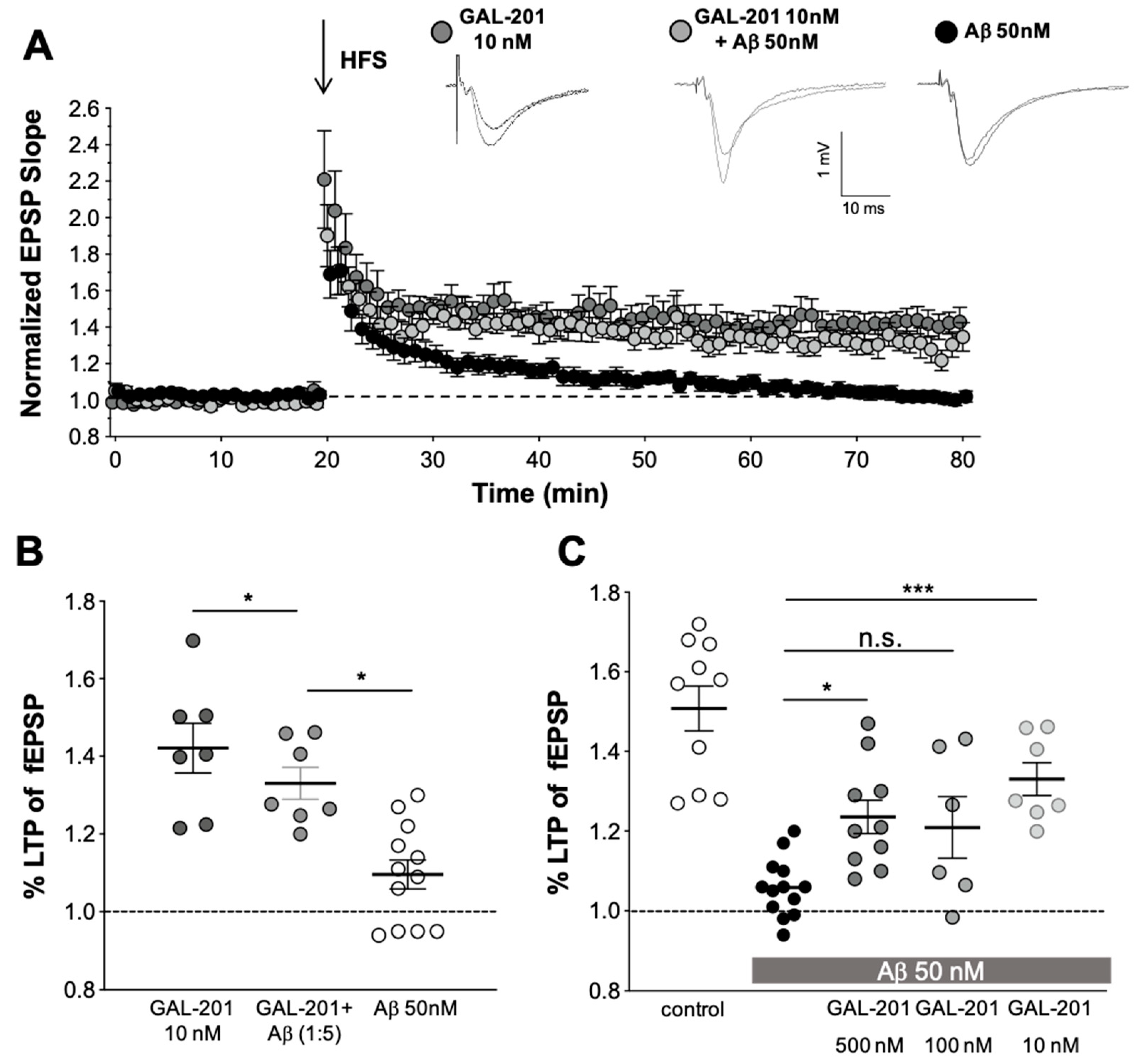
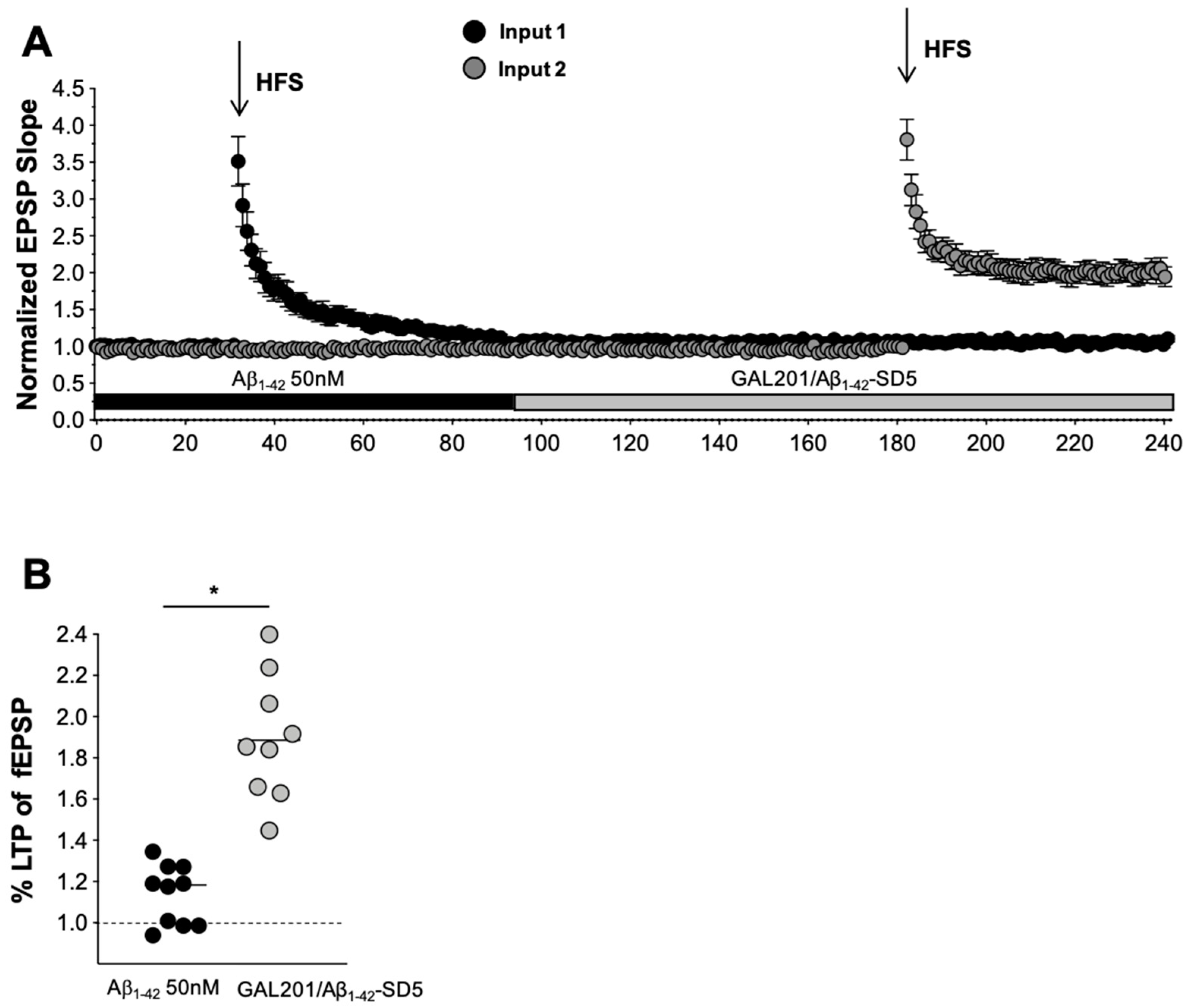
2.3. In Vitro LTP
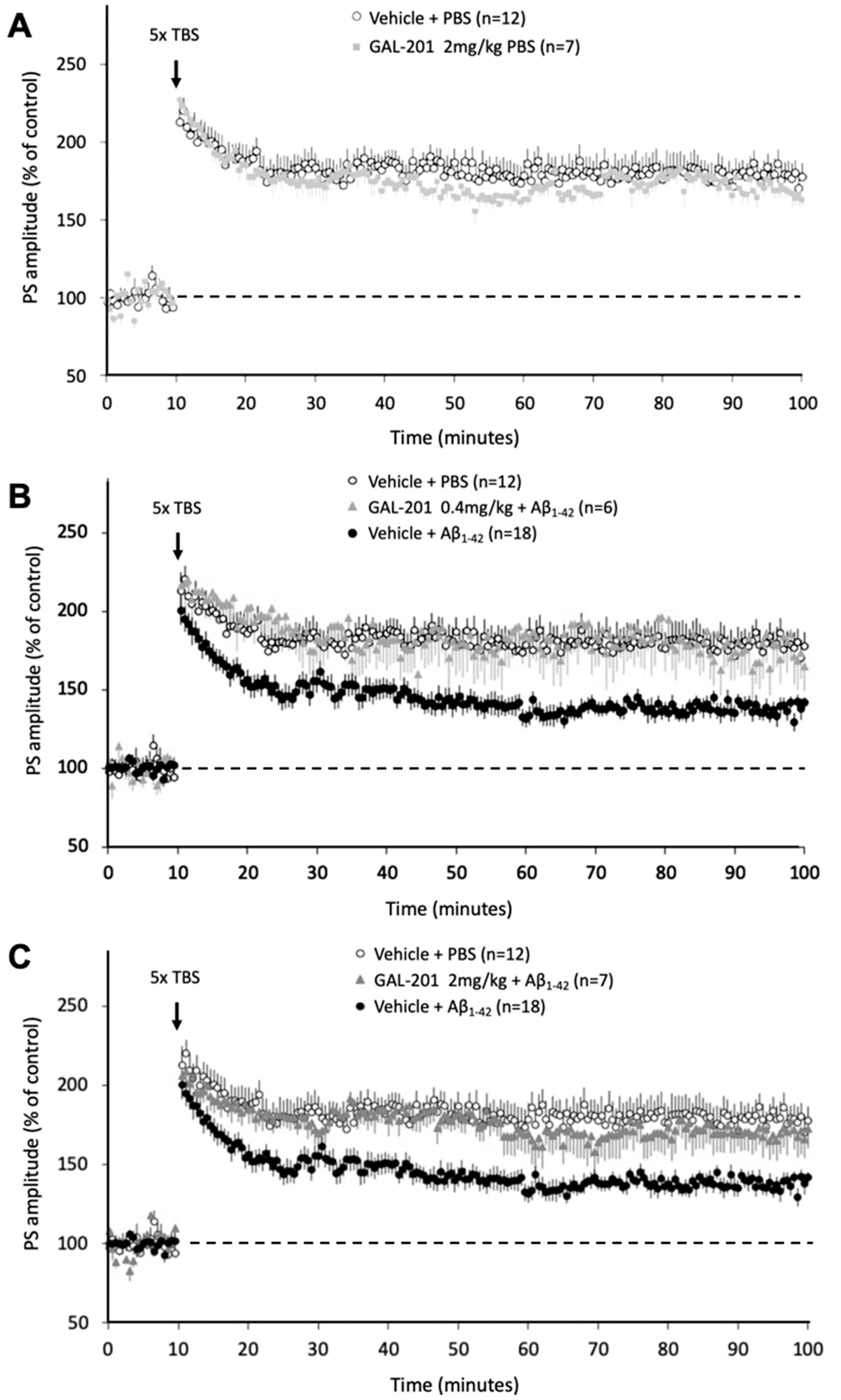
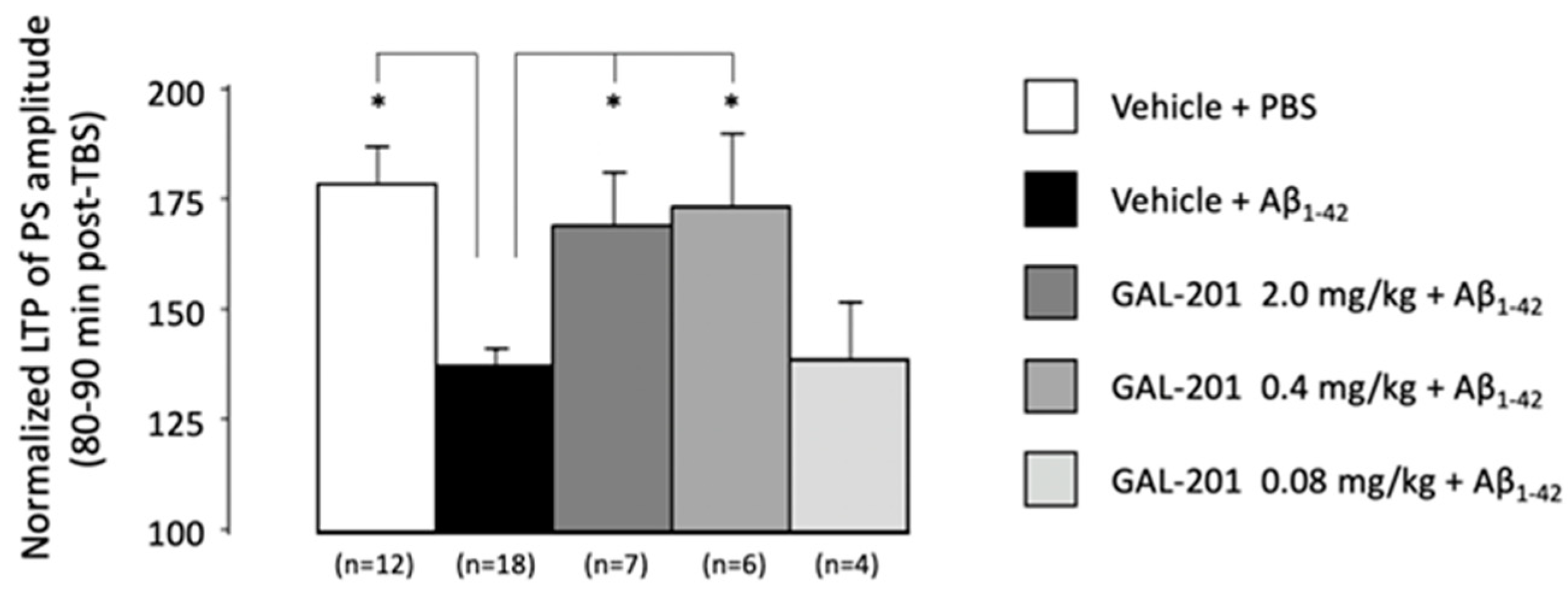
2.4. In Vivo LTP
3. Discussion
3.1. Targeting Aβ1-42 Oligomers
3.2. Availability of GAL-201 to Enter the Brain after Oral Application
3.3. Patch Clamp with Hippocampal Neurons
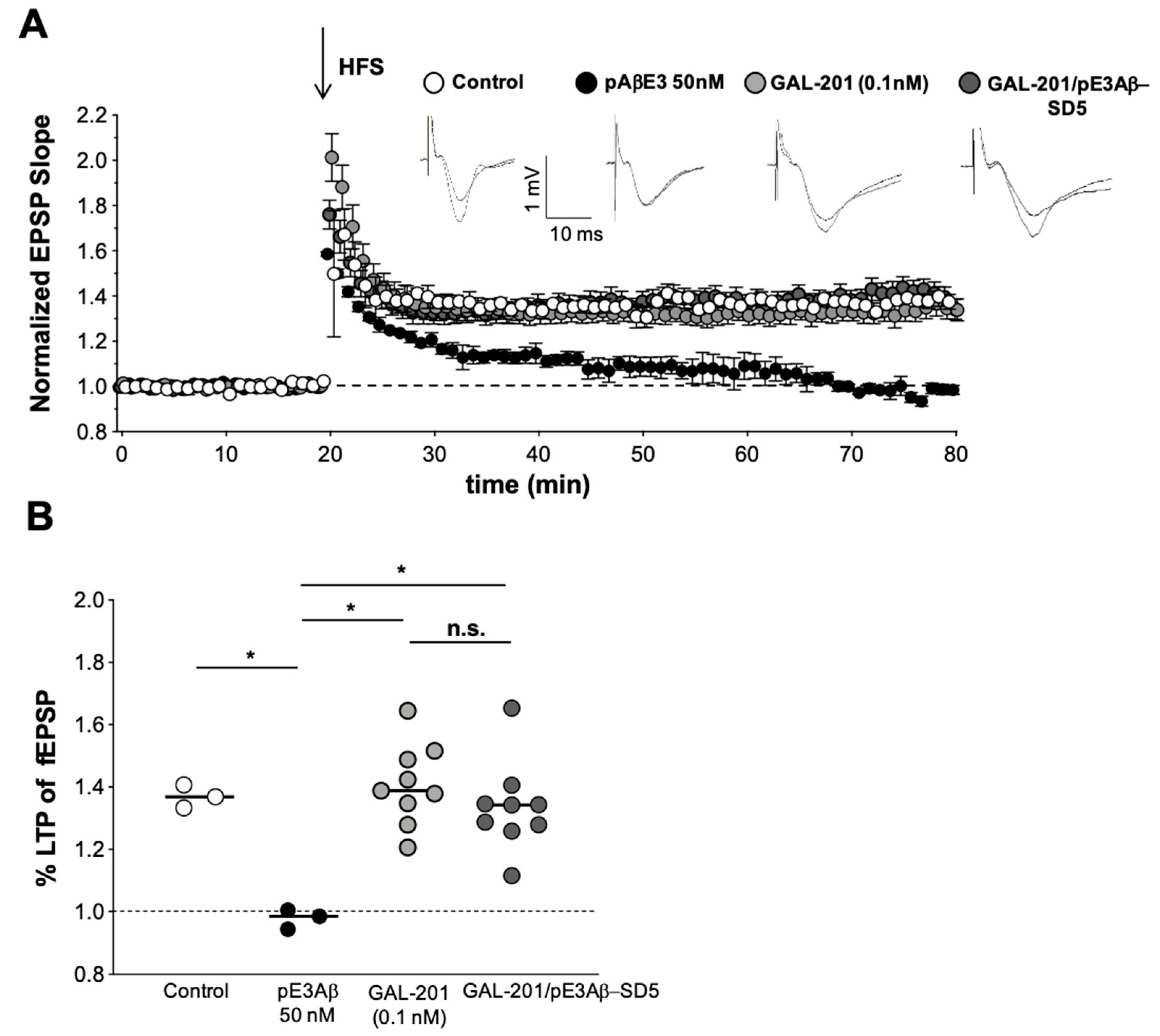
3.4. In Vitro LTP Using Aβ1-42 or AβpE3
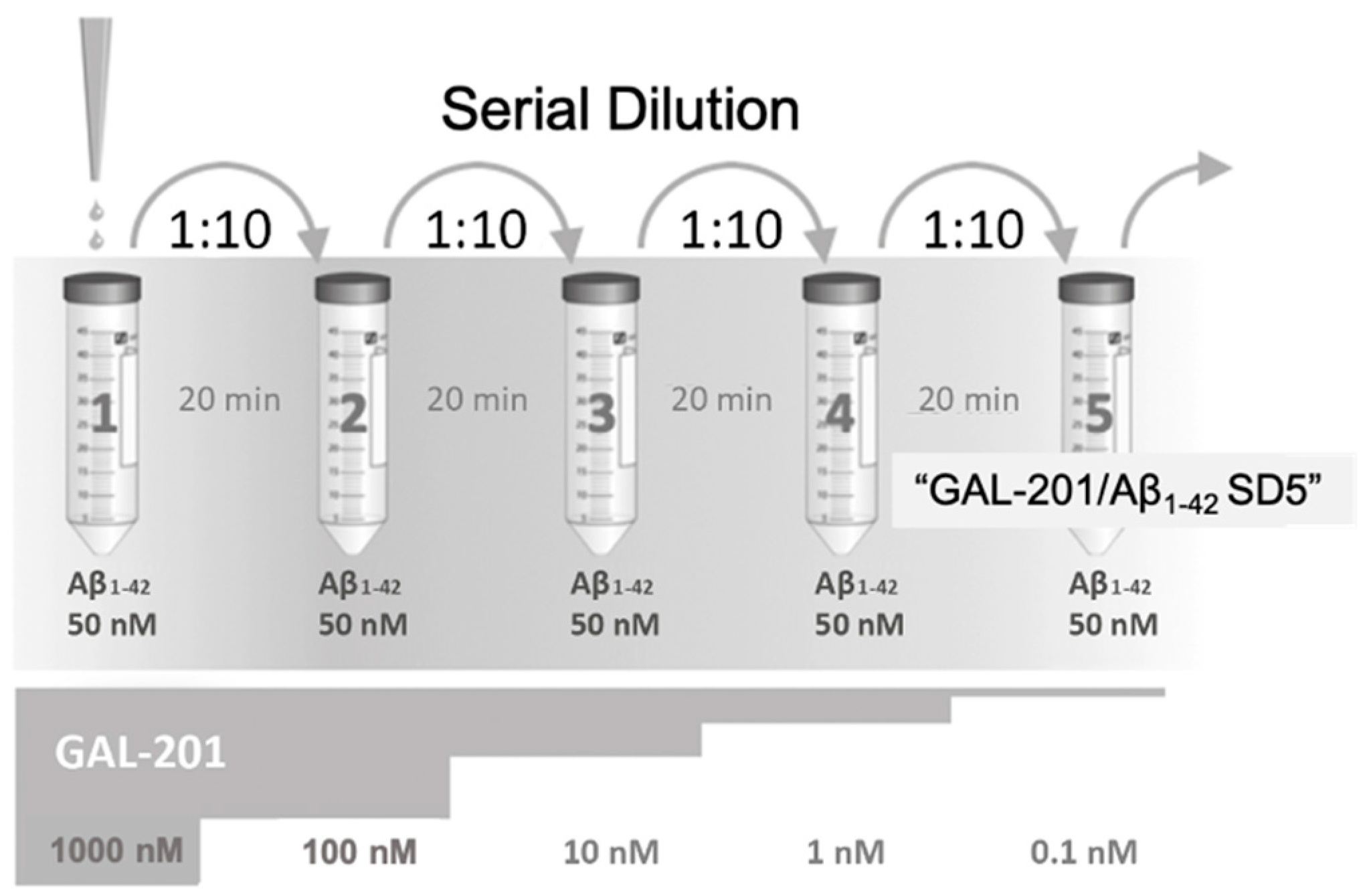
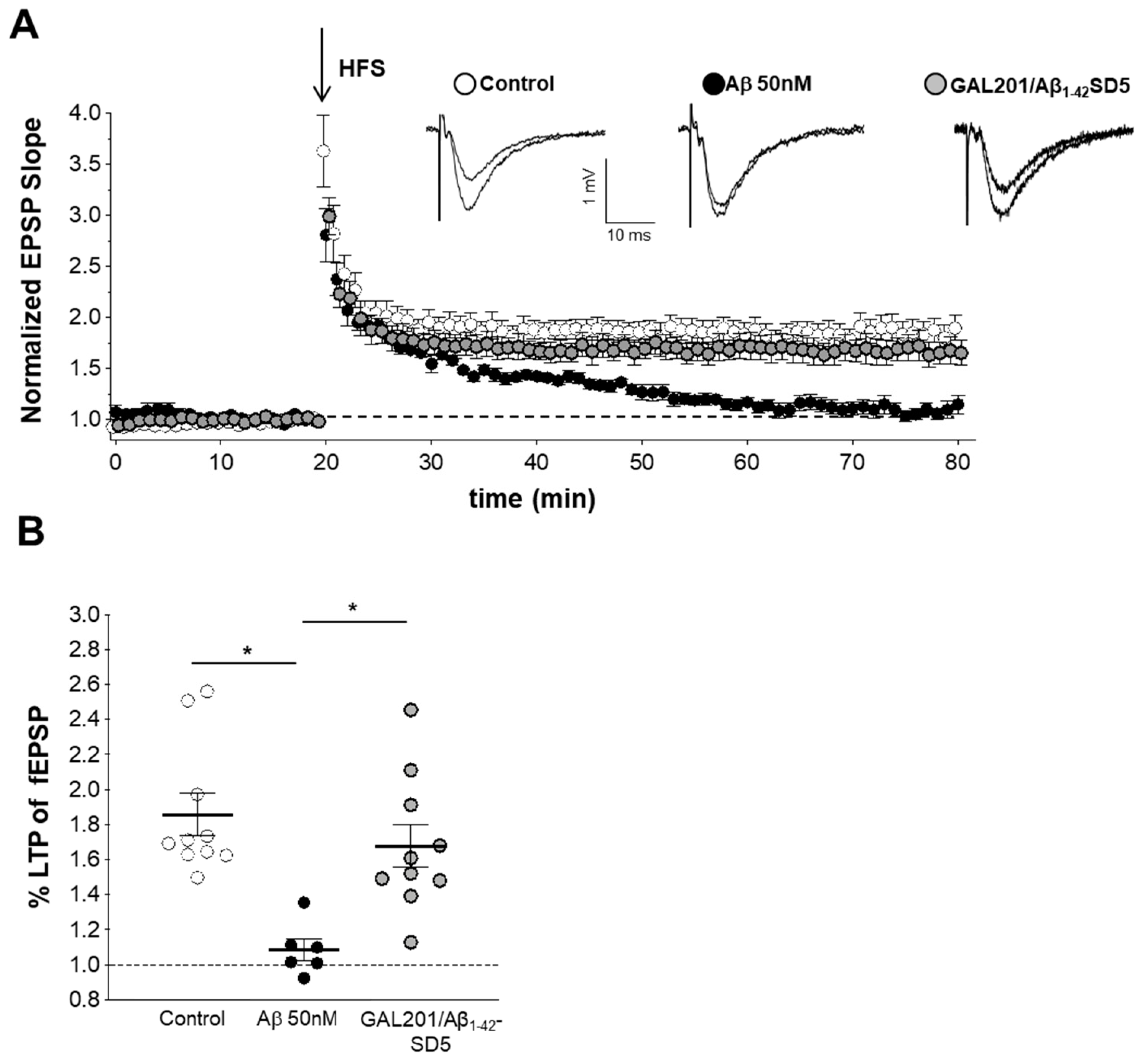
3.5. Self-Propagating Detoxification of Aβ1-42 Using Serial Dilation of GAL-201
3.6. Reversal of an Already Established Synaptotoxic Effect Caused by Aβ1-42
3.7. In Vivo Effects of GAL-201 Using LTP
3.8. Comparison of GAL-201 with Other Aβ Oligomer Targeting Drugs
4. Materials and Methods
4.1. Aβ1-42 and AβpE3
4.2. Binding Assay Using Surface Plasmon Resonance (SPR)
4.3. Pharmacokinetics in Rats
4.4. Electrophysiology
4.5. In Vitro Extracellular Recording
4.6. In Vivo LTP in the CA1 Hippocampal Region of the Anaesthetized Rat
4.7. Preparation of Test Compound and Oligomeric Aβ1-42
4.8. Administration of Test Compound and Oligomeric Aβ1-42
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Berthelot, K.; Cullin, C.; Lecomte, S. What does make an amyloid toxic: Morphology, structure or interaction with membrane? Biochimie 2013, 95, 12–19. [Google Scholar] [CrossRef]
- Williamson, R.; Sutherland, C. Neuronal membranes are key to the pathogenesis of Alzheimer’s disease: The role of both raft and non-raft membrane domains. Curr. Alzheimer Res. 2011, 8, 213–221. [Google Scholar] [CrossRef]
- Murakami, K.; Shimizu, T. Cytoplasmic superoxide radical: A possible contributing factor to intracellular Abeta oligomerization in Alzheimer disease. Commun. Integr. Biol. 2012, 5, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Ohyagi, Y. Intracellular amyloid beta-protein as a therapeutic target for treating Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 555–561. [Google Scholar] [CrossRef]
- Ohyagi, Y.; Tabira, T. Intracellular amyloid beta-protein and its associated molecules in the pathogenesis of Alzheimer’s disease. Mini Rev. Med. Chem. 2006, 6, 1075–1080. [Google Scholar] [CrossRef]
- Ul Islam, B.; Khan, M.S.; Jabir, N.R.; Kamal, M.A.; Tabrez, S. Elucidating Treatment of Alzheimer’s Disease via Different Receptors. Curr. Top. Med. Chem. 2017, 17, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Zamponi, G.W.; Ferguson, S.S. Glutamate receptors function as scaffolds for the regulation of beta-amyloid and cellular prion protein signaling complexes. Mol. Brain 2015, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Valles, A.S.; Borroni, M.V.; Barrantes, F.J. Targeting brain alpha7 nicotinic acetylcholine receptors in Alzheimer’s disease: Rationale and current status. CNS Drugs 2014, 28, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Ferreira, I.L.; Rego, A.C. Dysfunctional synapse in Alzheimer’s disease—A focus on NMDA receptors. Neuropharmacology 2014, 76, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Quirion, R. Group 1 metabotropic glutamate receptor function and its regulation of learning and memory in the aging brain. Front. Pharmacol. 2012, 3, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine—Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stravalaci, M.; Bastone, A.; Beeg, M.; Cagnotto, A.; Colombo, L.; Di Fede, G.; Tagliavini, F.; Cantu, L.; Del Favero, E.; Mazzanti, M.; et al. Specific recognition of biologically active amyloid-beta oligomers by a new Surface Plasmon Resonance-based immunoassay and an in vivo assay in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 27796–27805. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef] [Green Version]
- Tampi, R.R.; Forester, B.P.; Agronin, M. Aducanumab: Evidence from clinical trial data and controversies. Drugs Context 2021, 10. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimer Res. Ther. 2021, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Salt, T.E.; Luong, V.; Wood, N.; Cheung, W.; Maass, A.; Ferrari, G.; Russo-Marie, F.; Sillito, A.M.; Cheetham, M.E.; et al. Targeting amyloid-beta in glaucoma treatment. Proc. Natl. Acad. Sci. USA 2007, 104, 13444–13449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldblum, D.; Kipfer-Kauer, A.; Sarra, G.M.; Wolf, S.; Frueh, B.E. Distribution of amyloid precursor protein and amyloid-beta immunoreactivity in DBA/2J glaucomatous mouse retinas. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5085–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berisha, F.; Feke, G.T.; Trempe, C.L.; McMeel, J.W.; Schepens, C.L. Retinal abnormalities in early Alzheimer’s disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2285–2289. [Google Scholar] [CrossRef] [Green Version]
- Ning, A.; Cui, J.; To, E.; Ashe, K.H.; Matsubara, J. Amyloid-beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5136–5143. [Google Scholar] [CrossRef] [Green Version]
- Ghiso, J.A.; Doudevski, I.; Ritch, R.; Rostagno, A.A. Alzheimer’s disease and glaucoma: Mechanistic similarities and differences. J. Glaucoma 2013, 22 (Suppl. S5), S36. [Google Scholar] [CrossRef] [Green Version]
- Sivak, J.M. The aging eye: Common degenerative mechanisms between the Alzheimer’s brain and retinal disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Jawhar, S.; Wirths, O.; Bayer, T.A. Pyroglutamate amyloid-beta (Abeta): A hatchet man in Alzheimer disease. J. Biol. Chem. 2011, 286, 38825–38832. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.L.; Le, K.X.; Cynis, H.; Ekpo, E.; Kleinschmidt, M.; Palmour, R.M.; Ervin, F.R.; Snigdha, S.; Cotman, C.W.; Saido, T.C.; et al. Pyroglutamate-3 amyloid-beta deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am. J. Pathol. 2013, 183, 369–381. [Google Scholar] [CrossRef]
- Russo, C.; Violani, E.; Salis, S.; Venezia, V.; Dolcini, V.; Damonte, G.; Benatti, U.; D’Arrigo, C.; Patrone, E.; Carlo, P.; et al. Pyroglutamate-modified amyloid beta-peptides--AbetaN3(pE)--strongly affect cultured neuron and astrocyte survival. J. Neurochem. 2002, 82, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- Schilling, S.; Lauber, T.; Schaupp, M.; Manhart, S.; Scheel, E.; Bohm, G.; Demuth, H.U. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 2006, 45, 12393–12399. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.C.; Konig, S.; Roeber, S.; et al. Nitration of Tyrosine 10 Critically Enhances Amyloid beta Aggregation and Plaque Formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Rammes, G.; Parsons, C.G. The Abeta aggregation modulator MRZ-99030 prevents and even reverses synaptotoxic effects of Abeta1-42 on LTP even following serial dilution to a 500:1 stoichiometric excess of Abeta1-42, suggesting a beneficial prion-like seeding mechanism. Neuropharmacology 2020, 179, 108267. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Nichols, E.; Szoeke, C.E.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Murray, C.J. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fa, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef]
- Gulisano, W.; Melone, M.; Ripoli, C.; Tropea, M.R.; Li Puma, D.D.; Giunta, S.; Cocco, S.; Marcotulli, D.; Origlia, N.; Palmeri, A.; et al. Neuromodulatory Action of Picomolar Extracellular Abeta42 Oligomers on Presynaptic and Postsynaptic Mechanisms Underlying Synaptic Function and Memory. J. Neurosci. 2019, 39, 5986–6000. [Google Scholar] [CrossRef] [Green Version]
- Frydman-Marom, A.; Rechter, M.; Shefler, I.; Bram, Y.; Shalev, D.E.; Gazit, E. Cognitive-performance recovery of Alzheimer’s disease model mice by modulation of early soluble amyloidal assemblies. Angew. Chem. Int. Ed. Engl. 2009, 48, 1981–1986. [Google Scholar] [CrossRef]
- Halliday, G. Pathology and hippocampal atrophy in Alzheimer’s disease. Lancet Neurol. 2017, 16, 862–864. [Google Scholar] [CrossRef]
- Alberdi, E.; Sanchez-Gomez, M.V.; Cavaliere, F.; Perez-Samartin, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammes, G.; Gravius, A.; Ruitenberg, M.; Wegener, N.; Chambon, C.; Sroka-Saidi, K.; Jeggo, R.; Staniaszek, L.; Spanswick, D.; O’Hare, E.; et al. MRZ-99030—A novel modulator of Abeta aggregation: II—Reversal of Abeta oligomer-induced deficits in long-term potentiation (LTP) and cognitive performance in rats and mice. Neuropharmacology 2015, 92, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Lin, H.; Quist, A.P. Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim. Biophys. Acta 2007, 1768, 1966–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Rammes, G.; Seeser, F.; Mattusch, K.; Zhu, K.; Haas, L.; Kummer, M.; Heneka, M.; Herms, J.; Parsons, C.G. The NMDA receptor antagonist Radiprodil reverses the synaptotoxic effects of different amyloid-beta (Abeta) species on long-term potentiation (LTP). Neuropharmacology 2018, 140, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundqvist, M.; Qian, F.; O’Malley, T.; et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Abeta antibodies. Nat. Struct. Mol. Biol. 2020, 27, 1125–1133. [Google Scholar] [CrossRef]
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A. Elucidating the Abeta42 Anti-Aggregation Mechanism of Action of Tramiprosate in Alzheimer’s Disease: Integrating Molecular Analytical Methods, Pharmacokinetic and Clinical Data. CNS Drugs 2017, 31, 495–509. [Google Scholar] [CrossRef]
- Schemmert, S.; Camargo, L.C.; Honold, D.; Gering, I.; Kutzsche, J.; Willuweit, A.; Willbold, D. In Vitro and In Vivo Efficacies of the Linear and the Cyclic Version of an All-d-Enantiomeric Peptide Developed for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6553. [Google Scholar] [CrossRef]
- Russ, H.; Danysz, W.; Parsons, C.G. Interval therapy for the treatment of loss of eyesight in humans with glaucoma and other degenerative eye diseases. Patent WO2013189606A3, 27 December 2013. [Google Scholar]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin. Pharmacokinet. 2018, 57, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Schartmann, E.; Schemmert, S.; Niemietz, N.; Honold, D.; Ziehm, T.; Tusche, M.; Elfgen, A.; Gering, I.; Brener, O.; Shah, N.J.; et al. In Vitro Potency and Preclinical Pharmacokinetic Comparison of All-D-Enantiomeric Peptides Developed for the Treatment of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 859–873. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, A.; Luheshi, L.M.; Söllvander, S.; Pereira de Barros, T.; Macao, B.; Knowles, T.P.; Biverstål, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef] [Green Version]
- Ruitenberg, M. Binding of MRZ-14042-01 to Monomeric Aβ1-42. A Surface Plasmon Resonance (SPR) Study; Merz-31261; Merz Pharmaceuticals: Frankfurt, Germany, 26 November 2012; pp. 1–10. [Google Scholar]
- Seibenhener, M.L.; Wooten, M.W. Isolation and culture of hippocampal neurons from prenatal mice. J. Vis. Exp. JoVE 2012, 65, e3634. [Google Scholar] [CrossRef] [Green Version]
- Anderson, W.W.; Collingridge, G.L. The LTP Program: A data acquisition program for on-line analysis of long-term potentiation and other synaptic events. J. Neurosci. Methods 2001, 108, 71–83. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C.; Pennisi, M.; Topple, A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J. Neurosci. Methods 1985, 13, 139–143. [Google Scholar] [CrossRef]
- Frenguelli, B.; Lodge, D.; Rammes, G.; Collingridge, G.; Quack, G.; Jerecic, J.; Headley, M.; Danysz, W. A tribute to Chris Parsons. Neuropharmacology 2021, 195, 108633. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration GAL-201 | 500 nM | 100 nM | 10 nM |
| Stoichiometric ratio GAL-201:A1-42 | 10:1 | 2:1 | 1:5 |
| Relative effect size (blocking A1-42 toxicity) | 40.0% | 33.3% | 60.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russ, H.; Mazzanti, M.; Parsons, C.; Riemann, K.; Gebauer, A.; Rammes, G. The Small Molecule GAL-201 Efficiently Detoxifies Soluble Amyloid β Oligomers: New Approach towards Oral Disease-Modifying Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 5794. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105794
Russ H, Mazzanti M, Parsons C, Riemann K, Gebauer A, Rammes G. The Small Molecule GAL-201 Efficiently Detoxifies Soluble Amyloid β Oligomers: New Approach towards Oral Disease-Modifying Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(10):5794. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105794
Chicago/Turabian StyleRuss, Hermann, Michele Mazzanti, Chris Parsons, Katrin Riemann, Alexander Gebauer, and Gerhard Rammes. 2022. "The Small Molecule GAL-201 Efficiently Detoxifies Soluble Amyloid β Oligomers: New Approach towards Oral Disease-Modifying Treatment of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 10: 5794. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105794