
Recent Advances of m6A Demethylases Inhibitors and Their Biological Functions in Human Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
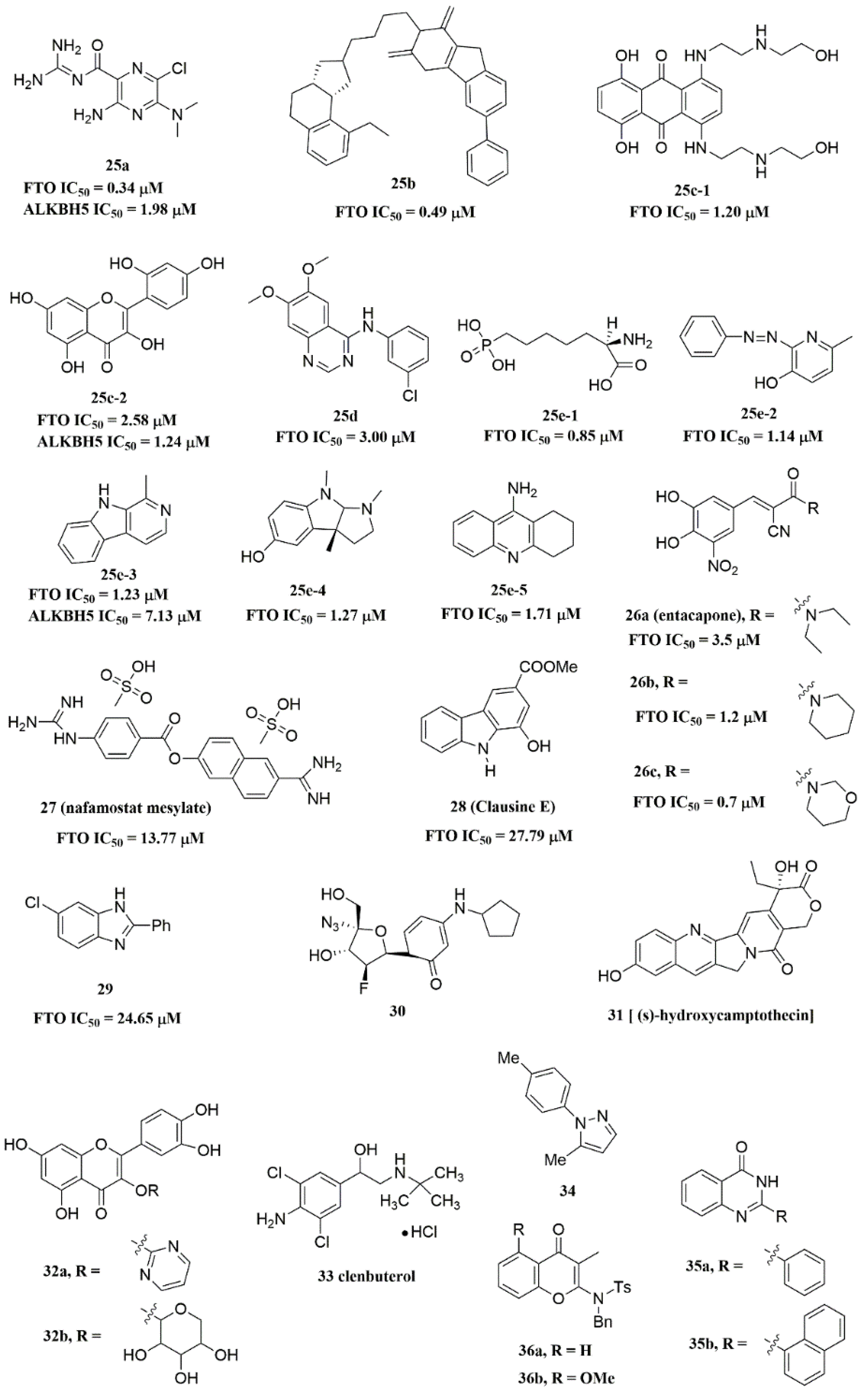{kind=link}
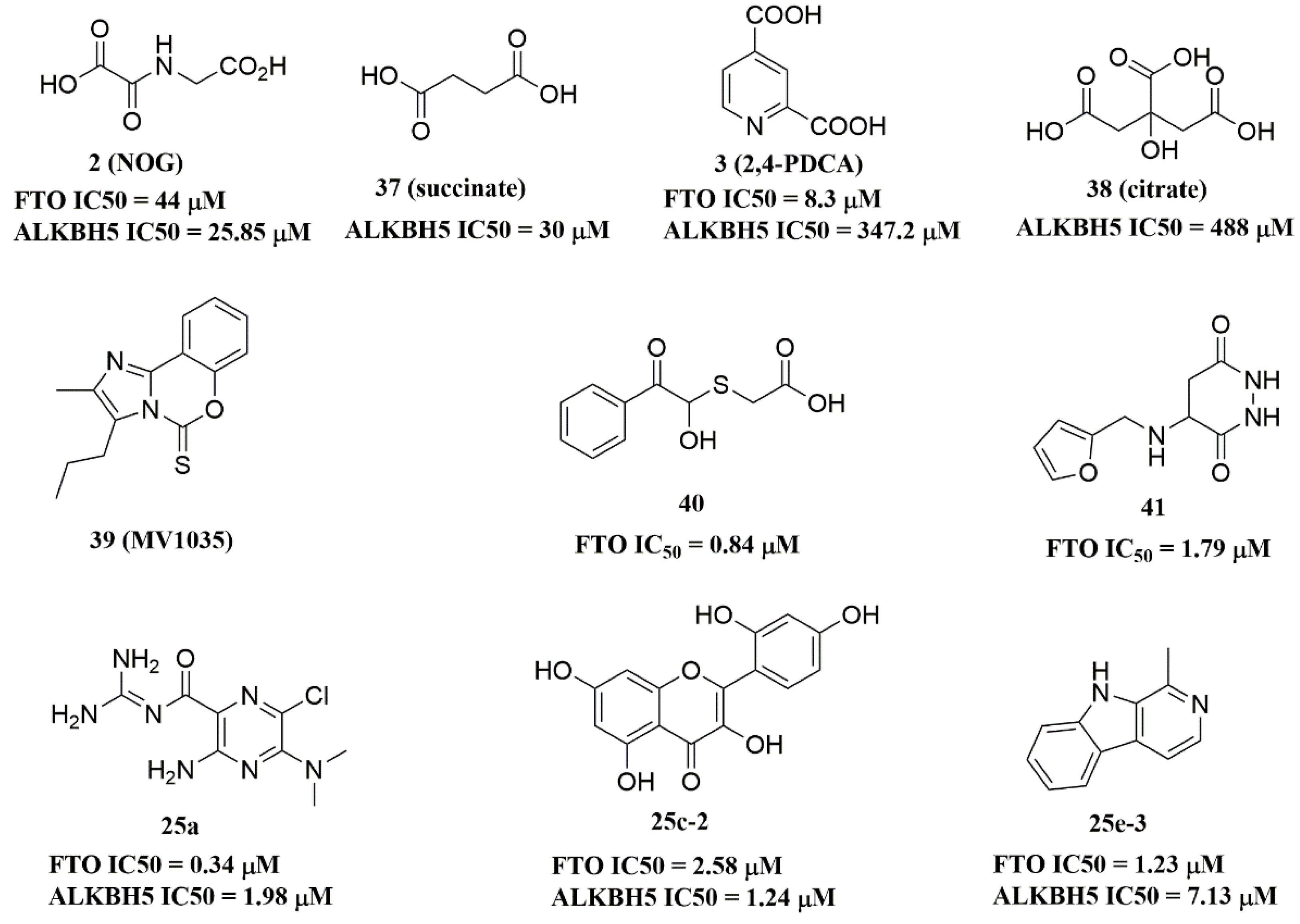{kind=link}
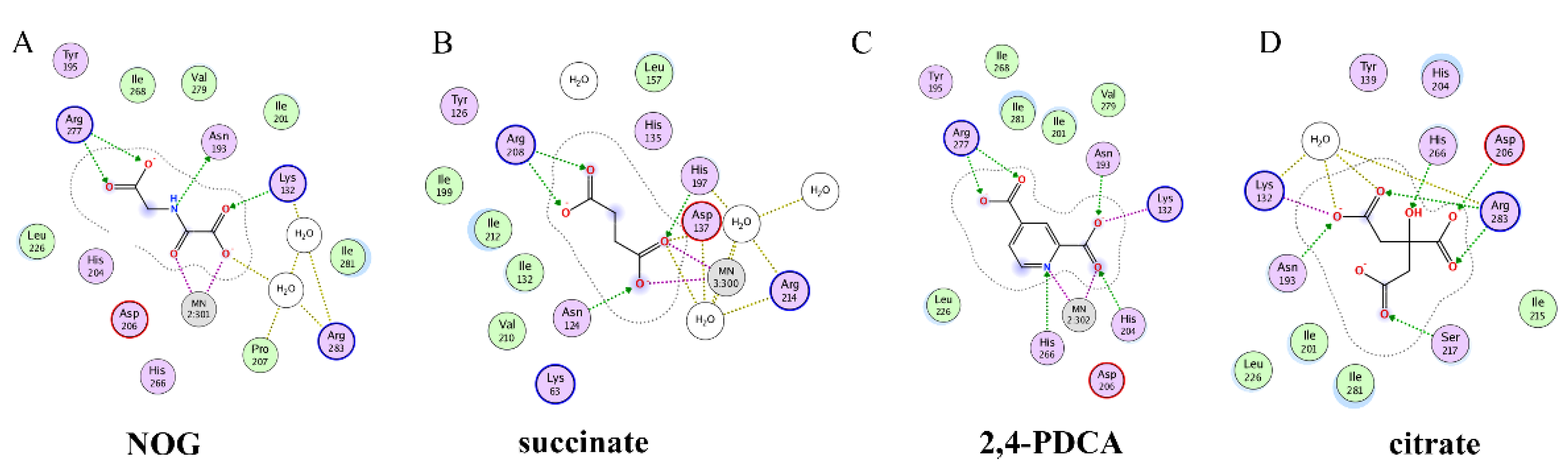{kind=link}
Abstract
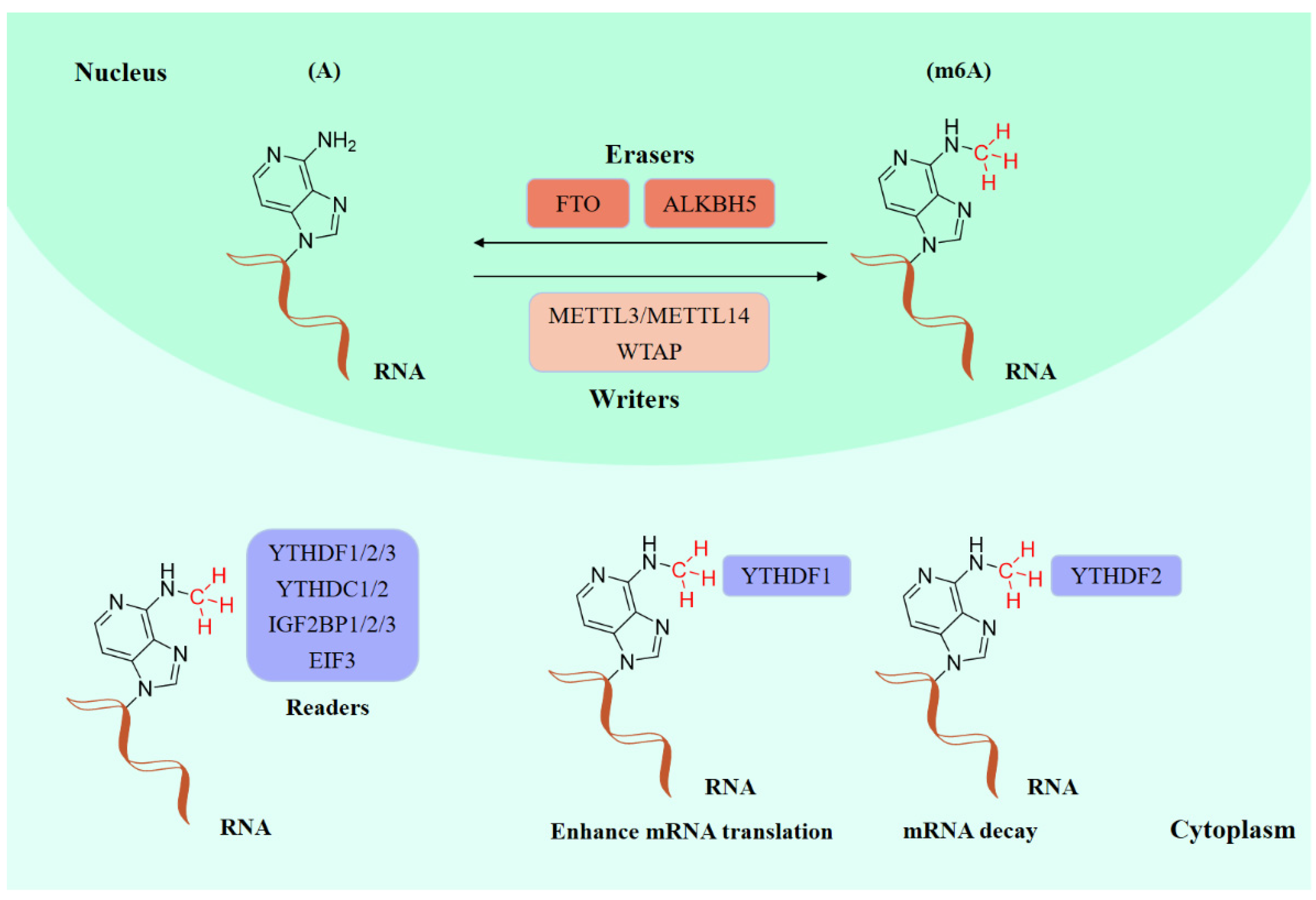
:1. Introduction
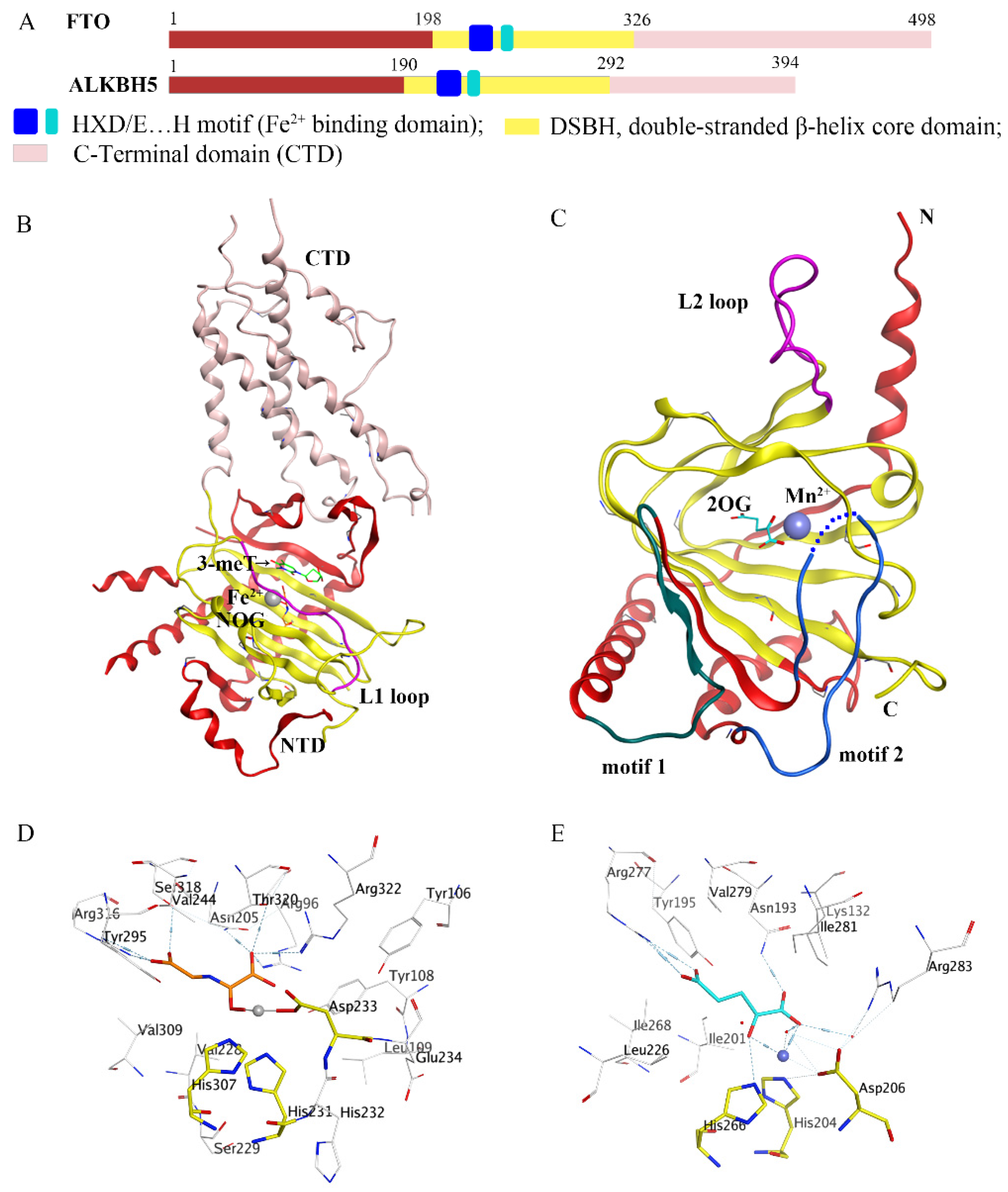
2. Structures of Fat-Mass- and Obesity-Associated Protein (FTO) and alkB Homolog 5 (ALKBH5)
2.1. Structures and Functions of FTO
2.2. Structures and Functions of ALKBH5
2.3. Structural Comparisons of FTO and ALKBH5
3. FTO and ALKBH5 in Human Diseases
3.1. FTO Is Involved in Regulating Various Diseases
3.1.1. FTO in Adipogenesis and Metabolism-Related Diseases
3.1.2. FTO in Heart Failure
3.1.3. FTO in Leukemia
3.1.4. FTO in Glioblastoma
3.1.5. FTO in Breast Cancer
3.1.6. FTO in Melanoma
3.1.7. FTO in Endometrial Cancer
3.1.8. FTO in Gastric Cancer
3.1.9. FTO in Bladder Cancer
3.1.10. FTO in Esophageal Squamous Cell Carcinoma (ESCC)
3.1.11. FTO in Multiple Myeloma (MM)
3.2. ALKBH5 Participates in the Biological Processes of Various Diseases
3.2.1. ALKBH5 in Breast Cancer
3.2.2. ALKBH5 in Glioblastoma
3.2.3. ALKBH5 in AML
3.2.4. ALKBH5 in Ischemic Heart Disease
3.2.5. ALKBH5 in Lung Cancer
3.2.6. ALKBH5 in Epithelial Ovarian Cancer
3.2.7. ALKBH5 Serves as Pancreatic Cancer (PC) Suppressor
3.2.8. ALKBH5 Suppresses Malignancy of Hepatocellular Carcinoma (HCC)
3.2.9. ALKBH5 in Osteogenesis and Osteosarcoma
3.2.10. ALKBH5 in Other Diseases
3.3. N6-methyladenosine (m6A) Demethylases Play an Essential Role in Drug Therapy Responses
3.3.1. m6A Demethylases in Chemotherapy Resistance
3.3.2. ALKBH5 in Cancer Immunity
4. Inhibitors
4.1. Strategies Used for Developing m6A Demethylases Inhibitors
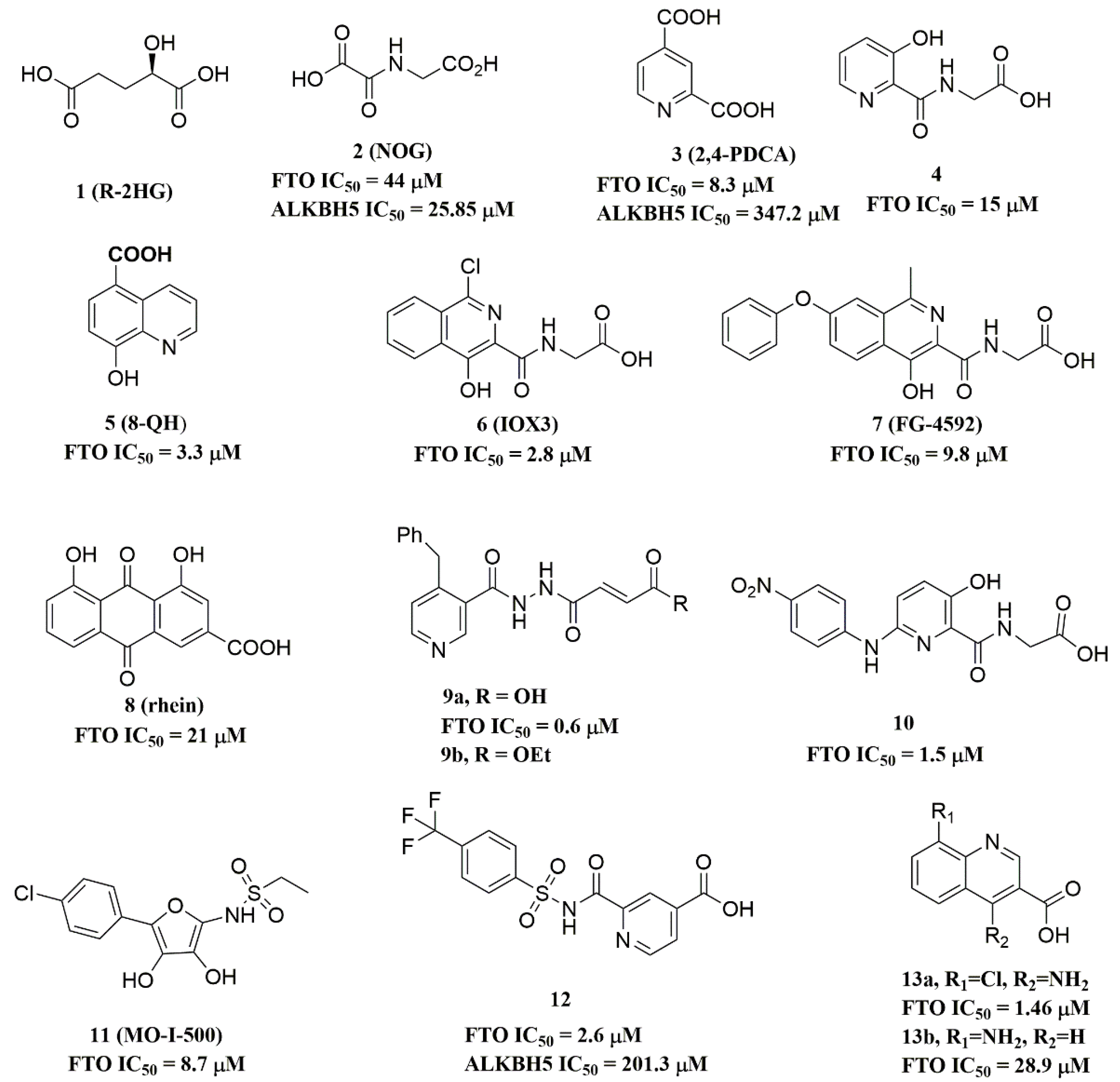
4.2. FTO Inhibitors
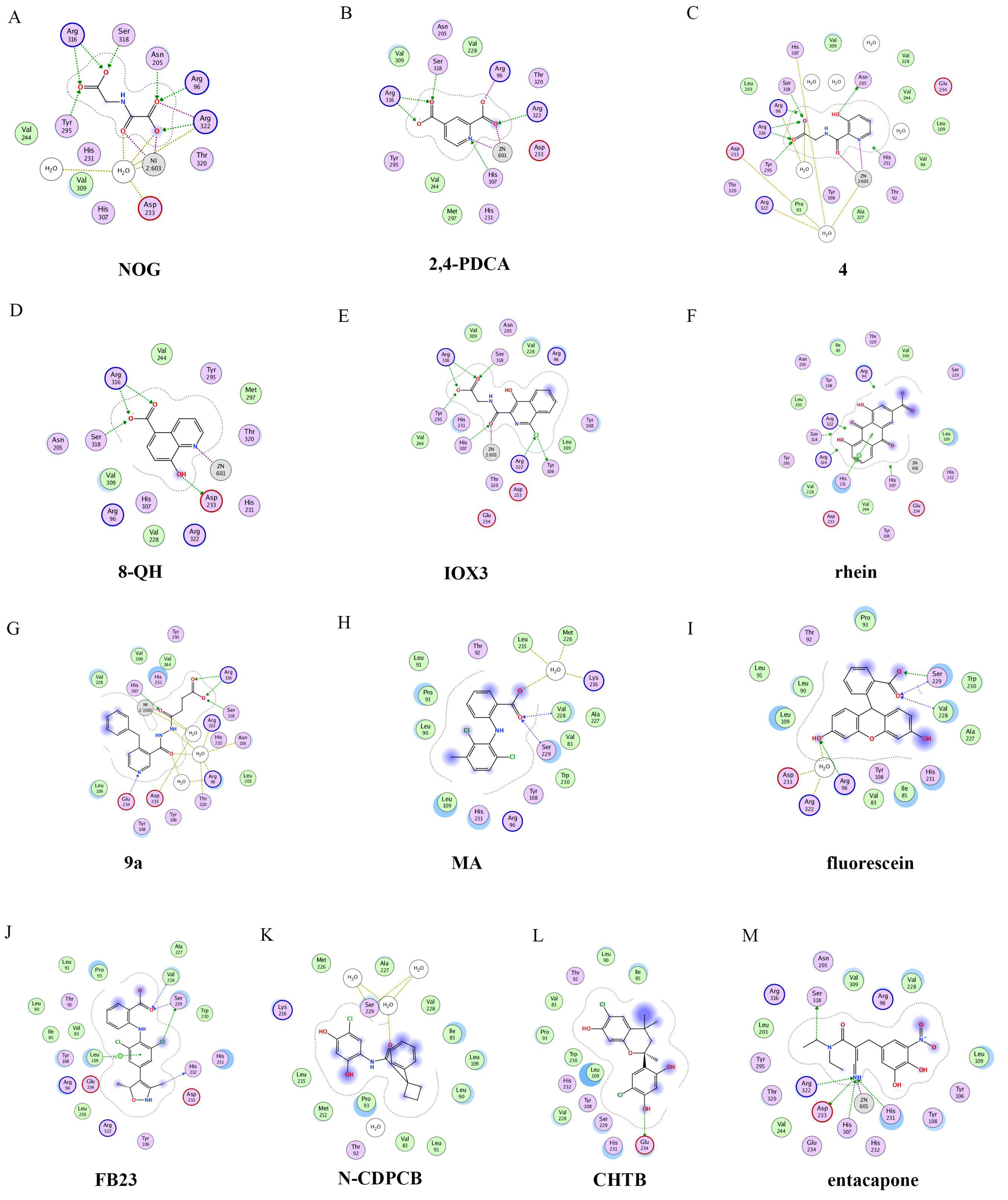
4.2.1. Metal-Chelating Inhibitors
4.2.2. Substrate Competitive Inhibitors
4.2.3. FTO Inhibitors with Other Scaffolds
4.3. ALKBH5 Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhao, X.; Wu, Y.S.; Li, M.M.; Wang, X.J.; Yang, Y.G. N6-methyl-adenosine (m6A) in RNA: An old modification with a novel epigenetic function. Genom. Proteom. Bioinform. 2013, 11, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Ma, S.; Chen, C.; Ji, X.; Liu, J.; Zhou, Q.; Wang, G.; Yuan, W.; Kan, Q.; Sun, Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 2019, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m(6)A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerken, T.; Girard, C.A.; Tung, Y.C.; Webby, C.J.; Saudek, V.; Hewitson, K.S.; Yeo, G.S.; McDonough, M.A.; Cunliffe, S.; McNeill, L.A.; et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007, 318, 1469–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalhammer, A.; Bencokova, Z.; Poole, R.; Loenarz, C.; Adam, J.; O’Flaherty, L.; Schödel, J.; Mole, D.; Giaslakiotis, K.; Schofield, C.J.; et al. Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1α (HIF-1α). PLoS ONE 2011, 6, e16210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Niu, T.; Chang, J.; Lei, X.; Zhao, M.; Wang, Q.; Cheng, W.; Wang, J.; Feng, Y.; Chai, J. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature 2010, 464, 1205–1209. [Google Scholar] [CrossRef]
- Aik, W.; Scotti, J.S.; Choi, H.; Gong, L.; Demetriades, M.; Schofield, C.J.; McDonough, M.A. Structure of human RNA N⁶-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014, 42, 4741–4754. [Google Scholar] [CrossRef]
- Feng, C.; Liu, Y.; Wang, G.; Deng, Z.; Zhang, Q.; Wu, W.; Tong, Y.; Cheng, C.; Chen, Z. Crystal structures of the human RNA demethylase Alkbh5 reveal basis for substrate recognition. J. Biol. Chem. 2014, 289, 11571–11583. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liu, K.; Tempel, W.; Demetriades, M.; Aik, W.; Schofield, C.J.; Min, J. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J. Biol. Chem. 2014, 289, 17299–17311. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Zhang, L.; Zheng, G.; Fu, Y.; Ji, Q.; Liu, F.; Chen, H.; He, C. Crystal structure of the RNA demethylase ALKBH5 from zebrafish. FEBS Lett. 2014, 588, 892–898. [Google Scholar] [CrossRef] [Green Version]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.J.; Mull, N.; Reagan, J.L.; Nemr, S.; Mitri, J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: A meta-analysis of observational studies. Blood 2012, 119, 4845–4850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, K.; Kitamoto, T.; Kitamoto, A.; Mizusawa, S.; Matsuo, T.; Nakata, Y.; Kamohara, S.; Miyatake, N.; Kotani, K.; Komatsu, R.; et al. Association of variations in the FTO, SCG3 and MTMR9 genes with metabolic syndrome in a Japanese population. J. Hum. Genet. 2011, 56, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaklamani, V.; Yi, N.; Sadim, M.; Siziopikou, K.; Zhang, K.; Xu, Y.; Tofilon, S.; Agarwal, S.; Pasche, B.; Mantzoros, C. The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med. Genet. 2011, 12, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iles, M.M.; Law, M.H.; Stacey, S.N.; Han, J.; Fang, S.; Pfeiffer, R.; Harland, M.; Macgregor, S.; Taylor, J.C.; Aben, K.K.; et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat. Genet. 2013, 45, 428–432, 432e1. [Google Scholar] [PubMed] [Green Version]
- Li, X.; Liang, L.; Zhang, M.; Song, F.; Nan, H.; Wang, L.E.; Wei, Q.; Lee, J.E.; Amos, C.I.; Qureshi, A.A.; et al. Obesity-related genetic variants, human pigmentation, and risk of melanoma. Hum. Genet. 2013, 132, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Delahanty, R.J.; Beeghly-Fadiel, A.; Xiang, Y.B.; Long, J.; Cai, Q.; Wen, W.; Xu, W.H.; Cai, H.; He, J.; Gao, Y.T.; et al. Association of obesity-related genetic variants with endometrial cancer risk: A report from the Shanghai Endometrial Cancer Genetics Study. Am. J. Epidemiol. 2011, 174, 1115–1126. [Google Scholar] [CrossRef]
- Fischer, J.; Koch, L.; Emmerling, C.; Vierkotten, J.; Peters, T.; Brüning, J.C.; Rüther, U. Inactivation of the Fto gene protects from obesity. Nature 2009, 458, 894–898. [Google Scholar] [CrossRef]
- Karra, E.; O’Daly, O.G.; Choudhury, A.I.; Yousseif, A.; Millership, S.; Neary, M.T.; Scott, W.R.; Chandarana, K.; Manning, S.; Hess, M.E.; et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J. Clin. Investig. 2013, 123, 3539–3551. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Peng, S.; Xiao, W.; Ju, D.; Sun, B.; Hou, N.; Liu, Q.; Wang, Y.; Zhao, H.; Gao, C.; Zhang, S.; et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci. Transl. Med. 2019, 11, eaau7116. [Google Scholar] [CrossRef]
- Nakae, J.; Cao, Y.; Oki, M.; Orba, Y.; Sawa, H.; Kiyonari, H.; Iskandar, K.; Suga, K.; Lombes, M.; Hayashi, Y. Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes 2008, 57, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, N.; Biwer, L.A.; Good, M.E.; Ruddiman, C.A.; Wolpe, A.G.; DeLalio, L.J.; Murphy, S.; Macal, E.H., Jr.; Ragolia, L.; Serbulea, V.; et al. Loss of Endothelial FTO Antagonizes Obesity-Induced Metabolic and Vascular Dysfunction. Circ. Res. 2020, 126, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berulava, T.; Buchholz, E.; Elerdashvili, V.; Pena, T.; Islam, M.R.; Lbik, D.; Mohamed, B.A.; Renner, A.; von Lewinski, D.; Sacherer, M.; et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 2020, 22, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Mathiyalagan, P.; Adamiak, M.; Mayourian, J.; Sassi, Y.; Liang, Y.; Agarwal, N.; Jha, D.; Zhang, S.; Kohlbrenner, E.; Chepurko, E.; et al. FTO-Dependent N(6)-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation 2019, 139, 518–532. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, H.; Wu, J.; Cai, Y.; Dong, Z.; Zhao, Y.; Hu, Q.; Hu, K.; Sun, A.; Ge, J. m6A demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct. Target. Ther. 2021, 6, 377. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Van Der Werf, I.; Jamieson, C. The Yin and Yang of RNA Methylation: An Imbalance of Erasers Enhances Sensitivity to FTO Demethylase Small-Molecule Targeting in Leukemia Stem Cells. Cancer Cell 2019, 35, 540–541. [Google Scholar] [CrossRef] [Green Version]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 2018, 172, 90–105.e23. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Al-Kali, A.; Zhang, Z.; Liu, J.; Pang, J.; Zhao, N.; He, C.; Litzow, M.R.; Liu, S. A dynamic N(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018, 28, 1062–1076. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.E.; Gholamalizadeh, M.; Doaei, S.; Mirsafa, F. FTO Gene Affects Obesity and Breast Cancer Through Similar Mechanisms: A New Insight into the Molecular Therapeutic Targets. Nutr. Cancer 2018, 70, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, Y.; Li, L.; Chen, M.; Zhang, C.; Zuo, X.B.; Zhou, F.S.; Liang, B.; Zhu, J.; Li, P.; et al. Association study of susceptibility loci with specific breast cancer subtypes in Chinese women. Breast Cancer Res. Treat. 2014, 146, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Thompson, D.J.; Dos Santos Silva, I.; Scott, C.; Tamimi, R.M.; Lindstrom, S.; Kraft, P.; Hazra, A.; Li, J.; Eriksson, L.; et al. Novel Associations between Common Breast Cancer Susceptibility Variants and Risk-Predicting Mammographic Density Measures. Cancer Res. 2015, 75, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Kinne, H.E.; Milligan, R.D.; Washburn, L.J.; Olsen, M.; Lucci, A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS ONE 2016, 11, e0159072. [Google Scholar]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.F.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef] [Green Version]
- Chourasia, A.H.; Macleod, K.F. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 2015, 11, 1937–1938. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wei, J.; Cui, Y.H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, D.; Lai, Y.; Liu, Y.; Tao, X.; Wang, Q.; Zhao, G.; Gu, H.; Liao, H.; Zhu, Y.; et al. Estrogen induces endometrial cancer cell proliferation and invasion by regulating the fat mass and obesity-associated gene via PI3K/AKT and MAPK signaling pathways. Cancer Lett. 2012, 319, 89–97. [Google Scholar] [CrossRef]
- Zhu, Y.; Shen, J.; Gao, L.; Feng, Y. Estrogen promotes fat mass and obesity-associated protein nuclear localization and enhances endometrial cancer cell proliferation via the mTOR signaling pathway. Oncol. Rep. 2016, 35, 2391–2397. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wan, Y.; Zhang, Z.; Jiang, Y.; Lang, J.; Cheng, W.; Zhu, L. FTO demethylates m6A modifications in HOXB13 mRNA and promotes endometrial cancer metastasis by activating the WNT signalling pathway. RNA Biol. 2020, 18, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shao, W.; Jiang, Y.; Wang, X.; Liu, Y.; Liu, X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol. Rep. 2017, 38, 2285–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Huang, J.; Hu, J. m(6)A RNA Methylation Regulators Contribute to Malignant Progression and Have Clinical Prognostic Impact in Gastric Cancer. Front. Oncol. 2019, 9, 1038. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhang, M.; Ge, S.; Huang, W.; Lin, X.; Gao, J.; Gong, J.; Shen, L. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019, 8, 4766–4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Jiang, X.; Zhang, Z.; Zhao, Z.; Xing, W.; Liu, Y.; Jiang, X.; Zhao, H. HDAC3-dependent transcriptional repression of FOXA2 regulates FTO/m6A/MYC signaling to contribute to the development of gastric cancer. Cancer Gene Ther. 2021, 28, 141–155. [Google Scholar] [CrossRef]
- Tao, L.; Mu, X.; Chen, H.; Jin, D.; Zhang, R.; Zhao, Y.; Fan, J.; Cao, M.; Zhou, Z. FTO modifies the m6A level of MALAT and promotes bladder cancer progression. Clin. Transl. Med. 2021, 11, e310. [Google Scholar] [CrossRef]
- Zhou, G.; Yan, K.; Liu, J.; Gao, L.; Jiang, X.; Fan, Y. FTO promotes tumour proliferation in bladder cancer via the FTO/miR-576/CDK6 axis in an m6A-dependent manner. Cell Death Discov. 2021, 7, 329. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, C.; Ma, S.; Li, Z.; Wang, W.; Li, Y.; Ma, Y.; Fang, J.; Wang, Y.; Cao, W.; et al. RNA m6A demethylase FTO-mediated epigenetic up-regulation of LINC00022 promotes tumorigenesis in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 294. [Google Scholar] [CrossRef]
- Xu, A.; Zhang, J.; Zuo, L.; Yan, H.; Chen, L.; Zhao, F.; Fan, F.; Xu, J.; Zhang, B.; Zhang, Y.; et al. FTO promotes multiple myeloma progression by posttranscriptional activation of HSF1 in an m(6)A-YTHDF2-dependent manner. Mol. Ther. 2021, 30, 1104–1118. [Google Scholar] [CrossRef]
- Kourtis, N.; Moubarak, R.S.; Aranda-Orgilles, B.; Lui, K.; Aydin, I.T.; Trimarchi, T.; Darvishian, F.; Salvaggio, C.; Zhong, J.; Bhatt, K.; et al. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat. Cell Biol. 2015, 17, 322–332. [Google Scholar] [CrossRef]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iv Santaliz-Ruiz, L.E.; Xie, X.; Old, M.; Teknos, T.N.; Pan, Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int. J. Cancer 2014, 135, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, K.; Banasavadi-Siddegowda, Y.; Mo, X.; Kim, S.H.; Mao, P.; Kig, C.; Nardini, D.; Sobol, R.W.; Chow, L.M.; Kornblum, H.I.; et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 2013, 31, 1051–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, A.H.; Wei, P.; Zhang, S.; Yao, J.; Yuan, Y.; Zhou, A.D.; Lang, F.F.; Heimberger, A.B.; Rao, G.; Huang, S. FoxM1 Drives a Feed-Forward STAT3-Activation Signaling Loop That Promotes the Self-Renewal and Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 2337–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski-Chauvel, A.; Lacore, M.G.; Arnauduc, F.; Delmas, C.; Toulas, C.; Cohen-Jonathan-Moyal, E.; Seva, C. The m6A RNA Demethylase ALKBH5 Promotes Radioresistance and Invasion Capability of Glioma Stem Cells. Cancers 2020, 13, 40. [Google Scholar] [CrossRef]
- Shen, C.; Sheng, Y.; Zhu, A.C.; Robinson, S.; Jiang, X.; Dong, L.; Chen, H.; Su, R.; Yin, Z.; Li, W.; et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020, 27, 64–80.e9. [Google Scholar] [CrossRef]
- Piekorz, R.P.; Hoffmeyer, A.; Duntsch, C.D.; McKay, C.; Nakajima, H.; Sexl, V.; Snyder, L.; Rehg, J.; Ihle, J.N. The centrosomal protein TACC3 is essential for hematopoietic stem cell function and genetically interfaces with p53-regulated apoptosis. EMBO J. 2002, 21, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Pu, J.; Wang, L.; Wu, L.; Xiao, J.; Liu, Q.; Chen, J.; Zhang, M.; Liu, Y.; Ni, M.; et al. ATG16L1 phosphorylation is oppositely regulated by CSNK2/casein kinase 2 and PPP1/protein phosphatase 1 which determines the fate of cardiomyocytes during hypoxia/reoxygenation. Autophagy 2015, 11, 1308–1325. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Feng, X.; Zhang, H.; Luo, Y.; Huang, J.; Lin, M.; Jin, J.; Ding, X.; Wu, S.; Huang, H.; et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 2019, 15, 1419–1437. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Shang, J.; Ji, W. ALKBH5-m(6)A-FOXM1 signaling axis promotes proliferation and invasion of lung adenocarcinoma cells under intermittent hypoxia. Biochem. Biophys. Res. Commun. 2020, 521, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, L.; Dong, Z.; Xiong, J. miR-149 suppresses human non-small cell lung cancer growth and metastasis by inhibiting the FOXM1/cyclin D1/MMP2 axis. Oncol. Rep. 2017, 38, 3522–3530. [Google Scholar] [PubMed]
- Zhu, Z.; Qian, Q.; Zhao, X.; Ma, L.; Chen, P. N(6)-methyladenosine ALKBH5 promotes non-small cell lung cancer progress by regulating TIMP3 stability. Gene 2020, 731, 144348. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Guo, J.; Wu, Y.; Yang, L.; Wang, X.; Du, J.; Dai, J.; Chen, W.; Gong, K.; Miao, S.; et al. m(6)A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol. Cancer 2020, 19, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Gan, X.; Jiang, X.; Diao, S.; Wu, H.; Hu, J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J. Exp. Clin. Cancer Res. 2019, 38, 163. [Google Scholar] [CrossRef] [Green Version]
- Nie, S.; Zhang, L.; Liu, J.; Wan, Y.; Jiang, Y.; Yang, J.; Sun, R.; Ma, X.; Sun, G.; Meng, H.; et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2021, 40, 284. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Mehta, P.; Ward, M.R.; Bregenzer, M.E.; Fleck, E.M.A.; Tan, L.; McLean, K.; Buckanovich, R.J.; Mehta, G. Personalized Medicine-Based Approach to Model Patterns of Chemoresistance and Tumor Recurrence Using Ovarian Cancer Stem Cell Spheroids. Clin. Cancer Res. 2017, 23, 6934–6945. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, F.; Du, C.; Guo, H.; Ma, L.; Liu, X.; Kornmann, M.; Tian, X.; Yang, Y. BRM/SMARCA2 promotes the proliferation and chemoresistance of pancreatic cancer cells by targeting JAK2/STAT3 signaling. Cancer Lett. 2017, 402, 213–224. [Google Scholar] [CrossRef]
- Yogev, O.; Almeida, G.S.; Barker, K.T.; George, S.L.; Kwok, C.; Campbell, J.; Zarowiecki, M.; Kleftogiannis, D.; Smith, L.M.; Hallsworth, A.; et al. In Vivo Modeling of Chemoresistant Neuroblastoma Provides New Insights into Chemorefractory Disease and Metastasis. Cancer Res. 2019, 79, 5382–5393. [Google Scholar] [CrossRef]
- Guo, X.; Li, K.; Jiang, W.; Hu, Y.; Xiao, W.; Huang, Y.; Feng, Y.; Pan, Q.; Wan, R. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol. Cancer 2020, 19, 91. [Google Scholar] [CrossRef]
- Tang, B.; Yang, Y.; Kang, M.; Wang, Y.; Wang, Y.; Bi, Y.; He, S.; Shimamoto, F. m(6)A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol. Cancer 2020, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhao, S.; Shen, J.; Guo, L.; Sun, Y.; Zhu, Y.; Ma, Z.; Zhang, X.; Hu, Y.; Xiao, W.; et al. The MiR-135b-BMAL1-YY1 loop disturbs pancreatic clockwork to promote tumourigenesis and chemoresistance. Cell Death Dis. 2018, 9, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhao, Y.; Chen, J.; Peng, C.; Zhang, Y.; Tong, R.; Cheng, Q.; Yang, B.; Feng, X.; Lu, Y.; et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m(6)A-guided epigenetic inhibition of LYPD1. Mol. Cancer 2020, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Kuang, M.J.; Han, S.J.; Wang, A.B.; Qiu, J.; Wang, F.; Tan, B.Y.; Wang, D.C. BMP2 Modified by the m(6)A Demethylation Enzyme ALKBH5 in the Ossification of the Ligamentum Flavum Through the AKT Signaling Pathway. Calcif. Tissue Int. 2020, 106, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Shen, L.; Liu, Y.; Ming, H.; Zhu, X.; Chu, M.; Lin, J. The m6A methyltransferase METTL3 cooperates with demethylase ALKBH5 to regulate osteogenic differentiation through NF-κB signaling. Mol. Cell Biochem. 2020, 463, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, L.; Wang, Y. ALKBH5-mediated m(6)A demethylation of lncRNA PVT1 plays an oncogenic role in osteosarcoma. Cancer Cell Int. 2020, 20, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derderian, C.; Orunmuyi, A.T.; Olapade-Olaopa, E.O.; Ogunwobi, O.O. PVT1 Signaling Is a Mediator of Cancer Progression. Front. Oncol. 2019, 9, 502. [Google Scholar] [CrossRef] [Green Version]
- Li, X.C.; Jin, F.; Wang, B.Y.; Yin, X.J.; Hong, W.; Tian, F.J. The m6A demethylase ALKBH5 controls trophoblast invasion at the maternal-fetal interface by regulating the stability of CYR61 mRNA. Theranostics 2019, 9, 3853–3865. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, S.; Piao, H.Y.; Wang, Y.; Wu, Y.; Meng, X.Y.; Yang, D.; Zheng, Z.C.; Zhao, Y. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J. Physiol. Biochem. 2019, 75, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Bai, Z.L.; Xia, D.; Zhao, Z.J.; Zhao, R.; Wang, Y.Y.; Zhe, H. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol. Carcinog. 2018, 57, 590–597. [Google Scholar] [CrossRef]
- Shriwas, O.; Priyadarshini, M.; Samal, S.K.; Rath, R.; Panda, S.; Das Majumdar, S.K.; Muduly, D.K.; Botlagunta, M.; Dash, R. DDX3 modulates cisplatin resistance in OSCC through ALKBH5-mediated m(6)A-demethylation of FOXM1 and NANOG. Apoptosis 2020, 25, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Yang, S.; Wang, S.; Wu, J.; Zheng, B.; Wang, K.; Shen, S.; Jeong, S.; Li, Z.; Zhu, Y.; et al. M(6)A Demethylase ALKBH5 Regulates PD-L1 Expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res. 2021, 81, 4778–4793. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Kang, Y.; Wang, L.; Huff, S.; Tang, R.; Hui, H.; Agrawal, K.; Gonzalez, G.M.; Wang, Y.; Patel, S.P.; et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 20159–20170. [Google Scholar] [CrossRef]
- Dong, F.; Qin, X.; Wang, B.; Li, Q.; Hu, J.; Cheng, X.; Guo, D.; Cheng, F.; Fang, C.; Tan, Y.; et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 5876–5888. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, L.; Ding, G.; Huang, H.; Cao, G.; Sun, X.; Lou, N.; Wei, Q.; Shen, T.; Xu, X.; et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J. Immunother. Cancer 2020, 8, e000308. [Google Scholar] [CrossRef] [Green Version]
- Aik, W.; Demetriades, M.; Hamdan, M.K.; Bagg, E.A.; Yeoh, K.K.; Lejeune, C.; Zhang, Z.; McDonough, M.A.; Schofield, C.J. Structural basis for inhibition of the fat mass and obesity associated protein (FTO). J. Med. Chem. 2013, 56, 3680–3688. [Google Scholar] [CrossRef]
- Chen, B.; Ye, F.; Yu, L.; Jia, G.; Huang, X.; Zhang, X.; Peng, S.; Chen, K.; Wang, M.; Gong, S.; et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J. Am. Chem. Soc. 2012, 134, 17963–17971. [Google Scholar] [CrossRef]
- Selberg, S.; Yu, L.Y.; Bondarenko, O.; Kankuri, E.; Seli, N.; Kovaleva, V.; Herodes, K.; Saarma, M.; Karelson, M. Small-Molecule Inhibitors of the RNA M6A Demethylases FTO Potently Support the Survival of Dopamine Neurons. Int. J. Mol. Sci. 2021, 22, 4537. [Google Scholar] [CrossRef]
- Wang, R.; Han, Z.; Liu, B.; Zhou, B.; Wang, N.; Jiang, Q.; Qiao, Y.; Song, C.; Chai, J.; Chang, J. Identification of Natural Compound Radicicol as a Potent FTO Inhibitor. Mol. Pharm. 2018, 15, 4092–4098. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef] [PubMed]
- Selberg, S.; Seli, N.; Kankuri, E.; Karelson, M. Rational Design of Novel Anticancer Small-Molecule RNA m6A Demethylase ALKBH5 Inhibitors. ACS Omega 2021, 6, 13310–13320. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yan, J.; Li, Q.; Li, J.; Gong, S.; Zhou, H.; Gan, J.; Jiang, H.; Jia, G.F.; Luo, C.; et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015, 43, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Svensen, N.; Jaffrey, S.R. Fluorescent RNA Aptamers as a Tool to Study RNA-Modifying Enzymes. Cell Chem. Biol. 2016, 23, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Yang, T.; Dong, J.; Prasetya, F.; Xie, Y.; Wong, K.H.Q.; Cheong, A.; Woon, E.C.Y. Multiprotein Dynamic Combinatorial Chemistry: A Strategy for the Simultaneous Discovery of Subfamily-Selective Inhibitors for Nucleic Acid Demethylases FTO and ALKBH3. Chem. Asian J. 2018, 13, 2854–2867. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.N.; Zhou, K.; Xu, Q.; Zhang, C.Y. Identification of Specific N(6)-Methyladenosine RNA Demethylase FTO Inhibitors by Single-Quantum-Dot-Based FRET Nanosensors. Anal. Chem. 2020, 92, 13936–13944. [Google Scholar] [CrossRef]
- Han, X.; Wang, N.; Li, J.; Wang, Y.; Wang, R.; Chang, J. Identification of nafamostat mesilate as an inhibitor of the fat mass and obesity-associated protein (FTO) demethylase activity. Chem. Biol. Interact. 2019, 297, 80–84. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Han, X.; Wang, N.; Song, C.; Wang, R.; Chang, J. Identification of Clausine E as an inhibitor of fat mass and obesity-associated protein (FTO) demethylase activity. J. Mol. Recognit. 2019, 32, e2800. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Han, X.; Wang, N.; Yu, W.; Wang, R.; Chang, J. The role of chlorine atom on the binding between 2-phenyl-1H-benzimidazole analogues and fat mass and obesity-associated protein. J. Mol. Recognit. 2019, 32, e2774. [Google Scholar] [CrossRef]
- Shishodia, S.; Demetriades, M.; Zhang, D.; Tam, N.Y.; Maheswaran, P.; Clunie-O’Connor, C.; Tumber, A.; Leung, I.K.H.; Ng, Y.M.; Leissing, T.M.; et al. Structure-Based Design of Selective Fat Mass and Obesity Associated Protein (FTO) Inhibitors. J. Med. Chem. 2021, 64, 16609–16625. [Google Scholar] [CrossRef]
- He, W.; Zhou, B.; Liu, W.; Zhang, M.; Shen, Z.; Han, Z.; Jiang, Q.; Yang, Q.; Song, C.; Wang, R.; et al. Identification of A Novel Small-Molecule Binding Site of the Fat Mass and Obesity Associated Protein (FTO). J. Med. Chem. 2015, 58, 7341–7348. [Google Scholar] [CrossRef] [PubMed]
- Huff, S.; Tiwari, S.K.; Gonzalez, G.M.; Wang, Y.; Rana, T.M. m(6)A-RNA Demethylase FTO Inhibitors Impair Self-Renewal in Glioblastoma Stem Cells. ACS Chem. Biol. 2021, 16, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Rose, N.R.; McDonough, M.A.; King, O.N.; Kawamura, A.; Schofield, C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc. Rev. 2011, 40, 4364–4397. [Google Scholar] [CrossRef] [PubMed]
- McMurray, F.; Demetriades, M.; Aik, W.; Merkestein, M.; Kramer, H.; Andrew, D.S.; Scudamore, C.L.; Hough, T.A.; Wells, S.; Ashcroft, F.M.; et al. Pharmacological inhibition of FTO. PLoS ONE 2015, 10, e0121829. [Google Scholar] [CrossRef] [Green Version]
- Toh, J.D.W.; Sun, L.; Lau, L.Z.M.; Tan, J.; Low, J.J.A.; Tang, C.W.Q.; Cheong, E.J.Y.; Tan, M.J.H.; Chen, Y.; Hong, W.; et al. A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N(6)-methyladenosine demethylase FTO. Chem. Sci. 2015, 6, 112–122. [Google Scholar] [CrossRef]
- Zheng, G.; Cox, T.; Tribbey, L.; Wang, G.Z.; Iacoban, P.; Booher, M.E.; Gabriel, G.J.; Zhou, L.; Bae, N.; Rowles, J.; et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem. Neurosci. 2014, 5, 658–665. [Google Scholar] [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e10. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Hong, T.; Huang, Y.; Su, H.; Wu, F.; Chen, Y.; Wei, L.; Huang, W.; Hua, X.; Xia, Y.; et al. Fluorescein Derivatives as Bifunctional Molecules for the Simultaneous Inhibiting and Labeling of FTO Protein. J. Am. Chem. Soc. 2015, 137, 13736–13739. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhou, B.; Zhang, M.; Liu, W.; Han, Z.; Song, C.; Yu, W.; Yang, Q.; Wang, R.; Wang, S.; et al. A Novel Inhibitor of the Obesity-Related Protein FTO. Biochemistry 2016, 55, 1516–1522. [Google Scholar] [CrossRef]
- Prakash, M.; Itoh, Y.; Fujiwara, Y.; Takahashi, Y.; Takada, Y.; Mellini, P.; Elboray, E.E.; Terao, M.; Yamashita, Y.; Yamamoto, C.; et al. Identification of Potent and Selective Inhibitors of Fat Mass Obesity-Associated Protein Using a Fragment-Merging Approach. J. Med. Chem. 2021, 64, 15810–15824. [Google Scholar] [CrossRef]
- Sun, K.; Du, Y.; Hou, Y.; Zhao, M.; Li, J.; Du, Y.; Zhang, L.; Chen, C.; Yang, H.; Yan, F.; et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m(6)A signaling. Theranostics 2021, 11, 5831–5846. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Z.; Wang, N.; Han, X.; Yu, W.; Wang, R.; Chang, J. Identification of the binding between three fluoronucleoside analogues and fat mass and obesity-associated protein by isothermal titration calorimetry and spectroscopic techniques. J. Pharm. Biomed. Anal. 2018, 149, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Wang, Z.; Zhang, L.; Wang, N.; Han, X.; Wang, R.; Chang, J. Study of the Binding between Camptothecin Analogs and FTO by Spectroscopy and Molecular Docking. J. Fluoresc. 2017, 27, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, N.; Han, X.; Wang, R.; Chang, J. Interaction of two flavonols with fat mass and obesity-associated protein investigated by fluorescence quenching and molecular docking. J. Biomol. Struct. Dyn. 2018, 36, 3388–3397. [Google Scholar] [CrossRef]
- Wang, Z.; Han, X.; Wang, N.; Wang, R.; Chang, J. Binding of clenbuterol to HSA and FTO: A spectroscopic analysis and molecular docking. Med. Chem. Res. 2018, 27, 944–953. [Google Scholar] [CrossRef]
- Yang, L.; Yu, W.; Li, Z.; Zhang, X.; Ren, T.; Zhang, L.; Wang, R.; Chang, J. Comparative study of the binding between FTO protein and five pyrazole derivatives by spectrofluorimetry. J. Mol. Liq. 2016, 218, 156–165. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, T.; Tian, X.; Wang, Z.; Yu, W.; Wang, R.; Chang, J. Investigation of the Interaction between 1,3-Diazaheterocyclic Compounds and the Fat Mass and Obesity-Associated Protein by Fluorescence Spectroscopy and Molecular Modeling. J. Fluoresc. 2017, 27, 369–378. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Z.; Ren, T.; Liu, H.; Wang, X.; Wang, R.; Chang, J. Investigation of the interaction between FTO and 3-substituted 2-aminochromones by spectroscopy and molecular modeling. Med. Chem. Res. 2017, 26, 1349–1358. [Google Scholar] [CrossRef]
- Malacrida, A.; Rivara, M.; Di Domizio, A.; Cislaghi, G.; Miloso, M.; Zuliani, V.; Nicolini, G. 3D proteome-wide scale screening and activity evaluation of a new ALKBH5 inhibitor in U87 glioblastoma cell line. Bioorg. Med. Chem. 2020, 28, 115300. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, Y.; Fu, Y.; Huang, M.; Shen, D.; Zhao, B.; Liu, H.; Zheng, Y.; Huang, L. Recent Advances of m6A Demethylases Inhibitors and Their Biological Functions in Human Diseases. Int. J. Mol. Sci. 2022, 23, 5815. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105815
You Y, Fu Y, Huang M, Shen D, Zhao B, Liu H, Zheng Y, Huang L. Recent Advances of m6A Demethylases Inhibitors and Their Biological Functions in Human Diseases. International Journal of Molecular Sciences. 2022; 23(10):5815. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105815
Chicago/Turabian StyleYou, Yazhen, Yundong Fu, Mingjie Huang, Dandan Shen, Bing Zhao, Hongmin Liu, Yichao Zheng, and Lihua Huang. 2022. "Recent Advances of m6A Demethylases Inhibitors and Their Biological Functions in Human Diseases" International Journal of Molecular Sciences 23, no. 10: 5815. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105815