
Identification of Genomic Regions Associated with Vine Growth and Plant Height of Soybean

 and
and
Abstract
:1. Introduction
2. Results
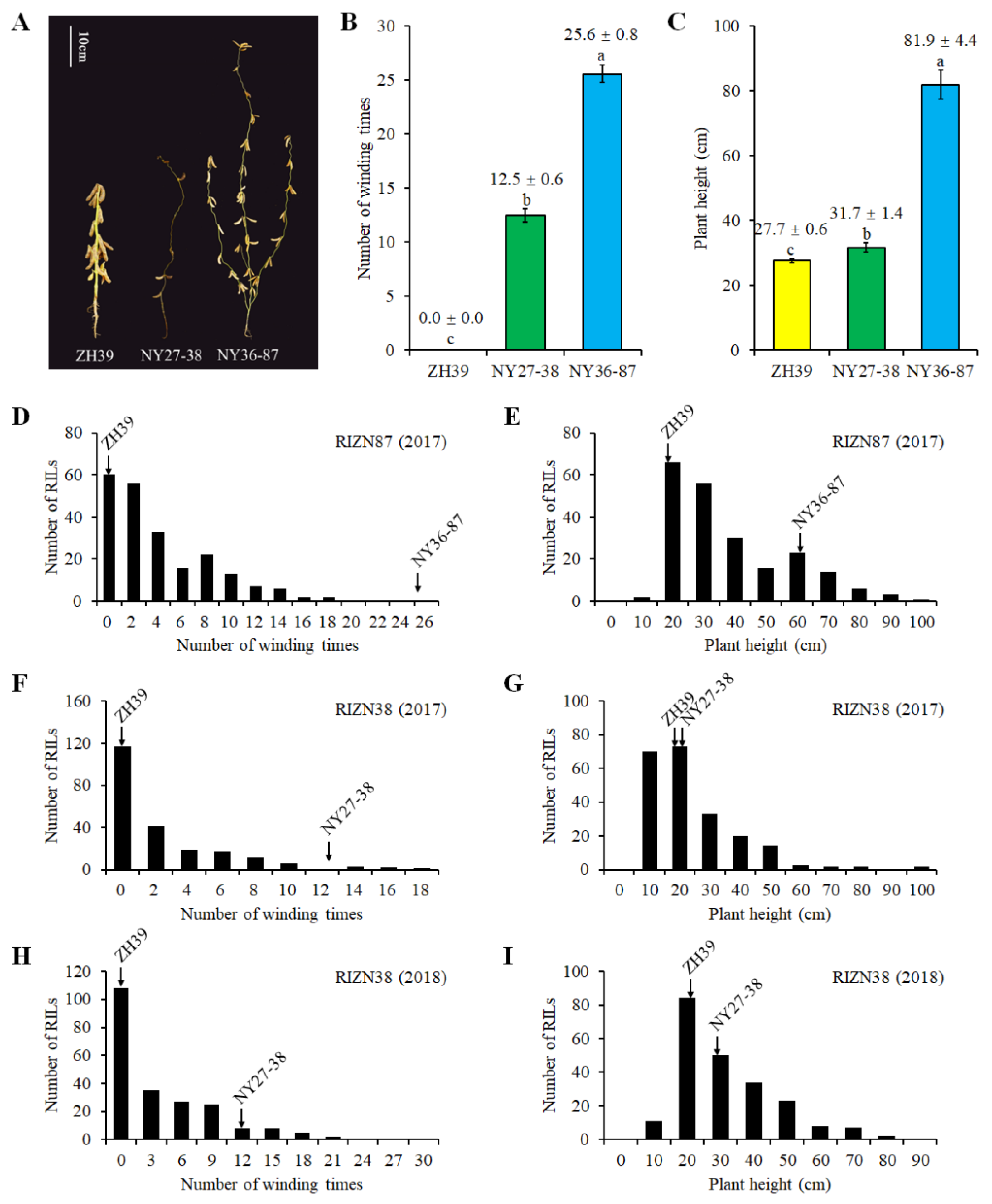
2.1. Plant Height and Vine-like Growth of Parents and RILs
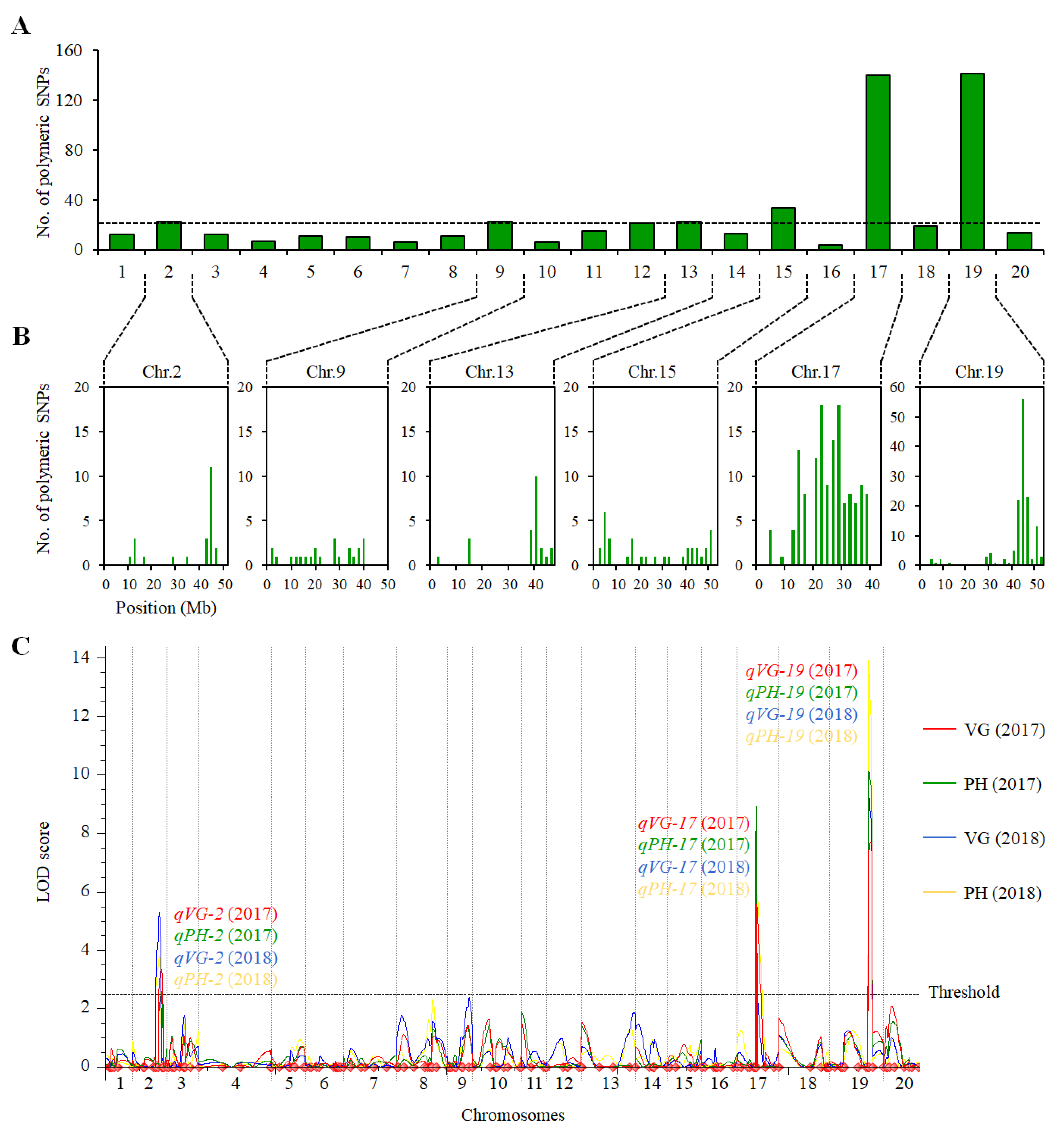
2.2. QTL Analysis of VG and PH in the ZH39 × NY27-38 Population
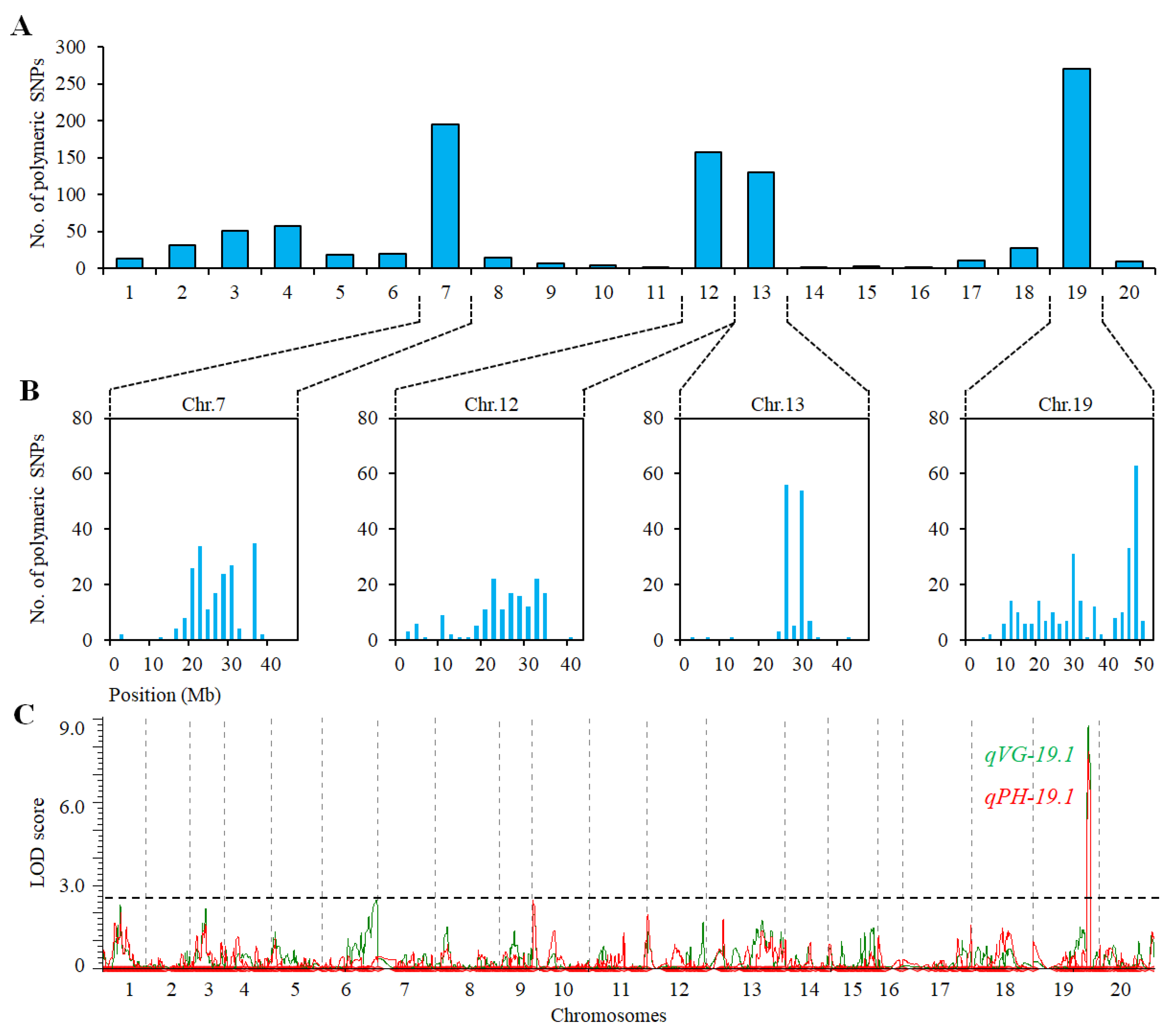
2.3. QTL Analysis for VG and PH in Population of ZH39 × NY36-87
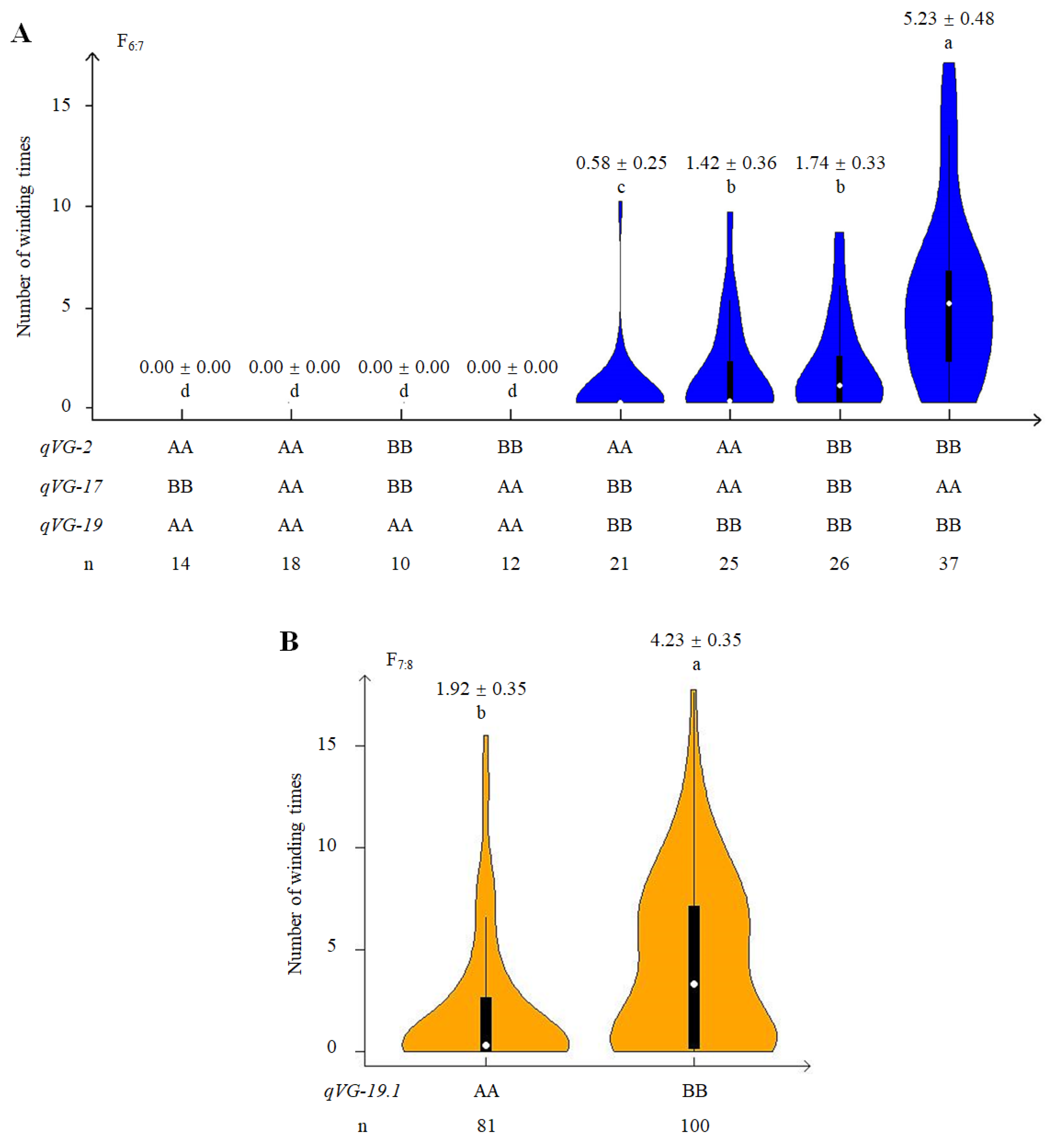
2.4. Phenotypic Effect of QTLs Associated with VG
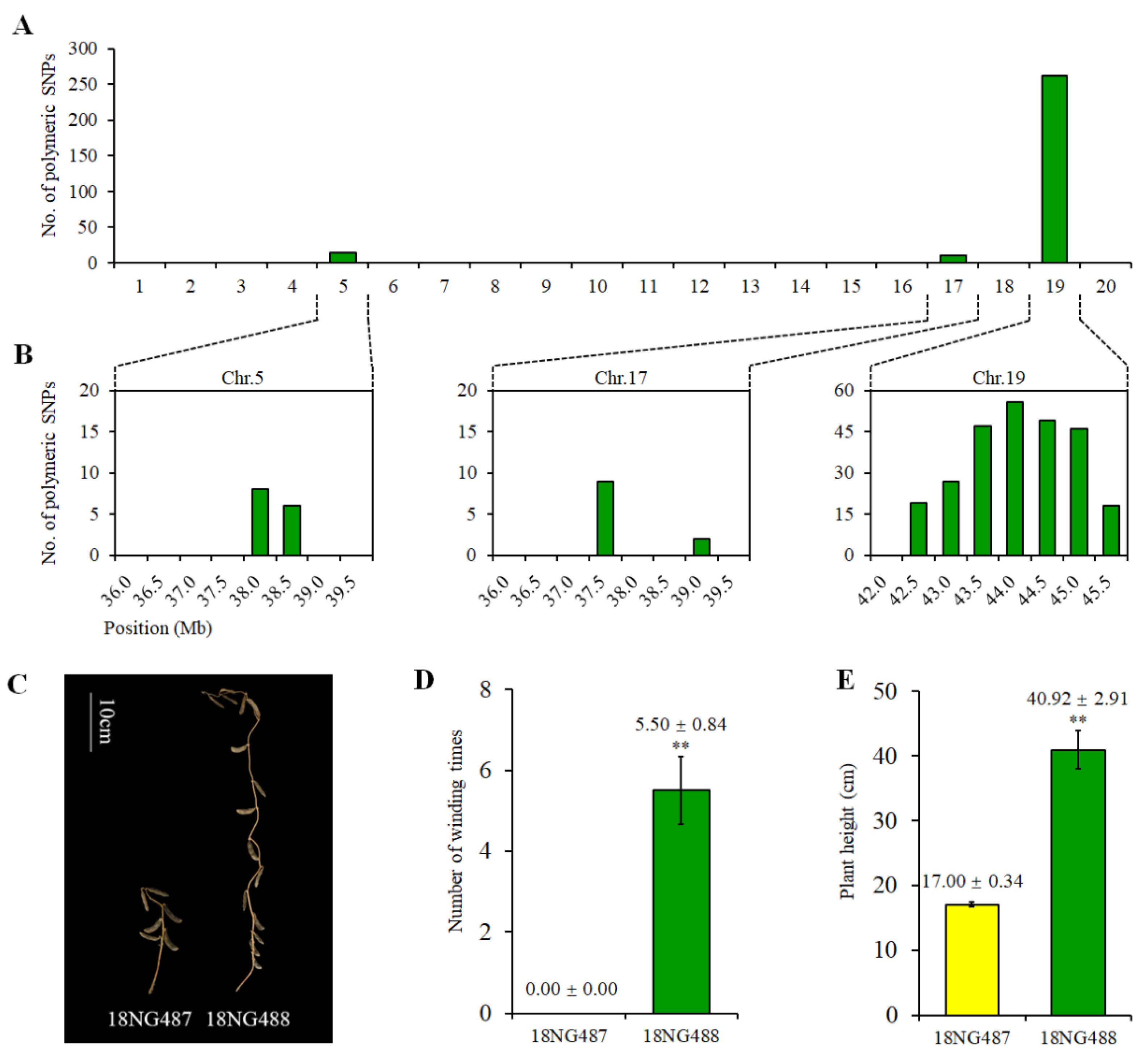
2.5. Genetic and Phenotypic Variation of Near Isogenic Lines (NILs)
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Phenotype Investigation
4.2. Genotyping DNA Bulks and Individuals with Extreme Phenotypes by SNP Array
4.3. Genotyping RIZN38 Individuals with SSR Markers
4.4. Development of Near Isogenic Lines (NILs)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, X.Y.; Jiang, J.H.; Liu, Y.; Yu, L.L.; Chang, R.Z.; Guan, R.X.; Qiu, L.J. Identification of a novel salt tolerance-related locus in wild soybean (Glycine soja Sieb. & Zucc.). Front. Plant Sci. 2021, 12, 791175. [Google Scholar]
- Jin, T.; Sun, Y.Y.; Shan, Z.; He, J.B.; Wang, N.; Gai, J.Y.; Li, Y. Natural variation in the promoter of GsERD15B affects salt tolerance in soybean. Plant Biotechnol. J. 2021, 19, 1155–1169. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, J.M.; Jung, J.; Shin, I.; Park, S.; Lee, J.S.; Jeong, S.C.; Lee, J.D.; Jung, J.K.; Ha, B.K.; et al. Fine-mapping and candidate gene analysis for the foxglove aphid resistance gene Raso2 from wild soybean PI 366121. Theor. Appl. Genet. 2021, 134, 2687–2698. [Google Scholar] [CrossRef]
- Patil, G.; Vuong, T.D.; Kale, S.; Valliyodan, B.; Deshmukh, R.; Zhu, C.S.; Wu, X.L.; Bai, Y.H.; Yungbluth, D.; Lu, F.; et al. Dissecting genomic hotspots underlying seed protein, oil, and sucrose content in an interspecific mapping population of soybean using high-density linkage mapping. Plant Biotechnol. J. 2018, 16, 1939–1953. [Google Scholar] [CrossRef]
- Qi, X.P.; Li, M.W.; Xie, M.; Liu, X.; Ni, M.; Shao, G.H.; Song, C.; Yim, A.K.; Tao, Y.; Wong, F.L.; et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat. Commun. 2014, 5, 4340. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.Y.; Wang, W.B.; He, Q.Y.; Xiang, S.H.; Tian, D.; Zhao, T.J.; Gai, J.Y. Identifying a wild allele conferring small seed size, high protein content and low oil content using chromosome segment substitution lines in soybean. Theor. Appl. Genet. 2019, 132, 2793–2807. [Google Scholar] [CrossRef]
- Carpenter, J.A.; Fehr, W.R. Genetic variability for desirable agronomic traits in populations containing Glycine soja germplasm. Crop Sci. 1986, 26, 681–686. [Google Scholar] [CrossRef]
- Wang, D.; Graef, G.L.; Procopiuk, A.M.; Diers, B.W. Identification of putative QTL that underlie yield in interspecific soybean backcross populations. Theor. Appl. Genet. 2004, 108, 458–467. [Google Scholar] [CrossRef]
- Liu, B.H.; Fujita, T.; Yan, Z.H.; Sakamoto, S.; Xu, D.H.; Abe, J. QTL mapping of domestication-related traits in soybean (Glycine max). Ann. Bot. 2007, 100, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Swarm, S.A.; Sun, L.J.; Wang, X.T.; Wang, W.D.; Brown, P.J.; Ma, J.X.; Nelson, R.L. Genetic dissection of domestication-related traits in soybean through genotyping-by-sequencing of two interspecific mapping populations. Theor. Appl. Genet. 2019, 132, 1195–1209. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kaga, A.; Tomooka, N.; Yano, H.; Takada, Y.; Kato, S.; Vaughan, D. QTL affecting fitness of hybrids between wild and cultivated soybeans in experimental fields. Ecol. Evol. 2013, 3, 2150–2168. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.K.; Liu, L.; Kong, J.J.; Xu, Z.Y.; Akhter, B.J.; Zhao, T.J. QTL architecture of vine growth habit and gibberellin oxidase gene diversity in wild soybean (Glycine soja). Sci. Rep. 2019, 9, 7393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, M.W.; Wong, F.L.; Luk, C.Y.; Chung, C.Y.; Yung, W.S.; Wang, Z.L.; Xie, M.; Song, S.K.; Chung, G.; et al. Increased copy number of gibberellin 2-oxidase 8 genes reduced trailing growth and shoot length during soybean domestication. Plant J. 2021, 107, 1739–1755. [Google Scholar] [CrossRef] [PubMed]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, G.M.; Kong, K.K.; Sharmin, R.A.; Kong, J.J.; Bhat, J.A.; Zhao, T.J. Characterization and rapid gene-mapping of leaf lesion mimic phenotype of spl-1 mutant in soybean (Glycine max (L.) Merr.). Int. J. Mol. Sci. 2019, 20, 2193. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Li, Z.; Liu, Z.X.; Guo, Y.; Qiu, L.J. Next-generation sequencing from bulked-segregant analysis accelerates the aimultaneous identification of two qualitative genes in soybean. Front. Plant Sci. 2017, 8, 919. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Li, R.C.; Jiang, H.W.; Zhang, Z.G.; Zhao, Y.Y.; Xie, J.G.; Wang, Q.; Zheng, H.Y.; Hou, L.L.; Xiong, X.; Xin, D.W.; et al. Combined linkage mapping and BSA to identify QTL and candidate genes for plant height and the number of nodes on the main stem in soybean. Int. J. Mol. Sci. 2019, 21, 42. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.T.; Wu, X.Y.; Wu, W.P.; Wu, M.; Chen, J.Q.; Wang, B. A bean common mosaic virus-resistance gene in the soybean variant V94-5152 was mapped to the Rsv4 locus conferring resistance to soybean mosaic virus. Theor. Appl. Genet. 2021, 134, 2367–2377. [Google Scholar] [CrossRef]
- Nadeem, M.; Chen, A.D.; Hong, H.L.; Li, D.D.; Li, J.J.; Zhao, D.; Wang, W.; Wang, X.B.; Qiu, L.J. GmMs1 encodes a kinesin-like protein essential for male fertility in soybean (Glycine max L.). J. Integr. Plant Biol. 2021, 63, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Kumar, V.; Gill, B.S.; Rathi, P.; Shukla, S.; Singh, R.K.; Husain, S.M. Linkage mapping of Mungbean yellow mosaic India virus (MYMIV) resistance gene in soybean. Breed Sci. 2017, 67, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, S.; Sl, K.; Kumar, V.; Singh, B.; Rao, A.R.; Mithra, S.A.; Rai, V.; Singh, A.K.; Singh, N.K. Mapping QTLs for salt tolerance in rice (Oryza sativa L.) by bulked segregant analysis of recombinant inbred lines using 50K SNP chip. PLoS ONE 2016, 11, e0153610. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.; Wang, Q.L.; Liu, S.J.; Huang, S.; Mu, J.M.; Zeng, Q.D.; Huang, L.L.; Han, D.J.; Kang, Z.S. Saturation mapping of a major effect QTL for stripe rust resistance on wheat chromosome 2B in cultivar Napo 63 using SNP genotyping arrays. Front. Plant Sci. 2017, 8, 653. [Google Scholar] [CrossRef] [Green Version]
- Broich, S.L.; Palmer, R.G. A cluster analysis of wild and domesticated soybean phenotypes. Euphytica 1980, 29, 23–32. [Google Scholar] [CrossRef]
- Broich, S.L.; Palmer, R.G. Evolutionary studies of the soybean: The frequency and distribution of alleles among collections of Glycine max and G. soja of various origin. Euphytica 1981, 30, 55–64. [Google Scholar] [CrossRef]
- Wu, J.H.; Wang, Q.L.; Xu, L.S.; Chen, X.M.; Li, B.; Mu, J.M.; Zeng, Q.D.; Huang, L.L.; Han, D.J.; Kang, Z.S. Combining single nucleotide polymorphism genotyping array with bulked segregant analysis to map a gene controlling adult plant resistance to stripe rust in wheat line 03031-1-5 H62. Phytopathology 2018, 108, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Magwene, P.M.; Willis, J.H.; Kelly, J.K. The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput. Biol. 2011, 7, e1002255. [Google Scholar] [CrossRef] [Green Version]
- Briggs, W.H.; Mcmullen, M.D.; Gaut, B.S.; Doebley, J. Linkage mapping of domestication loci in a large maize teosinte backcross resource. Genetics 2007, 177, 1915–1928. [Google Scholar] [CrossRef] [Green Version]
- Doebley, J.F.; Stec, A. Inheritance of the morphological differences between maize and teosinte: Comparison of results for two F2 populations. Genetics 1993, 134, 559–570. [Google Scholar] [CrossRef]
- Koinange, E.M.; Singh, S.P.; Gepts, P. Genetic control of the domestication syndrome in common bean. Crop Sci. 1996, 36, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Komatsuda, T.; Pourkheirandish, M.; He, C.F.; Azhaguvel, P.; Kanamori, H.; Perovic, D.; Stein, N.; Graner, A.; Wicker, T.; Tagiri, A.; et al. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 2007, 104, 1424–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, D.M.; Whipple, C.J.; Takuno, S.; Kursel, L.E.; Doebley, J.F. From many, one: Genetic control of prolificacy during maize domestication. PLoS Genet. 2013, 9, e1003604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Z.X.; Wang, X.B.; Lee, R.; Li, Y.H.; Specht, J.E.; Nelson, R.L.; McClean, P.E.; Qiu, L.J.; Ma, J.X. Artificial selection for determinate growth habit in soybean. Proc. Natl. Acad. Sci. USA 2010, 107, 8563–8568. [Google Scholar] [CrossRef] [Green Version]
- Bernard, R.L. Two genes affecting stem termination in soybean. Crop Sci 1972, 12, 235–239. [Google Scholar] [CrossRef]
- Studer, A.J.; Doebley, J.F. Do large effect QTL fractionate? A case study at the maize domestication QTL teosinte branched1. Genetics 2011, 188, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, Z.H.; Doebley, J.F. Genetic dissection of a genomic region with pleiotropic effects on domestication traits in maize reveals multiple linked QTL. Genetics 2014, 198, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Repinski, S.L.; Kwak, M.; Gepts, P. The common bean growth habit gene PvTFL1y is a functional homolog of Arabidopsis TFL1. Theor. Appl. Genet. 2012, 124, 1539–1547. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F. Harnessing crop wild diversity for climate change adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef]
- Cortés, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern strategies to assess and breed forest tree adaptation to changing climate. Front. Plant Sci. 2020, 11, 583323. [Google Scholar] [CrossRef]
- Shi, A.N.; Gepts, P.; Song, Q.J.; Xiong, H.Z.; Michaels, T.E.; Chen, S.Y. Genome-wide association study and genomic prediction for soybean cyst nematode resistance in USDA common bean (Phaseolus vulgaris) core collection. Front. Plant Sci. 2021, 12, 624156. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Bohra, A.; Roorkiwal, M.; Barmukh, R.; Cowling, W.A.; Chitikineni, A.; Lam, H.M.; Hickey, L.T.; Croser, J.S.; Bayer, P.E.; et al. Fast-forward breeding for a food-secure world. Trends Genet. 2021, 37, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Song, X.G.; Meng, X.B.; Guo, H.Y.; Cheng, Q.; Jing, Y.H.; Chen, M.J.; Liu, G.F.; Wang, B.; Wang, Y.H.; Li, J.Y.; et al. Targeting a gene regulatory element enhances rice grain yield by decoupling panicle number and size. Nat. Biotechnol. 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, T.; Meng, X.B.; Du, H.L.; Zhang, J.K.; Liu, G.F.; Chen, M.J.; Jing, Y.H.; Kou, L.Q.; Li, X.X.; et al. A route de novo domestication of wild allotetraploid rice. Cell 2021, 184, 1156–1170. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.J.; Sun, B.C.; Tian, Y.; Su, S.S.; Zhang, Y.; Zhang, W.H.; Wang, J.S.; Yu, P.; Guo, B.F.; Li, H.H.; et al. Dissection the practical soybean breeding pipeline by developing high throughput functional array ZDX1. Theor. Appl. Genet. 2022, 135, 1413–1427. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.H.; Zhang, L.Y.; Wang, J.K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Li, H.H.; Ye, G.Y.; Wang, J.K. A modified algorithm for the improvement of composite interval mapping. Genetics 2007, 175, 361–374. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | QTLs | Chr. | Marker Interval | Position (Mb) | LOD | PVE (%) | ADD |
|---|---|---|---|---|---|---|---|
| 2017 | qVG-2 | 2 | SSR_2_1540-SSR_2_1602 | 43.3–44.3 | 3.34 | 5.80 | −0.73 |
| qVG-17 | 17 | SSR_2_661-SSR_2_669 | 11.3–11.6 | 5.56 | 9.81 | 0.96 | |
| qVG-19 | 19 | SSR_19_1323-SSR_19_1335 | 45.1–45.3 | 7.69 | 15.30 | −1.12 | |
| 2018 | qVG-2 | 2 | SSR_2_1540-SSR_2_1602 | 43.3–44.3 | 5.31 | 9.84 | −1.54 |
| qVG-17 | 17 | SSR_2_661-SSR_2_669 | 11.3–11.6 | 2.63 | 4.01 | 1.00 | |
| qVG-19 | 19 | SSR_19_1323-SSR_19_1335 | 45.1–45.3 | 9.44 | 15.66 | −2.05 | |
| 2017 | qPH-2 | 2 | SSR_2_1540-SSR_2_1602 | 43.2–45.5 | 2.57 | 3.96 | −2.99 |
| qPH-17 | 17 | SSR_2_661-SSR_2_669 | 11.3–11.6 | 8.90 | 14.61 | 5.85 | |
| qPH-19 | 19 | SSR_19_1323-SSR_19_1335 | 45.1–45.3 | 10.13 | 17.43 | −6.54 | |
| 2018 | qPH-2 | 2 | SSR_2_1540-SSR_2_1602 | 43.3–44.3 | 3.82 | 6.33 | −3.78 |
| qPH-17 | 17 | SSR_2_661-SSR_2_669 | 11.3–11.6 | 5.95 | 8.52 | 4.45 | |
| qPH-19 | 19 | SSR_19_1323-SSR_19_1335 | 45.1–45.3 | 13.93 | 22.01 | −7.41 |
| Method | QTL | Chr. | Marker Interval | Position (Mb) | LOD | PVE (%) | ADD |
|---|---|---|---|---|---|---|---|
| SNP array | qVG-19.1 | 19 | Gm44946-Gm45005 | 44.7–45.4 | 8.79 | 56.04 | −4.38 |
| qPH-19.1 | 19 | Gm44946-Gm45005 | 44.7–45.4 | 7.85 | 52.26 | 15.23 | |
| BSA | qVG-19.1 | 19 | SSR_19_1323-SSR_19_1335 | 45.1–45.3 | 3.00 | 6.14 | −0.99 |
| qPH-19.1 | 19 | SSR_19_1323-SSR_19_1335 | 45.1–45.3 | 2.97 | 6.07 | −4.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Zhang, J.; Guo, X.; Chen, J.; Chang, R.; Guan, R.; Qiu, L. Identification of Genomic Regions Associated with Vine Growth and Plant Height of Soybean. Int. J. Mol. Sci. 2022, 23, 5823. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105823
Lu Y, Zhang J, Guo X, Chen J, Chang R, Guan R, Qiu L. Identification of Genomic Regions Associated with Vine Growth and Plant Height of Soybean. International Journal of Molecular Sciences. 2022; 23(10):5823. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105823
Chicago/Turabian StyleLu, Yipeng, Jiaming Zhang, Xiaoyang Guo, Jingjing Chen, Ruzhen Chang, Rongxia Guan, and Lijuan Qiu. 2022. "Identification of Genomic Regions Associated with Vine Growth and Plant Height of Soybean" International Journal of Molecular Sciences 23, no. 10: 5823. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105823