
The Role of IgG Fc Region N-Glycosylation in the Pathomechanism of Rheumatoid Arthritis

 , ,
, ,  and
and
Abstract
:1. Introduction
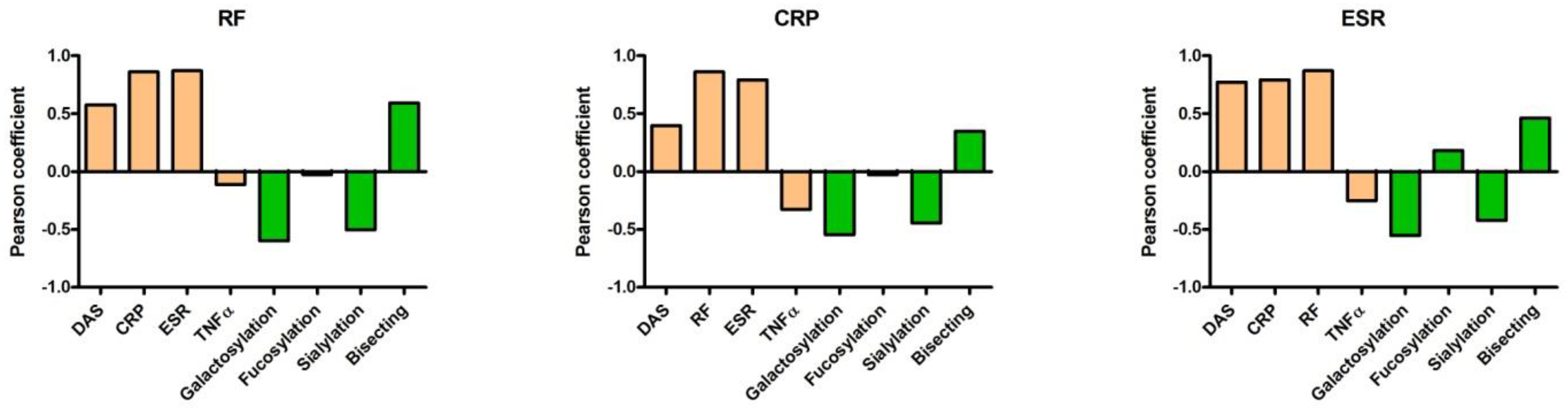
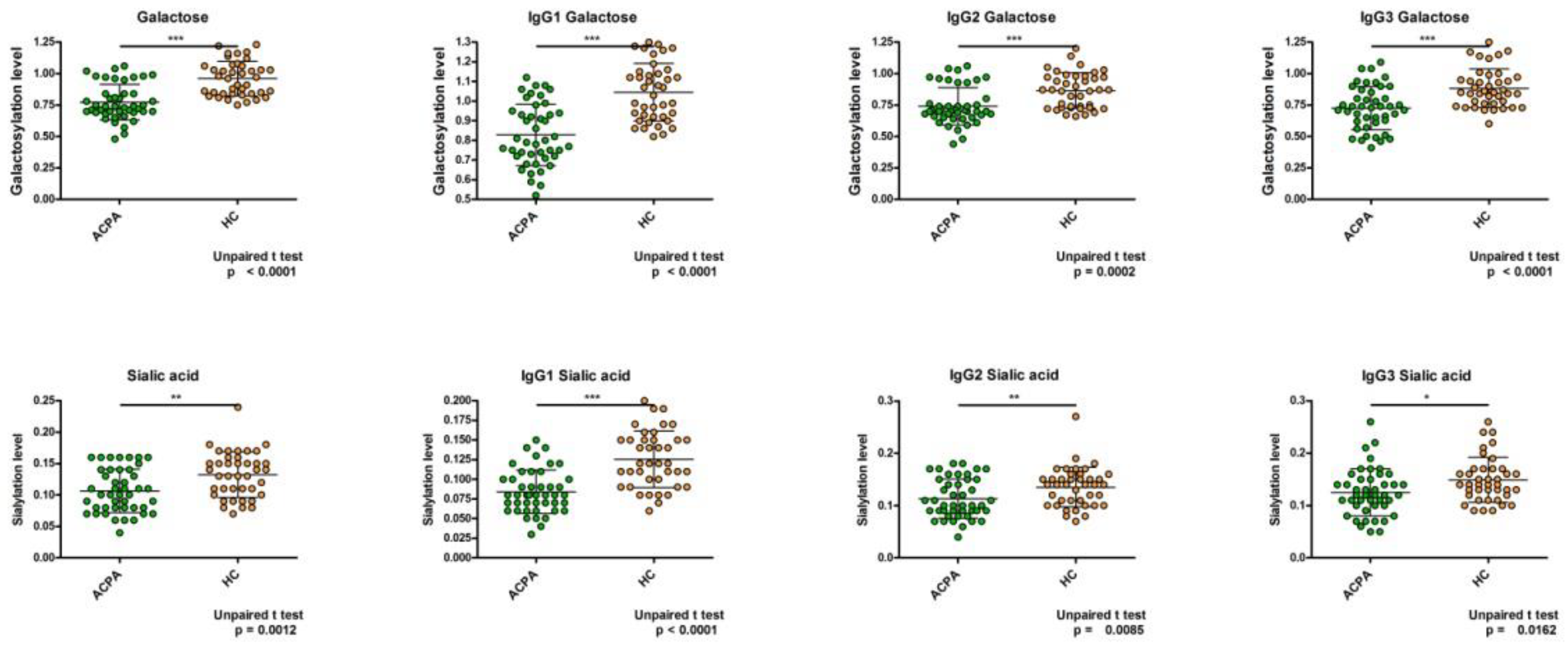
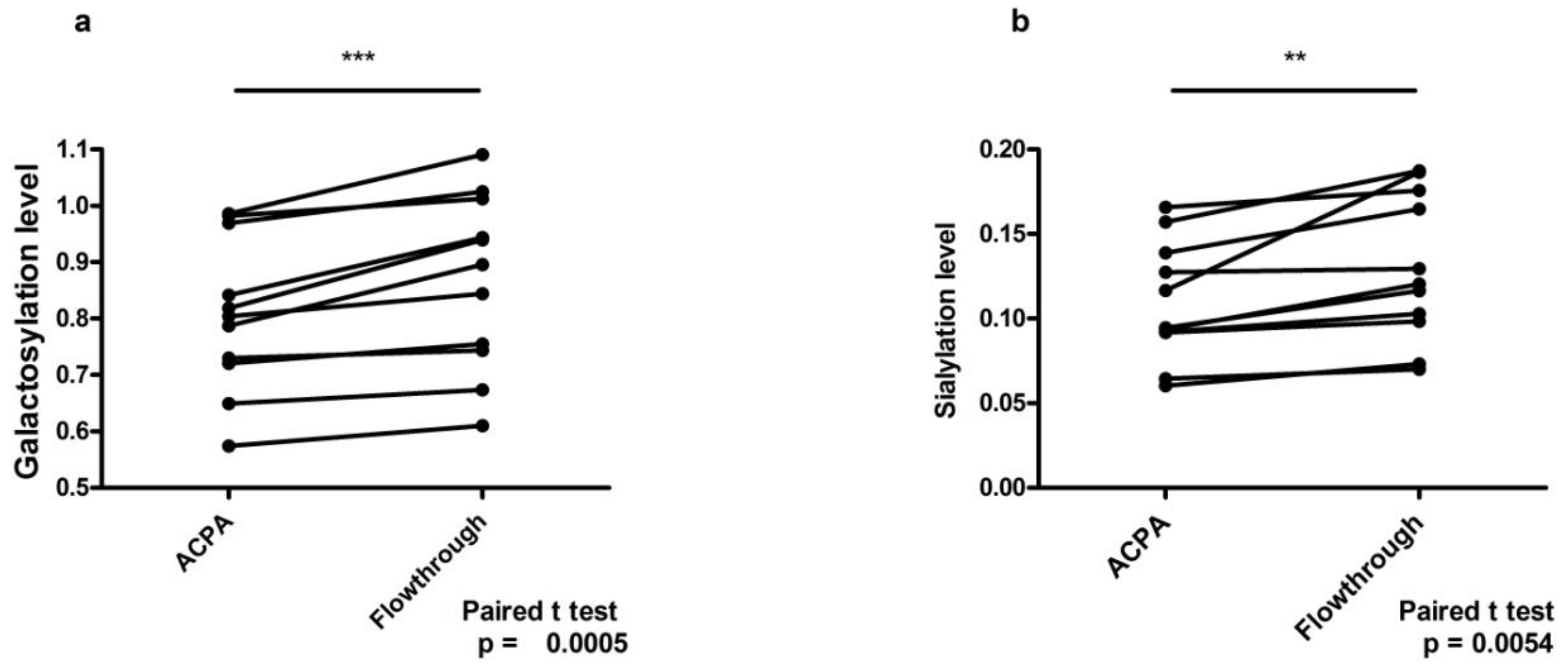
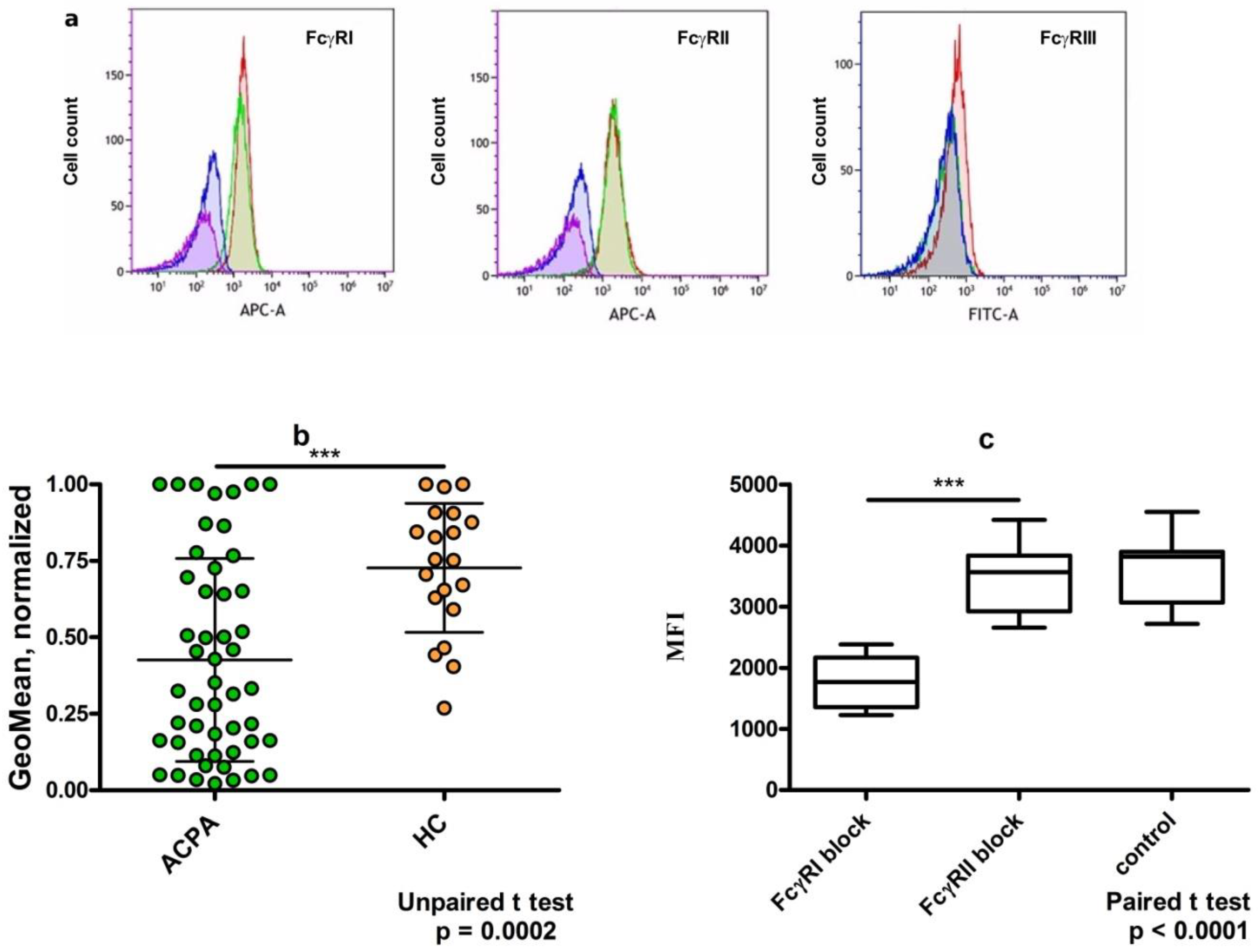
2. Results
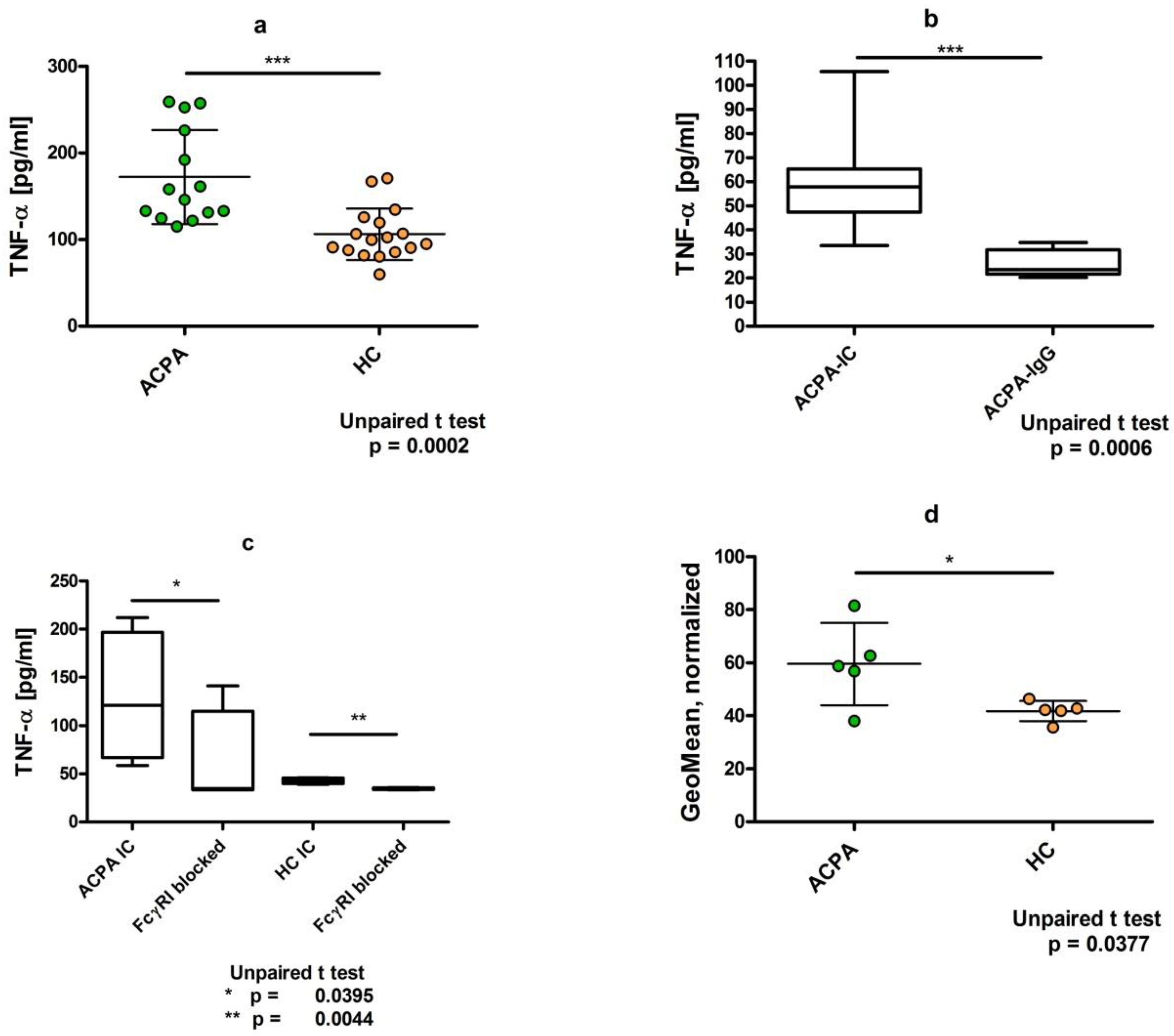
3. Dimeric Immune Complexes of ACPA-IgG and Control IgG Bind to FcγRI on U937 Cells
4. Dimeric ACPA Immune Complexes Induce Significantly Higher TNFα Release from U937 Cells Compared to Complexes Containing IgG from Healthy Individuals
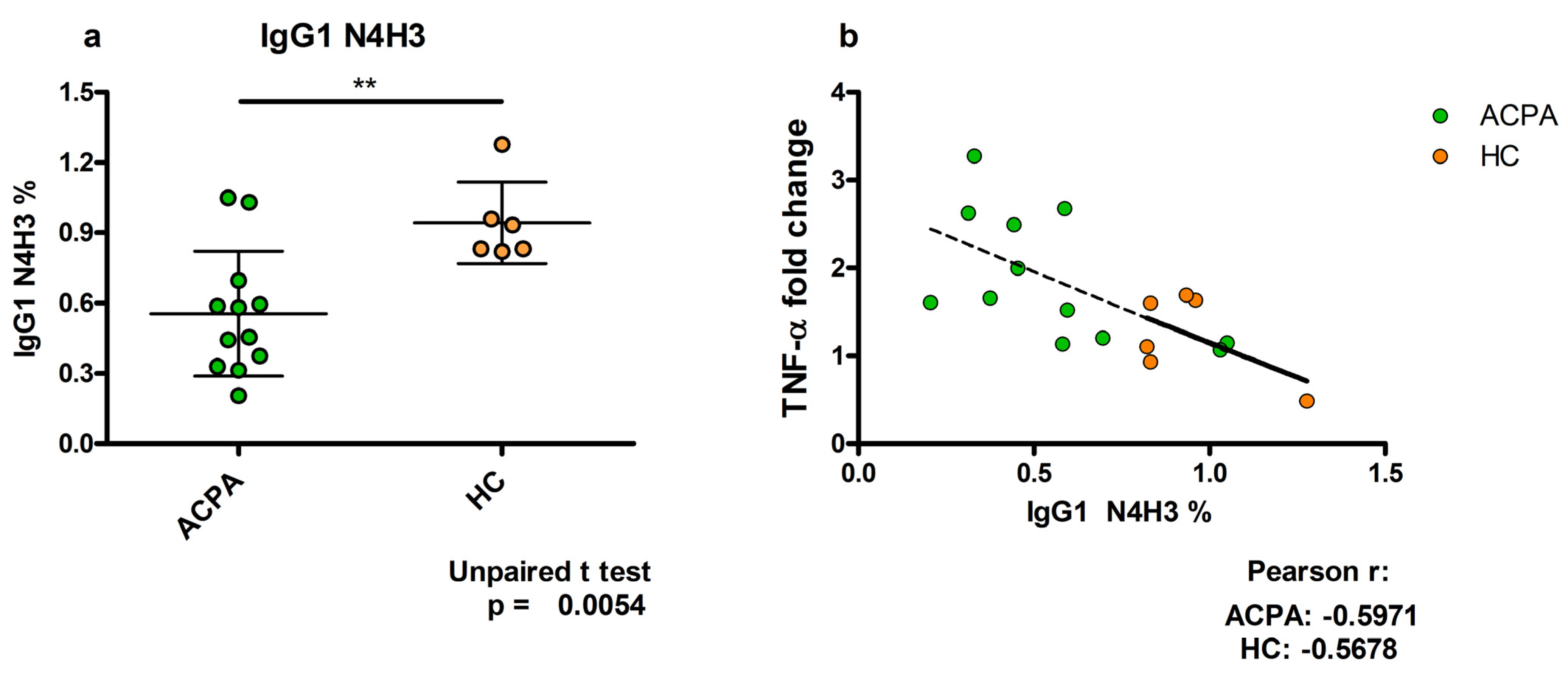
5. TNFα Production Induced by ACPA-Containing Immune Complexes Shows a Negative Correlation with the Relative Percentage of N4H3 Glycan of IgG Fc
6. Discussion
7. Conclusions
8. Materials and Methods
8.1. Blood Samples
8.2. Serum Preparation
8.3. Determination of Anti-Citrullinated Protein Antibodies (ACPA) in Sera by ELISA
8.4. Isolation of IgG
8.5. ACPA Isolation
8.6. Preparation of Immune Complexes
8.7. Immune Complex Binding Assay
8.8. Intracellular Staining to Monitor Inflammatory Cytokine TNFα Production
8.9. TNFα Detection in the Supernatant of IC-Treated U937 Cells
8.10. Glycosylation Analysis
8.11. Statistical Analysis
8.12. Further Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACPA | anti-citrullinated protein antibodies |
| CCP | cyclic citrullinated peptides |
| CRP | C-reactive protein |
| DAS | disease activity score |
| ESR | erythrocyte sedimentation rate |
| FcγR | IgG Fc receptor |
| GlcNAc | N-acetyl glucosamine |
| IC | immune complexes |
| PMA | phorbol myristate acetate |
| RA | rheumatoid arthritis |
| RF | rheuma factor |
| SDS-PAGE | sodium dodecyl sulphate polyacrylamide gel electrophoresis |
| TNFα | tumor necrosis factor α |
| Tfh | follicular helper T cells |
References
- Sparks, J.A. Rheumatoid Arthritis. Ann. Intern. Med. 2019, 170, ITC1–ITC16. [Google Scholar] [CrossRef] [PubMed]
- Derksen, V.; Huizinga, T.W.J.; van der Woude, D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 437–446. [Google Scholar] [CrossRef] [PubMed]
- van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H.U.; Haupl, T.; Burmester, G.R. The etiology of rheumatoid arthritis. J. Autoimmun. 2020, 110, 102400. [Google Scholar] [CrossRef]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008, 58, 678–688. [Google Scholar] [CrossRef]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011, 63, 53–62. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Toes, R.; Pisetsky, D.S. Pathogenic effector functions of ACPA: Where do we stand? Ann. Rheum. Dis. 2019, 78, 716–721. [Google Scholar] [CrossRef] [Green Version]
- Catrina, A.; Krishnamurthy, A.; Rethi, B. Current view on the pathogenic role of anti-citrullinated protein antibodies in rheumatoid arthritis. RMD Open 2021, 7, e001228. [Google Scholar] [CrossRef]
- Laurent, L.; Anquetil, F.; Clavel, C.; Ndongo-Thiam, N.; Offer, G.; Miossec, P.; Pasquali, J.L.; Sebbag, M.; Serre, G. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies. Ann. Rheum. Dis. 2015, 74, 1425–1431. [Google Scholar] [CrossRef]
- Dai, X.; Jayapal, M.; Tay, H.K.; Reghunathan, R.; Lin, G.; Too, C.T.; Lim, Y.T.; Chan, S.H.; Kemeny, D.M.; Floto, R.A.; et al. Differential signal transduction, membrane trafficking, and immune effector functions mediated by FcgammaRI versus FcgammaRIIa. Blood 2009, 114, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempers, A.C.; Nejadnik, M.R.; Rombouts, Y.; Ioan-Facsinay, A.; van Oosterhout, M.; Jiskoot, W.; Huizinga, T.W.J.; Toes, R.E.M.; Scherer, H.U. Fc gamma receptor binding profile of anti-citrullinated protein antibodies in immune complexes suggests a role for FcgammaRI in the pathogenesis of synovial inflammation. Clin. Exp. Rheumatol. 2018, 36, 284–293. [Google Scholar] [PubMed]
- Kao, D.; Danzer, H.; Collin, M.; Gross, A.; Eichler, J.; Stambuk, J.; Lauc, G.; Lux, A.; Nimmerjahn, F. A Monosaccharide Residue Is Sufficient to Maintain Mouse and Human IgG Subclass Activity and Directs IgG Effector Functions to Cellular Fc Receptors. Cell Rep. 2015, 13, 2376–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lux, A.; Yu, X.; Scanlan, C.N.; Nimmerjahn, F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J. Immunol. 2013, 190, 4315–4323. [Google Scholar] [CrossRef] [Green Version]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: A critical review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cobb, B.A. The history of IgG glycosylation and where we are now. Glycobiology 2020, 30, 202–213. [Google Scholar] [CrossRef]
- Parekh, R.B.; Dwek, R.A.; Sutton, B.J.; Fernandes, D.L.; Leung, A.; Stanworth, D.; Rademacher, T.W.; Mizuochi, T.; Taniguchi, T.; Matsuta, K.; et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 1985, 316, 452–457. [Google Scholar] [CrossRef]
- Zhou, X.; Motta, F.; Selmi, C.; Ridgway, W.M.; Gershwin, M.E.; Zhang, W. Antibody glycosylation in autoimmune diseases. Autoimmun. Rev. 2021, 20, 102804. [Google Scholar] [CrossRef]
- Rombouts, Y.; Ewing, E.; van de Stadt, L.A.; Selman, M.H.; Trouw, L.A.; Deelder, A.M.; Huizinga, T.W.; Wuhrer, M.; van Schaardenburg, D.; Toes, R.E.; et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 234–241. [Google Scholar] [CrossRef]
- Su, Z.; Xie, Q.; Wang, Y.; Li, Y. Abberant Immunoglobulin G Glycosylation in Rheumatoid Arthritis by LTQ-ESI-MS. Int. J. Mol. Sci. 2020, 21, 2045. [Google Scholar] [CrossRef] [Green Version]
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H.U.; Culemann, S.; Harre, U.; Ackermann, J.A.; Seefried, M.; Kleyer, A.; Uderhardt, S.; et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat. Immunol. 2017, 18, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F.; Miossec, P. Evolving concepts of the pathogenesis of rheumatoid arthritis with focus on the early and late stages. Curr. Opin. Rheumatol. 2020, 32, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Seeling, M.; Bruckner, C.; Nimmerjahn, F. Differential antibody glycosylation in autoimmunity: Sweet biomarker or modulator of disease activity? Nat. Rev. Rheumatol. 2017, 13, 621–630. [Google Scholar] [CrossRef]
- Vuckovic, F.; Kristic, J.; Gudelj, I.; Teruel, M.; Keser, T.; Pezer, M.; Pucic-Bakovic, M.; Stambuk, J.; Trbojevic-Akmacic, I.; Barrios, C.; et al. Association of systemic lupus erythematosus with decreased immunosuppressive potential of the IgG glycome. Arthritis Rheumatol. 2015, 67, 2978–2989. [Google Scholar] [CrossRef] [PubMed]
- Biermann, M.H.; Griffante, G.; Podolska, M.J.; Boeltz, S.; Sturmer, J.; Munoz, L.E.; Bilyy, R.; Herrmann, M. Sweet but dangerous—The role of immunoglobulin G glycosylation in autoimmunity and inflammation. Lupus 2016, 25, 934–942. [Google Scholar] [CrossRef]
- Simurina, M.; de Haan, N.; Vuckovic, F.; Kennedy, N.A.; Stambuk, J.; Falck, D.; Trbojevic-Akmacic, I.; Clerc, F.; Razdorov, G.; Khon, A.; et al. Glycosylation of Immunoglobulin G Associates With Clinical Features of Inflammatory Bowel Diseases. Gastroenterology 2018, 154, 1320–1333.e10. [Google Scholar] [CrossRef]
- Li, T.; DiLillo, D.J.; Bournazos, S.; Giddens, J.P.; Ravetch, J.V.; Wang, L.X. Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. USA 2017, 114, 3485–3490. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.A.; Giddens, J.; Pincetic, A.; Lomino, J.V.; Ravetch, J.V.; Wang, L.X.; Bjorkman, P.J. Structural characterization of anti-inflammatory immunoglobulin G Fc proteins. J. Mol. Biol. 2014, 426, 3166–3179. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, P.; Pincetic, A.; Maamary, J.; Lammens, K.; Ravetch, J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. USA 2013, 110, 9868–9872. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Baruah, K.; Harvey, D.J.; Vasiljevic, S.; Alonzi, D.S.; Song, B.D.; Higgins, M.K.; Bowden, T.A.; Scanlan, C.N.; Crispin, M. Engineering hydrophobic protein-carbohydrate interactions to fine-tune monoclonal antibodies. J. Am. Chem. Soc. 2013, 135, 9723–9732. [Google Scholar] [CrossRef]
- Subedi, G.P.; Barb, A.W. The immunoglobulin G1 N-glycan composition affects binding to each low affinity Fc gamma receptor. mAbs 2016, 8, 1512–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szarka, E.; Aradi, P.; Huber, K.; Pozsgay, J.; Vegh, L.; Magyar, A.; Gyulai, G.; Nagy, G.; Rojkovich, B.; Kiss, E.; et al. Affinity Purification and Comparative Biosensor Analysis of Citrulline-Peptide-Specific Antibodies in Rheumatoid Arthritis. Int. J. Mol. Sci. 2018, 19, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, C.; Grau, S.; Jager, C.; Sondermann, P.; Brunker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Sun, Z.; Zhang, L.; Cai, Y.; Peng, Y.; Cao, T.; Zhang, Y.; Lu, H. Chemical labeling for fine mapping of IgG N-glycosylation by ETD-MS. Chem. Sci. 2019, 10, 9302–9307. [Google Scholar] [CrossRef]
- Wang, T.T. IgG Fc Glycosylation in Human Immunity. Curr. Top. Microbiol. Immunol. 2019, 423, 63–75. [Google Scholar] [CrossRef]
- Sokolove, J.; Johnson, D.S.; Lahey, L.J.; Wagner, C.A.; Cheng, D.; Thiele, G.M.; Michaud, K.; Sayles, H.; Reimold, A.M.; Caplan, L.; et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody-mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 813–821. [Google Scholar] [CrossRef]
- Hodoniczky, J.; Zheng, Y.Z.; James, D.C. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol. Prog. 2005, 21, 1644–1652. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Yeh, Y.C.; Yang, K.D. Functions and therapeutic targets of Siglec-mediated infections, inflammations and cancers. J. Formos. Med. Assoc. Taiwan Yi Zhi 2021, 120, 5–24. [Google Scholar] [CrossRef]
- Ulyanova, T.; Blasioli, J.; Woodford-Thomas, T.A.; Thomas, M.L. The sialoadhesin CD33 is a myeloid-specific inhibitory receptor. Eur. J. Immunol. 1999, 29, 3440–3449. [Google Scholar] [CrossRef]
- Bordron, A.; Morel, M.; Bagacean, C.; Dueymes, M.; Pochard, P.; Harduin-Lepers, A.; Jamin, C.; Pers, J.O. Hyposialylation Must Be Considered to Develop Future Therapies in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 3402. [Google Scholar] [CrossRef] [PubMed]
- Bondt, A.; Hafkenscheid, L.; Falck, D.; Kuijper, T.M.; Rombouts, Y.; Hazes, J.M.W.; Wuhrer, M.; Dolhain, R. ACPA IgG galactosylation associates with disease activity in pregnant patients with rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Bondt, A.; Selman, M.H.; Deelder, A.M.; Hazes, J.M.; Willemsen, S.P.; Wuhrer, M.; Dolhain, R.J. Association between galactosylation of immunoglobulin G and improvement of rheumatoid arthritis during pregnancy is independent of sialylation. J. Proteome Res. 2013, 12, 4522–4531. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Pozsgay, J.; Szarka, E.; Huber, K.; Babos, F.; Magyar, A.; Hudecz, F.; Sarmay, G. Synthetic Peptide-Based ELISA and ELISpot Assay for Identifying Autoantibody Epitopes. Methods Mol. Biol. 2016, 1352, 223–233. [Google Scholar] [CrossRef]
- Szarka, E.; Babos, F.; Magyar, A.; Huber, K.; Szittner, Z.; Papp, K.; Prechl, J.; Pozsgay, J.; Neer, Z.; Adori, M.; et al. Recognition of new citrulline-containing peptide epitopes by autoantibodies produced in vivo and in vitro by B cells of rheumatoid arthritis patients. Immunology 2014, 141, 181–191. [Google Scholar] [CrossRef]
- Turiak, L.; Ozohanics, O.; Marino, F.; Drahos, L.; Vekey, K. Digestion protocol for small protein amounts for nano-HPLC-MS(MS) analysis. J. Proteom. 2011, 74, 942–947. [Google Scholar] [CrossRef]
- Ozohanics, O.; Turiak, L.; Puerta, A.; Vekey, K.; Drahos, L. High-performance liquid chromatography coupled to mass spectrometry methodology for analyzing site-specific N-glycosylation patterns. J. Chromatogr. A 2012, 1259, 200–212. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic and Clinical Parameters of Patients | Citrulline Peptide Specificities | |||||||
|---|---|---|---|---|---|---|---|---|
| Age (Years) | DAS | RF IU/mL | CCP IU/mL | CRP mg/mL | ESR mm/h | Multi-Pitope | Collagen | Filaggrin |
| 5.86 | 7.8 | 8.53 | ||||||
| 73 | 2.10 | 93.7 | 871 | 12.6 | 12 | 4.21 | 6.12 | 2.03 |
| 47 | 5.07 | 55 | 3200 | 0.67 | 20 | 1.45 | 1.27 | 4.22 |
| 72 | 7.20 | 1695 | 2424 | 34 | 95 | 2.48 | 1.48 | 3.24 |
| 65 | 3.70 | 53 | 3200 | 3 | 8.81 | 2.4 | 3.74 | |
| 67 | 6.90 | 105 | 8.6 | 48 | 1.85 | 1.15 | 3.3 | |
| 72 | 3.80 | 392 | 906 | 13.1 | 17 | 1.96 | 1.97 | 3.62 |
| 65 | 3.30 | 16.3 | 510 | 1.82 | 7 | 0.6 | 4.58 | 4.41 |
| 54 | 4.00 | 74 | 3090 | 5.9 | 14 | 4.42 | 5.87 | 4.29 |
| 65 | 3.70 | 53 | 2.2 | 12 | 1.98 | 5.87 | 4.29 | |
| 67 | 5.20 | 63 | 653 | 0.3 | 38 | 1.05 | 1.39 | 1.91 |
| 57 | 2.10 | 34 | 3200 | 1.5 | 3 | 8.53 | 4.21 | 10.18 |
| 66 | 3.80 | 70 | 8.4 | 19 | 0.99 | 1.13 | 1.24 | |
| 53 | 4.40 | 346 | 50 | 2.5 | 23 | 1.85 | 0.97 | 0.65 |
| 73 | 5.20 | 974 | 700 | 30 | 89 | 2.84 | 20.67 | 16.1 |
| 44 | 3.10 | 51.8 | 281 | 16.88 | 14 | 2.32 | 4.07 | 1.41 |
| 66 | 5.10 | 262 | 1633 | 10.5 | 24 | 4.38 | 5.67 | 3.19 |
| Synthetic Citrulline-Containing Peptides | Sequences (X Stands for Citrulline) * |
|---|---|
| Filaggrin19 (306–326) | Ac-SHQESTXGXSXGRSGRSGSK-NH2 |
| Collagen (359–369) | Ac-AXGLTGXPGDA-NH2 |
| Multi-Epitope | H-Ttds **-AXAXGSGSGXGXG-NH2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyebrovszki, B.; Ács, A.; Szabó, D.; Auer, F.; Novozánszki, S.; Rojkovich, B.; Magyar, A.; Hudecz, F.; Vékey, K.; Drahos, L.; et al. The Role of IgG Fc Region N-Glycosylation in the Pathomechanism of Rheumatoid Arthritis. Int. J. Mol. Sci. 2022, 23, 5828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105828
Gyebrovszki B, Ács A, Szabó D, Auer F, Novozánszki S, Rojkovich B, Magyar A, Hudecz F, Vékey K, Drahos L, et al. The Role of IgG Fc Region N-Glycosylation in the Pathomechanism of Rheumatoid Arthritis. International Journal of Molecular Sciences. 2022; 23(10):5828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105828
Chicago/Turabian StyleGyebrovszki, Balázs, András Ács, Dániel Szabó, Felícia Auer, Soma Novozánszki, Bernadette Rojkovich, Anna Magyar, Ferenc Hudecz, Károly Vékey, László Drahos, and et al. 2022. "The Role of IgG Fc Region N-Glycosylation in the Pathomechanism of Rheumatoid Arthritis" International Journal of Molecular Sciences 23, no. 10: 5828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105828