
Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism


Abstract
:1. Introduction
2. Specific Brain Regions Are Involved in Gaucher Disease
3. Neuropathological Involvement Is also Noted in “Non-Neuronopathic” GD1
4. Neuropathologic Findings in Neuronopathic GD
5. Neuropathology of Gaucher-Associated Parkinsonism
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brady, R.O.; Kanfer, J.N.; Bradley, R.M.; Shapiro, D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J. Clin. Investig. 1966, 45, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, E. Gaucher disease: Complexity in a “simple” disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Sevigny, J.; Rolfs, A.; Davies, E.H.; Goker-Alpan, O.; Abdelwahab, M.; Vellodi, A.; Mengel, E.; Lukina, E.; Yoo, H.; et al. The definition of neuronopathic Gaucher disease. J. Inherit. Metab. Dis. 2020, 43, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Mignot, C.; Doummar, D.; Maire, I.; De Villemeur, T.B. French Type 2 Gaucher Disease Study G. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006, 28, 39–48. [Google Scholar] [CrossRef]
- Roshan Lal, T.; Seehra, G.K.; Steward, A.M.; Poffenberger, C.N.; Ryan, E.; Tayebi, N.; Lopez, G.; Sidransky, E. The natural history of type 2 Gaucher disease in the 21st century: A retrospective study. Neurology 2020, 95, e2119–e2130. [Google Scholar] [CrossRef]
- Poffenberger, C.N.; Inati, S.; Tayebi, N.; Stubblefield, B.K.; Ryan, E.; Schiffmann, R.; Sidransky, E.; Lopez, G. EEG abnormalities in patients with chronic neuronopathic Gaucher disease: A retrospective review. Mol. Genet. Metab. 2020, 131, 358–363. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Wiggs, E.A.; Eblan, M.J.; Benko, W.; Ziegler, S.G.; Sidransky, E.; Schiffman, R. Cognitive outcome in treated patients with chronic neuronopathic Gaucher disease. J. Pediatr. 2008, 153, 89–94. [Google Scholar] [CrossRef]
- Benko, W.; Ries, M.; Wiggs, E.A.; Brady, R.O.; Schiffmann, R.; Fitzgibbon, E.J. The saccadic and neurological deficits in type 3 Gaucher disease. PLoS ONE 2011, 6, e22410. [Google Scholar] [CrossRef] [Green Version]
- Tayebi, N.; Walker, J.; Stubblefield, B.; Orvisky, E.; LaMarca, M.E.; Wong, K.; Rosenbaum, H.; Schiffman, R.; Bembi, B.; Sidransky, E. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 2003, 79, 104–109. [Google Scholar] [CrossRef]
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Schiffmann, R.; LaMarca, M.E.; Nussbaum, R.L.; McInerney-Leo, A.; Sidransky, E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef] [Green Version]
- Lwin, A.; Orvisky, E.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004, 81, 70–73. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Duran, R.; Lopez, G.; Kurzawa-Akanbi, M.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M.; Theuns, J.; Crosiers, D.; Cras, P.; et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013, 70, 727–735. [Google Scholar] [CrossRef]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cells Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Bultron, G.; Kacena, K.; Pearson, D.; Boxer, M.; Yang, R.; Sathe, S.; Pastores, G.; Mistry, P.K. The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Westbroek, W.; Sidransky, E. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron 2017, 93, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Soffer, D.; Yamanaka, T.; Wenger, D.A.; Suzuki, K.; Suzuki, K. Central nervous system involvement in adult-onset Gaucher’s disease. Acta Neuropathol. 1980, 49, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hulkova, H.; Ledvinova, J.; Poupetova, H.; Kohout, A.; Malinova, V.; Elleder, M. Autopsy case of Gaucher disease type I in a patient on enzyme replacement therapy. Comments on the dynamics of persistent storage process. J. Inherit. Metab. Dis. 2009, 32, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Kaye, E.M.; Ullman, M.D.; Wilson, E.R.; Barranger, J.A. Type 2 and type 3 Gaucher disease: A morphological and biochemical study. Ann. Neurol. 1986, 20, 223–230. [Google Scholar] [CrossRef]
- Lee, R.E. The pathology of Gaucher disease. Prog. Clin Biol. Res. 1982, 95, 177–217. [Google Scholar]
- Burrow, T.A.; Sun, Y.; Prada, C.E.; Bailey, L.; Zhang, W.; Brewer, A.; Wu, S.W.; Setchell, K.D.R.; Witte, D.; Cohen, M.B.; et al. CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11 years of therapy: Clinical, histopathologic, and biochemical findings. Mol. Genet. Metab. 2015, 114, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Conradi, N.; Kyllerman, M.; Mansson, J.E.; Percy, A.K.; Svennerholm, L. Late-infantile Gaucher disease in a child with myoclonus and bulbar signs: Neuropathological and neurochemical findings. Acta Neuropathol. 1991, 82, 152–157. [Google Scholar] [CrossRef]
- Kaga, K.; Ono, M.; Yakumaru, K.; Owada, M.; Mizutani, T. Brainstem pathology of infantile Gaucher’s disease with only wave I and II of auditory brainstem response. J. Laryngol. Otol. 1998, 112, 1069–1073. [Google Scholar] [CrossRef]
- Verghese, J.; Goldberg, R.F.; Desnick, R.J.; Grace, M.E.; Goldman, J.E.; Lee, S.C.; Dickson, D.W.; Rapin, I. Myoclonus from selective dentate nucleus degeneration in type 3 Gaucher disease. Arch. Neurol. 2000, 57, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Winkelman, M.D.; Banker, B.Q.; Victor, M.; Moser, H.W. Non-infantile neuronopathic Gaucher’s disease: A clinicopathologic study. Neurology 1983, 33, 994–1008. [Google Scholar] [CrossRef]
- Conradi, N.G.; Sourander, P.; Nilsson, O.; Svennerholm, L.; Erikson, A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol. 1984, 65, 99–109. [Google Scholar] [CrossRef]
- Civita, P.; Valerio, O.; Naccarato, A.G.; Gumbleton, M.; Pilkington, G.J. Satellitosis, a Crosstalk between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour. Cancers 2020, 12, 3720. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of alpha-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2018, 97, 92–107.e10. [Google Scholar] [CrossRef] [Green Version]
- Goker-Alpan, O.; Lopez, G.; Vithayathil, J.; Davis, J.; Hallett, M.; Sidransky, E. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch. Neurol. 2008, 65, 1353–1357. [Google Scholar] [CrossRef]
- Oeda, T.; Umemura, A.; Mori, Y.; Tomita, S.; Kohsaka, M.; Park, K.; Inoue, K.; Fujimura, H.; Hasegawa, H.; Sugiyama, H.; et al. Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol. Aging 2015, 36, 3306–3313. [Google Scholar] [CrossRef] [Green Version]
- Petrucci, S.; Ginevrino, M.; Trezzi, I.; Monfrini, E.; Ricciardi, L.; Albanese, A.; Avenali, M.; Barone, P.; Bentivoglio, A.R.; Bonifati, V.; et al. GBA-Related Parkinson’s Disease: Dissection of Genotype-Phenotype Correlates in a Large Italian Cohort. Mov. Disord. 2020, 35, 2106–2111. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132 Pt 7, 1783–1794. [Google Scholar] [CrossRef]
- Clark, L.N.; Kartsaklis, L.A.; Wolf Gilbert, R.; Dorado, B.; Ross, B.M.; Kisselev, S.; Verbitsky, M.; Mejia-Santana, H.; Cote, L.J.; Andrews, H.; et al. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch. Neurol. 2009, 66, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, C.H.; Beach, T.G.; Shill, H.A.; Caviness, J.N.; Driver-Dunckley, E.; Sabbagh, M.N.; Patel, A.; Sue, L.I.; Serrano, G.; Jacobson, S.A.; et al. GBA mutations in Parkinson disease: Earlier death but similar neuropathological features. Eur. J. Neurol. 2017, 24, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Pletnikova, O.; Geiger, J.T.; Murphy, N.A.; Abramzon, Y.; Rudow, G.; Mamais, A.; Sabir, M.S.; Crain, B.; Ahmed, S.; et al. Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol. Aging 2019, 76, 214.e1–214.e9. [Google Scholar] [CrossRef]
- Sklerov, M.; Kang, U.J.; Liong, C.; Clark, L.; Marder, K.; Pauciulo, M.; Nichols, W.C.; Chung, W.K.; Honig, L.S.; Cortes, E.; et al. Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov. Disord. Clin Pract. 2017, 4, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.-P.; Thomas, A.; et al. Dementia with Lewy bodies: An update and outlook. Mol. Neurodegener. 2019, 14, 5. [Google Scholar] [CrossRef]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.; Stefanis, L.; Attems, J. Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies—Current issues and future directions. J. Neurochem. 2019, 150, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Nishioka, K.; Ross, O.A.; Vilarino-Guell, C.; Cobb, S.A.; Kachergus, J.M.; Mann, D.M.; Snowden, J.; Richardson, A.M.T.; Neary, D.; Robinson, C.A.; et al. Glucocerebrosidase mutations in diffuse Lewy body disease. Parkinsonism Relat. Disord. 2011, 17, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A.; Attems, J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006, 112, 253–260. [Google Scholar] [CrossRef]
- Liu, A.K.L.; Chau, T.W.; Lim, E.J.; Ahmed, I.; Chang, R.C.; Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K.B. Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta Neuropathol. Commun. 2019, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Parkkinen, L.; Neumann, J.; O’Sullivan, S.S.; Holton, J.L.; Revesz, T.; Hardy, J.; Lees, A.J. Glucocerebrosidase mutations do not cause increased Lewy body pathology in Parkinson’s disease. Mol. Genet. Metab. 2011, 103, 410–412. [Google Scholar] [CrossRef]
- Berger-Sieczkowski, E.; Lutz, M.I.; Auff, E.; Kovacs, G.G. Gaucher cells are not associated with alpha-synuclein neuropathology in infants. Clin Neuropathol. 2016, 35, 122–128. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Goker-Alpan, O.; Stubblefield, B.K.; Giasson, B.I.; Sidransky, E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010, 120, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef]
- Huebecker, M.; Moloney, E.B.; van der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernick, A.I.; Walton, R.L.; Koga, S.; Soto-Beasley, A.I.; Heckman, M.G.; Gan-Or, Z.; Ren, Y.; Rademakers, R.; Uitti, R.J.; Wszolek, Z.K.; et al. GBA variation and susceptibility to multiple system atrophy. Parkinsonism Relat. Disord. 2020, 77, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Matsukawa, T.; Sasaki, H.; Yabe, I.; Matsushima, M.; Dürr, A.; Brice, A.; Takashima, H.; Kikuchi, A.; Aoki, M.; et al. Variants associated with Gaucher disease in multiple system atrophy. Ann. Clin Transl. Neurol. 2015, 2, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural. Transm. 2019, 126, 423–431. [Google Scholar] [CrossRef]
- Kon, T.; Tomiyama, M.; Wakabayashi, K. Neuropathology of Lewy body disease: Clinicopathological crosstalk between typical and atypical cases. Neuropathology 2020, 40, 30–39. [Google Scholar] [CrossRef]



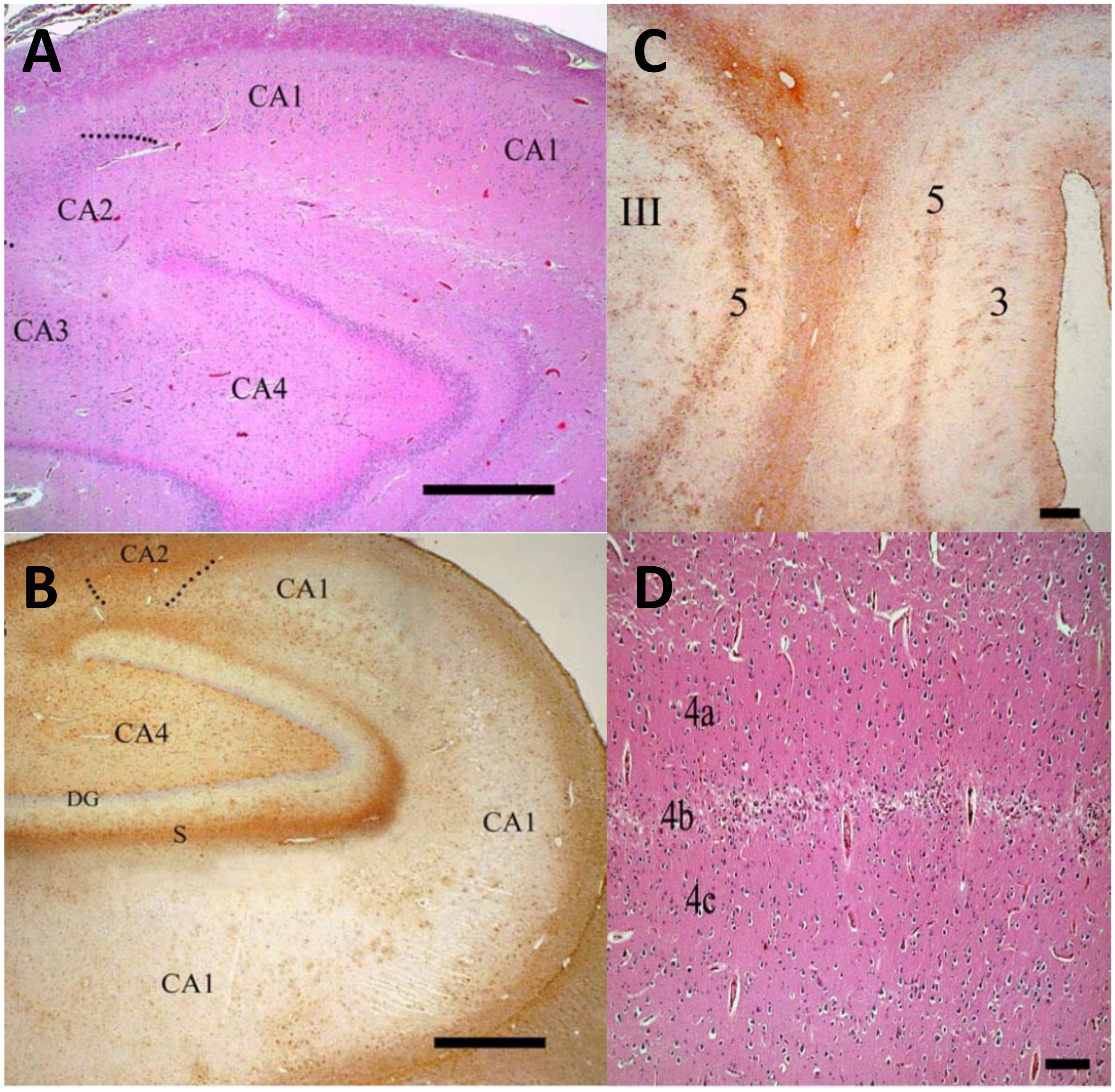{kind=link}
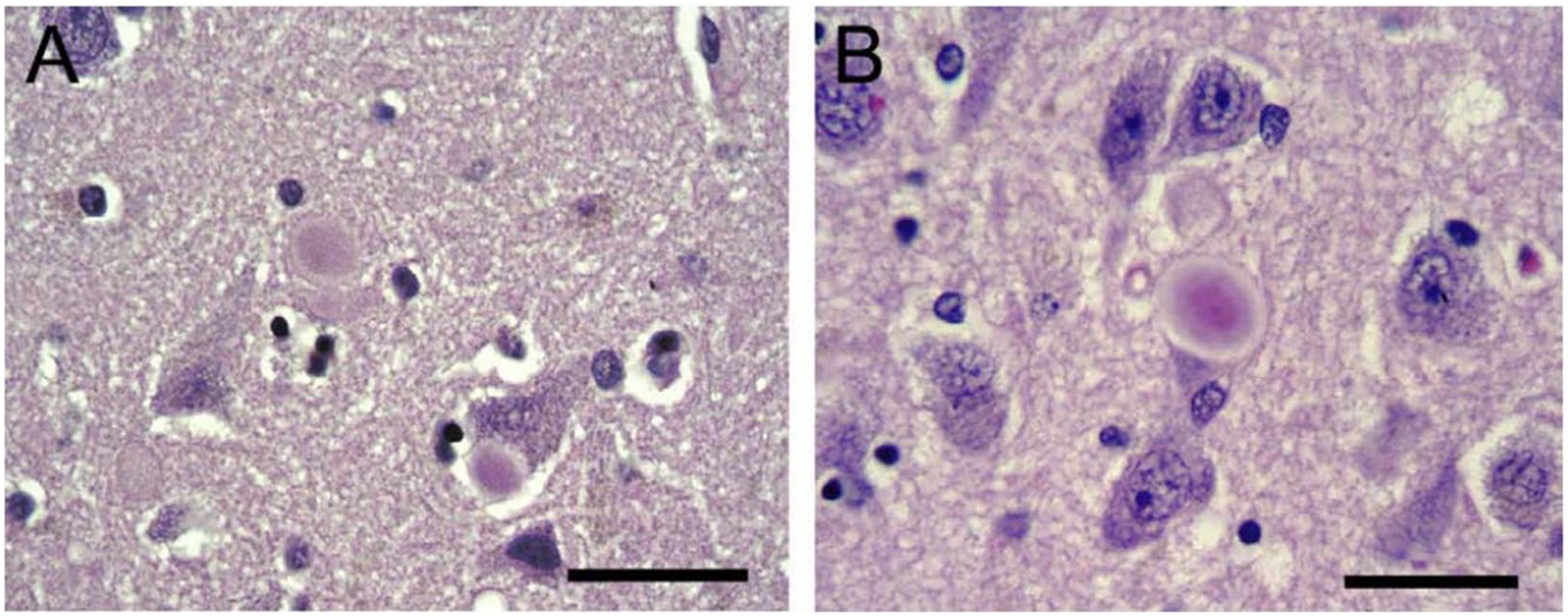{kind=link}
| Report | Gender/ Age of Death | Gaucher Diagnosis | Genotype | Neuronal Loss in SN | LB Pathology | Clinical Features |
|---|---|---|---|---|---|---|
| Wong et al., 2004 [22] | F/62 | GD1 | N370S/unknown | Yes | Diffuse cortical LB | Parkinsonism and dementia |
| Wong et al., 2004 [22] | F/53 | GD1 | D409H/L444P+duplication | Yes | Brainstem LB in hippocampal CA2–4 | Parkinsonism and dementia, supranuclear gaze palsy |
| Wong et al., 2004 [22] | M/75 | GD1 | N370S/N370S | Yes | Brainstem LB in SN | Parkinsonism and dementia |
| Wong et al., 2004 [22] | M/54 | GD1 | N370S/N370S | Yes | Brainstem LB in hippocampal CA2–4 | Parkinsonism and dementia |
| Adler et al., 2017 [42] | F/73 | Unknown | N370S/N370S | Unknown | Unknown | Parkinsonism and dementia |
| Blauwendraat et al., 2019 [47] | F/~90 | Un-diagnosed | N370S/N370S | Yes | No | Parkinsonism |
| Sklerov et al., 2017 [48] | ?/63 | Un-diagnosed | N370S/N370S | Yes | No | Orthostasis, parkinsonism, cognitive deficits |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furderer, M.L.; Hertz, E.; Lopez, G.J.; Sidransky, E. Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism. Int. J. Mol. Sci. 2022, 23, 5842. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105842
Furderer ML, Hertz E, Lopez GJ, Sidransky E. Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism. International Journal of Molecular Sciences. 2022; 23(10):5842. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105842
Chicago/Turabian StyleFurderer, Makaila L., Ellen Hertz, Grisel J. Lopez, and Ellen Sidransky. 2022. "Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism" International Journal of Molecular Sciences 23, no. 10: 5842. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105842