
The Role of Transposable Elements of the Human Genome in Neuronal Function and Pathology


Abstract
:1. Mobile Genetic Elements in the Human Genome and Their Regulation
1.1. Transposon Classification and Types of Transposable Elements in Our Genome
1.2. Inactive TEs in the Human Genome
1.3. L1 Elements: Their Structure and Mechanism of Mobilization
1.4. Trans RNA Targets Mobilized by L1 Elements and Gene Retrocopying
1.5. Alu Elements
1.6. SVA Elements

1.7. Molecular Mechanisms Suppressing TE Activity and Specific Cases of TE Unsilencing
1.7.1. DNA Methylation
1.7.2. Histone Methylation
1.7.3. Krüppel-Associated Box Domain Zinc Finger Proteins
1.7.4. RNA Interference
1.7.5. Other Mechanisms
1.7.6. “Self-Restriction” Inherent for Many TEs Is Absent in L1 Elements
1.7.7. TE Derepression during Cell Stress and Its Possible Explanations
1.8. Mechanisms and the Most Well-Known Examples of TE Exaptation
1.8.1. TEs as Modulators of Gene Expression Rate
1.8.2. TEs as a Source of Regulatory DNA Sequences
1.8.3. Transcription Factors Binding Sites within TEs
1.8.4. TE Insertions Modifying Coding Regions and Causing Formation of New Genes
1.8.5. Exonization of TEs Leading to the Formation of Novel Transcripts
1.8.6. TEs Introducing Alternative Polyadenylation Sites
1.8.7. RNA A-I Editing
1.8.8. Other TE-Related Mechanisms Altering the Fate of Transcripts
1.8.9. TE-Encoded Proteins and Non-Coding RNAs Used by the Host Cell
1.8.10. TEs Participating in DNA Repair and Chromosome Maintenance
2. The Role of TEs in the Normal Function of Neuronal Tissue
2.1. Somatic Mosaicism in Neurons: Its Sources and Possible Functions
2.2. Transposable Elements Are an Important Source of Neuronal Genetic Mosaicism
2.3. RNA A-I Editing in TE Sequences Contributes to Neuronal Somatic Mosaicism on RNA Level
2.4. TEs and Cell Differentiation in Neurogenesis
2.5. Specific TE Regulation in the Hippocampus: Is There a Role for TEs in Learning and Memory?
2.6. Is It Possible That Neurons Use TEs for RNA-Templated DNA Damage Repair?
2.7. Specific Examples of Domesticated Mobile Elements in Neurons
2.7.1. Neuron-Specific Transcription Regulation Provided by Exapted TEs
2.7.2. Neuron-Specific Proteins Encoded by TE-Derived Genes
2.7.3. Neuronal Non-Coding RNAs Encoded by TE-Derived Genes
2.8. TEs in the Human Brain Evolution
2.8.1. An overview of TE Evolution in Primate Lineage
2.8.2. TE-Mediated Recombinations in Human Brain Evolution
2.8.3. Multiple New Regulatory Elements Evolved from TEs in the Human Lineage
2.8.4. Alu Elements Participated in Our Brain Evolution in Diverse Ways
2.8.5. Human-Specific TE Insertions within Neuron-Associated Genes
3. The Role of Neuronal TEs in Pathology
3.1. TE-Related Mechanisms of Neuropathology
3.1.1. Known Mechanisms of TE-Associated Disorders in General
3.1.2. Neuropathologies Associated with TE Activation
3.1.3. TEs in Neurons and Stress
3.1.4. TEs in Neurons during Normal Aging
3.1.5. TEs, Autoimmunity and Neuroinflammation
3.1.6. TEs and Mitochondrial Dysfunction in the Context of Neurodegeneration
3.2. Specific Neurological Diseases with Reported Changes in TE Activity
3.2.1. Alzheimer’s Disease and Tauopathy
3.2.2. Parkinson’s Disease
3.2.3. Huntington’s Disease
3.2.4. Ataxia Telangiectasia
3.2.5. Spinal Muscular Atrophy
3.2.6. Amyotrophic Lateral Sclerosis and Fronto-Temporal Lobar Degeneration
3.2.7. Fragile X-Associated Tremor/Ataxia Syndrome
3.2.8. Multiple Sclerosis
3.2.9. Aicardi-Goutières Syndrome
3.2.10. Glioblastoma
3.2.11. Autism Spectrum Disorders
3.2.12. Rett Syndrome
3.2.13. Schizophrenia and Bipolar Disorder
3.2.14. Major Depressive Disorder
3.2.15. Post-Traumatic Stress Disorder
3.2.16. Drug Addiction and Alcoholism
3.2.17. Creutzfeldt-Jakob Disease
3.2.18. Neurofibromatosis Type I
3.2.19. Age-Related Macular Degeneration
3.2.20. X-Linked Dystonia-Parkinsonism
3.2.21. Ravine Encephalopathy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| 7SL RNA | signal recognition particle 7S RNA |
| ABCD1 | ATP binding cassette subfamily D member 1 |
| AD | Alzheimer’s disease |
| ADAR | adenosine deaminase that acts on RNA |
| ADNP | activity-dependent neuroprotector homeobox protein |
| AGS | Aicardi–Goutières syndrome |
| ALS | amyotrophic lateral sclerosis |
| AluI | Arthrobacter luteus endonuclease |
| AMD | age-related macular degeneration |
| AmnSINE1 | Amniota SINE1 |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| APOBEC3 | apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3 |
| APOE | apolipoprotein E |
| APP | amyloid β precursor protein |
| ARC | activity-regulated cytoskeleton-associated protein |
| ARE | adenylate-uridylate-rich elements |
| ASC | anthropoid-specific constrained region |
| ASD | autism spectrum disorders |
| AT | ataxia telangiectasia |
| ATM | ataxia telangiectasia mutated |
| AVPR1A | arginine vasopressin receptor 1A |
| Aβ | amyloid β |
| BCYRN1 | brain cytoplasmic RNA 1 |
| BD | bipolar disorder |
| BDNF | brain derived neurotrophic factor |
| BER | base excision repair |
| bp | base pair |
| C19MC | chromosome 19 microRNA cluster |
| C9ORF72 | guanine nucleotide exchange factor C9orf72 |
| CCFDN | congenital cataracts facial dysmorphism neuropathy |
| CENP-B | centromere-associated protein B |
| ChIP-PCR | chromatin immunoprecipitation-polymerase chain reaction |
| ChIP-Seq | chromatin immunoprecipitation sequencing |
| CHRNA9 | cholinergic receptor nicotinic α9 subunit |
| CJD | Creutzfeldt–Jakob disease |
| CMAH | cytidine monophospho-N-acetylneuraminic acid hydroxylase |
| CNNM2 | cyclin and CBS domain divalent metal cation transport mediator 2 |
| CNS | central nervous system |
| CNTN5 | contactin 5 |
| CNV | copy number variation |
| CREB | cAMP responsive element binding protein 1 |
| CRISPR/Cas | clustered regularly interspaced short palindromic repeats/CRISPR-associated proteins |
| CSF | cerebrospinal fluid |
| CTCF | CCCTC-binding factor |
| CYP20A1 | cytochrome P450 family 20 subfamily A member 1 |
| DBH | dopamine β-hydroxylase |
| DDP | DNA-dependent DNA polymerase |
| ddPCR | droplet digital polymerase chain reaction |
| DG | dentate gyrus |
| DHS | DNAse I-hypersensitive site |
| DLPFC | dorsolateral prefrontal cortex |
| DNMT | DNA methyltransferase |
| DNMT3L | DNA methyltransferase 3-like |
| DR2 | direct repeats of RGKTCA motifs separated by 2 bp |
| DR4 | direct repeats of RGKTCA motifs separated by 4 bp |
| DSB | DNA double-strand break |
| dsDNA | double-stranded DNA |
| dsRNA | double-stranded RNA |
| EGFP | enhanced green fluorescent protein |
| EGR1 | early growth response 1 |
| EN | engrailed homeobox |
| ENi | endonuclease-independent |
| Env | envelope protein |
| ERCC2 | ERCC excision repair 2, TFIIH core complex helicase subunit |
| ERV | endogenous retrovirus |
| ESCs | embryonic stem cells |
| eSINE | enhancer SINEs |
| EV | extracellular vehicle |
| FACS | fluorescent-activated cell sorting |
| FGF | fibroblast growth factor |
| FHIT | fragile histidine triad diadenosine triphosphatase |
| FMR1 | fragile X messenger ribonucleoprotein 1 |
| FOS | FBJ osteosarcoma oncogene |
| FOX | forkhead box |
| FOXO3 | forkhead box O3 |
| FOXP2 | forkhead box P2 |
| FRMD4A | FERM domain containing 4A |
| FTD | fronto-temporal dementia |
| FTLD | fronto-temporal lobar degeneration |
| FUS | fused in sarcoma |
| FXTAS | fragile X-associated tremor/ataxia syndrome |
| GABA | γ-aminobutyric acid |
| GABRB1 | γ-aminobutyric acid type A receptor subunit β1 |
| GADD45B | growth arrest and DNA damage inducible β |
| Gag | group-specific antigen |
| GFP | green fluorescent protein |
| GH | growth hormone |
| GluA2 | glutamate ionotropic receptor AMPA type subunit 2 |
| GO | gene ontology |
| GPR56 | G-protein-coupled receptor 56 |
| HCN | hippocampus neural stem cells |
| HD | Huntington’s disease |
| HDAC1 | histone deacetylase 1 |
| HERV | human endogenous retrovirus |
| hESCs | human embryonic stem cells |
| HetA | healing transposon |
| HFA | human fetal astrocyte |
| HIV1-Tat | human immunodeficiency virus type 1 trans-activator of transcription |
| hnRNPA1 | heterogeneous nuclear ribonucleoprotein A1 |
| HOX | homeobox |
| HR | homologous recombination |
| HSCs | hematopoietic stem cells |
| HTT | huntingtin |
| hYRNA | human RNA of the Y family |
| ID | intellectual disability |
| IFN | interferon |
| IL | interleukin |
| ILF3 | interleukin enhancer-binding factor 3 |
| iPSCs | induced pluripotent stem cells |
| IRES | internal ribosome entry site |
| IRF1 | interferon regulatory factor 1 |
| ISL1 | ISL LIM homeobox 1 |
| KAP1 | KRAB-associated protein 1 |
| kb | thousand base pairs |
| KD | knock-down |
| KDM4B | lysine demethylase 4B |
| KO | knock-out |
| KRAB-ZFP | Krüppel-associated box domain zinc finger protein |
| KZNF | Krüppel-associated box domain zinc finger protein |
| L1Hs | L1 human specific |
| lacZ | β-galactosidase |
| LDOC1 | LDOC1 regulator of NFKB signaling |
| LF-SINE | ‘living fossil’ SINE |
| lincRNA | long intergenic non-coding RNA |
| LINE | long interspersed nucleotide element |
| lncRNA | long non-coding RNA |
| LTM | long-term memory |
| LTR | long terminal repeat |
| LXREα | liver X receptor α |
| MAP2 | microtubule-associated protein 2 |
| MAPT | microtubule-associated protein tau |
| MART | mammalian retrotransposon transcript |
| Mb | million base pairs |
| mDa | mesencephalic dopaminergic neurons |
| MDD | major depressive disorder |
| MDM | monocyte-derived macrophage |
| MeCP2 | methyl-CpG-binding protein 2 |
| MER | medium reiterated repeat |
| MERVL | mouse endogenous retrovirus type L |
| MethylCap-Seq | Methyl-CpG binding domain-based capture and sequencing |
| MILI | mouse PIWI-like |
| MIR | mammalian-wide interspersed repeat |
| miRNA | microRNA |
| MITF-M | melanoma specific microphthalmia-associated transcription factor |
| MMR | DNA mismatch repair |
| Mov10 | Mov10 RISC complex RNA helicase |
| MPP+ | 1-methyl-4-phenylpyridinium |
| mRNP | messenger RNA |
| mRNP | messenger ribonucleoprotein |
| MS | multiple sclerosis |
| MSCs | mesenchymal stem cells |
| MTH1 | methylated purine nucleoside triphosphate hydrolase |
| My | million years |
| MYC | MYC proto-oncogene, bHLH transcription factor |
| MyD88 | myeloid differentiation primary response 88 |
| NAc | nucleus accumbens |
| NCAI | non-classical Alu insertion |
| NCLI | non-classical L1 insertions |
| NDUFS2 | NADH:ubiquinone oxidoreductase core subunit S2 |
| NEE | novel enriched environmental conditions |
| NER | nucleotide excision repair |
| Neu5Gc | N-glycolylneuraminic acid |
| NeuroD | neuronal differentiation |
| Neurog | neurogenin |
| NF1 | neurofibromatosis type I |
| NF90 | nuclear factor of activated T-cells, 90 kD |
| NFI | nuclear factor I |
| NF-κB | nuclear factor kappa B |
| NF-κB1 | nuclear factor kappa B subunit 1 |
| NHEJ | non-homologous end joining |
| NLS | nuclear localization signal |
| NPAS1 | neuronal PAS domain protein 1 |
| NPCs | neural precursor cells |
| NRIF | neurotrophin receptor-interacting factor |
| NRSF | neuron-restrictive silencer factor |
| NSCs | neural stem cells |
| NUDT1 | nudix hydrolase 1 |
| OPA1 | optic atrophy protein 1 |
| ORF | open reading frame |
| OXT | oxytocin/neurophysin I prepropeptide |
| p75NTR | nerve growth factor receptor |
| PA | passive avoidance |
| PARK2 | Parkinson juvenile disease protein 2 |
| PARK7 | Parkinsonism-associated deglycase |
| PBMCs | peripheral blood mononuclear cells |
| PCBP2 | poly(RC) binding protein 2 |
| PD | Parkinson’s disease |
| PDHA1 | pyruvate dehydrogenase E1 subunit α1 |
| PEG | paternally expressed |
| piRNA | PIWI-interacting RNA |
| PIT-1 | pituitary-specific positive transcription factor 1 |
| PIWI | P-element induced wimpy testis |
| PKR | protein kinase R |
| POGZ | pogo transposable element with zinc finger domain |
| pol II | RNA polymerase II |
| pol III | RNA polymerase III |
| POMC | proopiomelanocortin |
| PRC2 | Polycomb repressive complex 2 |
| PRKN | parkin RBR E3 ubiquitin protein ligase |
| PrP | prion protein |
| PTSD | post-traumatic stress disorder |
| qPCR | quantitative polymerase chain reaction |
| RAD52 | RAD52 homolog, DNA repair protein |
| RC-L1 | retrotransposition-competent L1 elements |
| RC-Seq | retrotransposon capture sequencing |
| rDNA | ribosomal DNA |
| RDP | RNA-dependent DNA polymerase |
| REST | RE1-silencing transcription factor |
| RHOXF2 | Rhox homeobox family member 2 |
| RNP | ribonucleoprotein particle |
| ROS | reactive oxygen species |
| RPA1 | replication protein A1 |
| RPE | retinal pigmented epithelium |
| rRNA | ribosomal RNA |
| RT | reverse transcription |
| RTL | retrotransposon Gaglike |
| RT-qPCR | quantitative reverse transcription polymerase chain reaction |
| RTT | Rett syndrome |
| RUNX3 | RUNX family transcription factor 3 |
| RYR3 | ryanodine receptor 3 |
| SAMHD1 | SAM domain and HD domain-containing protein 1 |
| SCAN | SRE-ZBP, CTfin51, AW-1 and number 18 cDNA |
| SEFL | stress-enhanced fear learning |
| SETDB1 | SET domain bifurcated histone lysine methyltransferase 1 |
| SETMAR | SET domain and mariner transposase fusion gene |
| sgRNA | single guide RNA |
| SINE | short interspersed nucleotide element |
| SINEUP | SINE element-containing translation up-regulator |
| SIRH | sushi-ichi-related retrotransposon-homolog |
| siRNA | small interfering RNA |
| SIRT6 | sirtuin 6 |
| SLAV | somatic L1-associated variants |
| SLC7A2 | solute carrier family 7 member 2 |
| SMA | spinal muscular atrophy |
| SMN1 | survival of motor neuron 1, telomeric |
| SMN2 | survival of motor neuron 2, centromeric |
| SNAR | small NF90 (ILF3) associated RNA |
| SNpc | substantia nigra pars compacta |
| SNV | single-nucleotide variant |
| SOD1 | superoxide dismutase |
| SOX | SRY-associated high mobility group box |
| SRY | sex-determining region Y |
| ssDNA | single-stranded DNA |
| ssRNA | single-stranded RNA |
| STAT | signal transducer and activator of transcription |
| STG | superior temporal gyrus |
| STM | short-term memory |
| SUV39H1 | suppressor of variegation 3–9 homolog 1 |
| SVA | SINE-R, VNTR, and Alu |
| SVZ | subventricular zone |
| SZ | schizophrenia |
| Ta | transcriptionally active |
| TACR3 | tachykinin receptor 3 |
| TAD | topologically associating domain |
| TAF1 | TATA-box binding protein associated factor 1 |
| TAHRE | telomere associated and HeTA-related |
| TART | telomere associated retrotransposon |
| TBX | T-box |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TC-NER | transcription-coupled nucleotide excision repair |
| TDP-43 | TAR DNA-binding protein 43 |
| TE | transposable element |
| TERT | telomerase reverse transcriptase |
| TF | transcription factor |
| TFIIIC | general transcription factor IIIC |
| TFR | transposon-free region |
| TLR4 | toll-like receptor 4 |
| TNPO1 | transportin 1 |
| TPRT | target-site primed reverse transcription |
| TR | telomerase RNA |
| TREX1 | three-prime repair exonuclease 1 |
| TRIM28 | tripartite motif containing 28 |
| tRNA | transfer RNA |
| TRPV3 | transient receptor potential cation channel subfamily V member 3 |
| Ty | transposons of yeast |
| U snRNA | uridine-rich small nuclear RNA |
| UCHL1 | ubiquitin carboxy-terminal hydrolase L1 |
| UTR | untranslated region |
| UV | ultraviolet |
| VNTR | variable number of tandem repeats |
| Wnt3a | wingless-type MMTV integration site family member 3a |
| WT | wild type |
| XPD | X-linked dystonia-parkinsonism |
| YY1 | Yin Yang-1 |
| ZCCHC16 | zinc finger CCHC domain-containing protein 16 |
| ZNF | zinc finger protein |
References
- McClintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344. [Google Scholar] [CrossRef] [Green Version]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as regulators of gene expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedli, M.; Trono, D. The Developmental Control of Transposable Elements and the Evolution of Higher Species. Annu. Rev. Cell. Dev. Biol. 2015, 31, 429–451. [Google Scholar] [CrossRef] [PubMed]
- van’t Hof, A.E.; Campagne, P.; Rigden, D.J.; Yung, C.J.; Lingley, J.; Quail, M.A.; Hall, N.; Darby, A.C.; Saccheri, I.J. The industrial melanism mutation in British peppered moths is a transposable element. Nature 2016, 534, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Linker, S.B.; Marchetto, M.C.; Narvaiza, I.; Denli, A.M.; Gage, F.H. Examining non-LTR retrotransposons in the context of the evolving primate brain. BMC Biol. 2017, 15, 68. [Google Scholar] [CrossRef] [Green Version]
- Kramerov, D.A.; Vassetzky, N.S. SINEs. Wiley Interdiscip. Rev. RNA 2011, 2, 772–786. [Google Scholar] [CrossRef]
- Ferrari, R.; Grandi, N.; Tramontano, E.; Dieci, G. Retrotransposons as Drivers of Mammalian Brain Evolution. Life 2021, 11, 376. [Google Scholar] [CrossRef]
- Dieci, G.; Conti, A.; Pagano, A.; Carnevali, D. Identification of RNA polymerase III-transcribed genes in eukaryotic genomes. Biochim. Biophys. Acta 2013, 1829, 296–305. [Google Scholar] [CrossRef]
- Walters, R.D.; Kugel, J.F.; Goodrich, J.A. InvAluable junk: The cellular impact and function of Alu and B2 RNAs. IUBMB Life 2009, 61, 831–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermant, C.; Torres-Padilla, M.-E. TFs for TEs: The transcription factor repertoire of mammalian transposable elements. Genes Dev. 2021, 35, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.N.; Vandewege, M.W.; Ray, D.A. Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Eickbush, T.H.; Jamburuthugoda, V.K. The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res. 2008, 134, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [Green Version]
- de Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.K. Human transposable elements in Repbase: Genomic footprints from fish to humans. Mob. DNA 2018, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.L.; Bubb, V.J.; Breen, G.; Quinn, J.P. Characterisation of the potential function of SVA retrotransposons to modulate gene expression patterns. BMC Evol. Biol. 2013, 13, 101. [Google Scholar] [CrossRef] [Green Version]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, R.N.; Mangum, S.F.; Ray, D.A. Pinpointing the vesper bat transposon revolution using the Miniopterus natalensis genome. Mob. DNA 2016, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, A.; Caizzi, R.; Moschetti, R.; Marsano, R.M. What Have We Learned in 30 Years of Investigations on Bari Transposons? Cells 2022, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [Green Version]
- Jern, P.; Coffin, J.M. Effects of Retroviruses on Host Genome Function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef] [Green Version]
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nat. Rev. Immunol. 2016, 16, 207–219. [Google Scholar] [CrossRef]
- Dolei, A.; Perron, H. The multiple sclerosis-associated retrovirus and its HERV-W endogenous family: A biological interface between virology, genetics, and immunology in human physiology and disease. J. Neurovirol. 2009, 15, 4–13. [Google Scholar] [CrossRef]
- Löwer, R.; Boller, K.; Hasenmaier, B.; Korbmacher, C.; Müller-Lantzsch, N.; Löwer, J.; Kurth, R. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc. Natl. Acad. Sci. USA 1993, 90, 4480–4484. [Google Scholar] [CrossRef] [Green Version]
- Boller, K.; König, H.; Sauter, M.; Mueller-Lantzsch, N.; Löwer, R.; Löwer, J.; Kurth, R. Evidence that HERV-K is the endogenous retrovirus sequence that codes for the human teratocarcinoma-derived retrovirus HTDV. Virology 1993, 196, 349–353. [Google Scholar] [CrossRef]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Frank, O.; Giehl, M.; Zheng, C.; Hehlmann, R.; Leib-Mösch, C.; Seifarth, W. Human Endogenous Retrovirus Expression Profiles in Samples from Brains of Patients with Schizophrenia and Bipolar Disorders. J. Virol. 2005, 79, 10890–10901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröger, V.; Emmer, A.; Staege, M.S.; Cynis, H. Endogenous Retroviruses in Nervous System Disorders. Pharmaceuticals 2021, 14, 70. [Google Scholar] [CrossRef] [PubMed]
- Cardelli, M.; Giacconi, R.; Malavolta, M.; Provinciali, M. Endogenous Retroelements in Cellular Senescence and Related Pathogenic Processes: Promising Drug Targets in Age-Related Diseases. Curr. Drug Targets 2016, 17, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.R.; Garcia-Perez, J.L.; Badge, R.M.; Moran, J.V. LINE-1 elements in structural variation and disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 187–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoye, J.P. Endogenous retroviruses: Still active after all these years? Curr. Biol. 2001, 11, R914–R916. [Google Scholar] [CrossRef] [Green Version]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef]
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta 2007, 1775, 138–162. [Google Scholar] [CrossRef]
- Gilbert, N.; Labuda, D. CORE-SINEs: Eukaryotic short interspersed retroposing elements with common sequence motifs. Proc. Natl. Acad. Sci. USA 1999, 96, 2869–2874. [Google Scholar] [CrossRef] [Green Version]
- Jacob-Hirsch, J.; Eyal, E.; Knisbacher, B.A.; Roth, J.; Cesarkas, K.; Dor, C.; Farage-Barhom, S.; Kunik, V.; Simon, A.J.; Gal, M.; et al. Whole-genome sequencing reveals principles of brain retrotransposition in neurodevelopmental disorders. Cell Res. 2018, 28, 187–203. [Google Scholar] [CrossRef] [Green Version]
- Han, J.S.; Szak, S.T.; Boeke, J.D. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 2004, 429, 268–274. [Google Scholar] [CrossRef]
- Brouha, B.; Schustak, J.; Badge, R.M.; Lutz-Prigge, S.; Farley, A.H.; Moran, J.V.; Kazazian, H.H., Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 2003, 100, 5280–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnegan, D.J. Retrotransposons. Curr. Biol. 2012, 22, R432–R437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscione, S.W.; Theodosakis, N.; Micevic, G.; Cornish, T.C.; Burns, K.H.; Neretti, N.; Rodić, N. Genome-wide characterization of human L1 antisense promoter-driven transcripts. BMC Genom. 2016, 17, 463. [Google Scholar] [CrossRef] [Green Version]
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol. Cell. Biol. 2001, 21, 1973–1985. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef]
- Singer, T.; McConnell, M.J.; Marchetto, M.C.; Coufal, N.G.; Gage, F.H. LINE-1 retrotransposons: Mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010, 33, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Bodak, M.; Yu, J.; Ciaudo, C. Regulation of LINE-1 in mammals. Biomol. Concepts 2014, 5, 409–428. [Google Scholar] [CrossRef] [Green Version]
- Denli, A.M.; Narvaiza, I.; Kerman, B.E.; Pena, M.; Benner, C.; Marchetto, M.C.; Diedrich, J.K.; Aslanian, A.; Ma, J.; Moresco, J.J.; et al. Primate-specific ORF0 contributes to retrotransposon-mediated diversity. Cell 2015, 163, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Lavie, L.; Maldener, E.; Brouha, B.; Meese, E.U.; Mayer, J. The human L1 promoter: Variable transcription initiation sites and a major impact of upstream flanking sequence on promoter activity. Genome Res. 2004, 14, 2253–2260. [Google Scholar] [CrossRef] [Green Version]
- Mita, P.; Wudzinska, A.; Sun, X.; Andrade, J.; Nayak, S.; Kahler, D.J.; Badri, S.; LaCava, J.; Ueberheide, B.; Yun, C.Y.; et al. LINE-1 protein localization and functional dynamics during the cell cycle. Elife 2018, 7, e30058. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.T.; Sokolowski, M.; Roy-Engel, A.M.; Smither, M.E.; Belancio, V.P. Identification of charged amino acids required for nuclear localization of human L1 ORF1 protein. Mob. DNA 2019, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Idica, A.; Sevrioukov, E.A.; Zisoulis, D.G.; Hamdorf, M.; Daugaard, I.; Kadandale, P.; Pedersen, I.M. MicroRNA miR-128 represses LINE-1 (L1) retrotransposition by down-regulating the nuclear import factor TNPO1. J. Biol. Chem. 2017, 292, 20494–20508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, L.; Guzman, H.; Sevrioukov, E.; Idica, A.; Park, E.; Bochnakian, A.; Daugaard, I.; Jury, D.; Mortazavi, A.; Zisoulis, D.G.; et al. miR-128 Restriction of LINE-1 (L1) Retrotransposition Is Dependent on Targeting hnRNPA1 mRNA. Int. J. Mol. Sci. 2019, 20, 1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cost, G.J.; Feng, Q.; Jacquier, A.; Boeke, J.D. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002, 21, 5899–5910. [Google Scholar] [CrossRef]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef] [Green Version]
- Gasior, S.L.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Marchetto, M.C.N.; Muotri, A.R.; Mu, Y.; Carson, C.T.; Macia, A.; Moran, J.V.; Gage, F.H. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20382–20387. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Yamaguchi, K.; Kajikawa, M.; Ichiyanagi, K.; Adachi, N.; Koyama, H.; Takeda, S.; Okada, N. Genetic evidence that the non-homologous end-joining repair pathway is involved in LINE retrotransposition. PLoS Genet. 2009, 5, e1000461. [Google Scholar] [CrossRef] [Green Version]
- Gasior, S.L.; Roy-Engel, A.M.; Deininger, P.L. ERCC1/XPF limits L1 retrotransposition. DNA Repair 2008, 7, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.K.; Huang, C.T.; Han, K.; Batzer, M.A. Endonuclease-independent insertion provides an alternative pathway for L1 retrotransposition in the human genome. Nucleic Acids Res. 2007, 35, 3741–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szak, S.T.; Pickeral, O.K.; Makalowski, W.; Boguski, M.S.; Landsman, D.; Boeke, J.D. Molecular archeology of L1 insertions in the human genome. Genome Biol. 2002, 3, research0052.0051. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, E.M.; Kazazian, H.H., Jr. Twin priming: A proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 2001, 11, 2059–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, T.A.; Gilbert, N.; Myers, J.S.; Vincent, B.J.; Stamato, T.D.; Taccioli, G.E.; Batzer, M.A.; Moran, J.V. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet. 2002, 31, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Maestre, J.; Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 2000, 24, 363–367. [Google Scholar] [CrossRef]
- Sultana, T.; van Essen, D.; Siol, O.; Bailly-Bechet, M.; Philippe, C.; Zine El Aabidine, A.; Pioger, L.; Nigumann, P.; Saccani, S.; Andrau, J.-C.; et al. The Landscape of L1 Retrotransposons in the Human Genome is Shaped by Pre-insertion Sequence Biases and Post-insertion Selection. Mol. Cell 2019, 74, 555–570.e557. [Google Scholar] [CrossRef]
- Gilbert, N.; Lutz-Prigge, S.; Moran, J.V. Genomic Deletions Created upon LINE-1 Retrotransposition. Cell 2002, 110, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, N.; Lutz, S.; Morrish, T.A.; Moran, J.V. Multiple Fates of L1 Retrotransposition Intermediates in Cultured Human Cells. Mol. Cell. Biol. 2005, 25, 7780. [Google Scholar] [CrossRef] [Green Version]
- Graham, T.; Boissinot, S. The genomic distribution of L1 elements: The role of insertion bias and natural selection. J. Biomed. Biotechnol. 2006, 2006, 75327. [Google Scholar] [CrossRef]
- Erwin, J.A.; Paquola, A.C.M.; Singer, T.; Gallina, I.; Novotny, M.; Quayle, C.; Bedrosian, T.A.; Alves, F.I.A.; Butcher, C.R.; Herdy, J.R.; et al. L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat. Neurosci. 2016, 19, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Boissinot, S.; Davis, J.; Entezam, A.; Petrov, D.; Furano, A.V. Fitness cost of LINE-1 (L1) activity in humans. Proc. Natl. Acad. Sci. USA 2006, 103, 9590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodea, G.O.; McKelvey, E.G.Z.; Faulkner, G.J. Retrotransposon-induced mosaicism in the neural genome. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewing, A.D.; Ballinger, T.J.; Earl, D.; Harris, C.C.; Ding, L.; Wilson, R.K.; Haussler, D. Retrotransposition of gene transcripts leads to structural variation in mammalian genomes. Genome Biol. 2013, 14, R22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaessmann, H.; Vinckenbosch, N.; Long, M. RNA-based gene duplication: Mechanistic and evolutionary insights. Nat. Rev. Genet. 2009, 10, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, B.; Sisu, C.; Frankish, A.; Howald, C.; Habegger, L.; Mu, X.J.; Harte, R.; Balasubramanian, S.; Tanzer, A.; Diekhans, M.; et al. The GENCODE pseudogene resource. Genome Biol. 2012, 13, R51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheetham, S.W.; Faulkner, G.J.; Dinger, M.E. Overcoming challenges and dogmas to understand the functions of pseudogenes. Nat. Rev. Genet. 2020, 21, 191–201. [Google Scholar] [CrossRef]
- Ding, W.; Lin, L.; Chen, B.; Dai, J. L1 elements, processed pseudogenes and retrogenes in mammalian genomes. IUBMB Life 2006, 58, 677–685. [Google Scholar] [CrossRef]
- Wei, W.; Gilbert, N.; Ooi, S.L.; Lawler, J.F.; Ostertag, E.M.; Kazazian, H.H.; Boeke, J.D.; Moran, J.V. Human L1 retrotransposition: Cis preference versus trans complementation. Mol. Cell. Biol. 2001, 21, 1429–1439. [Google Scholar] [CrossRef] [Green Version]
- Kopera, H.C.; Moldovan, J.B.; Morrish, T.A.; Garcia-Perez, J.L.; Moran, J.V. Similarities between long interspersed element-1 (LINE-1) reverse transcriptase and telomerase. Proc. Natl. Acad. Sci. USA 2011, 108, 20345. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Ewing, A.D.; Hancks, D.C.; Kazazian, H.H., Jr. Enrichment of processed pseudogene transcripts in L1-ribonucleoprotein particles. Hum. Mol. Genet. 2013, 22, 3730–3748. [Google Scholar] [CrossRef] [Green Version]
- Briggs, E.M.; McKerrow, W.; Mita, P.; Boeke, J.D.; Logan, S.K.; Fenyö, D. RIP-seq reveals LINE-1 ORF1p association with p-body enriched mRNAs. Mob. DNA 2021, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Grechishnikova, D.; Poptsova, M. Conserved 3’ UTR stem-loop structure in L1 and Alu transposons in human genome: Possible role in retrotransposition. BMC Genom. 2016, 17, 992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Perez, J.L.; Doucet, A.J.; Bucheton, A.; Moran, J.V.; Gilbert, N. Distinct mechanisms for trans-mediated mobilization of cellular RNAs by the LINE-1 reverse transcriptase. Genome Res. 2007, 17, 602–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.R.; Salvador-Palomeque, C.; Faulkner, G.J. Diversity through duplication: Whole-genome sequencing reveals novel gene retrocopies in the human population. Bioessays 2014, 36, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Emerman, M.; Malik, H.S.; McLaughlin, R.N.J. Retrocopying expands the functional repertoire of APOBEC3 antiviral proteins in primates. Elife 2020, 9, e58436. [Google Scholar] [CrossRef]
- Zhang, L.; Richards, A.; Barrasa, M.I.; Stephen, H.H.; Richard, A.Y.; Jaenisch, R. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2105968118. [Google Scholar] [CrossRef]
- Ostertag, E.M.; Prak, E.T.; DeBerardinis, R.J.; Moran, J.V.; Kazazian, H.H., Jr. Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res. 2000, 28, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Grover, D.; Mukerji, M.; Bhatnagar, P.; Kannan, K.; Brahmachari, S.K. Alu repeat analysis in the complete human genome: Trends and variations with respect to genomic composition. Bioinformatics 2004, 20, 813–817. [Google Scholar] [CrossRef] [Green Version]
- Houck, C.M.; Rinehart, F.P.; Schmid, C.W. A ubiquitous family of repeated DNA sequences in the human genome. J. Mol. Biol. 1979, 132, 289–306. [Google Scholar] [CrossRef]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullu, E.; Tschudi, C. Alu sequences are processed 7SL RNA genes. Nature 1984, 312, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Häsler, J.; Samuelsson, T.; Strub, K. Useful ‘junk’: Alu RNAs in the human transcriptome. Cell. Mol. Life Sci. 2007, 64, 1793–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lütcke, H. Signal Recognition Particle (SRP), a Ubiquitous Initiator of Protein Translocation. Eur. J. Biochem. 1995, 228, 531–550. [Google Scholar] [CrossRef]
- Martignetti, J.A.; Brosius, J. BC200 RNA: A neural RNA polymerase III product encoded by a monomeric Alu element. Proc. Natl. Acad. Sci. USA 1993, 90, 11563–11567. [Google Scholar] [CrossRef] [Green Version]
- Li, T.H.; Schmid, C.W. Differential stress induction of individual Alu loci: Implications for transcription and retrotransposition. Gene 2001, 276, 135–141. [Google Scholar] [CrossRef]
- Weiner, A.M. SINEs and LINEs: The art of biting the hand that feeds you. Curr. Opin. Cell Biol. 2002, 14, 343–350. [Google Scholar] [CrossRef]
- Ohshima, K. RNA-Mediated Gene Duplication and Retroposons: Retrogenes, LINEs, SINEs, and Sequence Specificity. Int. J. Evol. Biol. 2013, 2013, 424726. [Google Scholar] [CrossRef]
- Kriegs, J.O.; Churakov, G.; Jurka, J.; Brosius, J.; Schmitz, J. Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet. 2007, 23, 158–161. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker. Available online: https://github.com/rmhubley/RepeatMasker/blob/master/repeatmasker.help (accessed on 15 April 2022).
- Hancks, D.C.; Kazazian, H.H., Jr. Active human retrotransposons: Variation and disease. Curr. Opin. Genet. Dev. 2012, 22, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Ostertag, E.M.; Goodier, J.L.; Zhang, Y.; Kazazian, H.H., Jr. SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet. 2003, 73, 1444–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancks, D.C.; Kazazian, H.H., Jr. SVA retrotransposons: Evolution and genetic instability. Semin. Cancer Biol. 2010, 20, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xing, J.; Grover, D.; Hedges, D.; Han, K.; Walker, J.A.; Batzer, M. SVA elements: A hominid-specific retroposon family. J. Mol. Biol. 2005, 354, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Raiz, J.; Damert, A.; Chira, S.; Held, U.; Klawitter, S.; Hamdorf, M.; Löwer, J.; Strätling, W.H.; Löwer, R.; Schumann, G.G. The non-autonomous retrotransposon SVA is trans -mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012, 40, 1666–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodier, J.L.; Mandal, P.K.; Zhang, L.; Kazazian, H.H., Jr. Discrete subcellular partitioning of human retrotransposon RNAs despite a common mechanism of genome insertion. Hum. Mol. Genet. 2010, 19, 1712–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damert, A.; Raiz, J.; Horn, A.V.; Löwer, J.; Wang, H.; Xing, J.; Batzer, M.A.; Löwer, R.; Schumann, G.G. 5’-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res. 2009, 19, 1992–2008. [Google Scholar] [CrossRef] [Green Version]
- Gianfrancesco, O.; Bubb, V.J.; Quinn, J.P. SVA retrotransposons as potential modulators of neuropeptide gene expression. Neuropeptides 2017, 64, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, A.L.; Wilm, T.P.; Khursheed, K.; Shatunov, A.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; Smith, B.; Breen, G.; Al-Chalabi, A.; et al. An evaluation of a SVA retrotransposon in the FUS promoter as a transcriptional regulator and its association to ALS. PLoS ONE 2014, 9, e90833. [Google Scholar] [CrossRef] [Green Version]
- Miskey, C.; Izsvák, Z.; Kawakami, K.; Ivics, Z. DNA transposons in vertebrate functional genomics. Cell. Mol. Life Sci. 2005, 62, 629–641. [Google Scholar] [CrossRef]
- Rosser, J.M.; An, W. L1 expression and regulation in humans and rodents. Front. Biosci. 2012, 4, 2203–2225. [Google Scholar] [CrossRef]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapp, H.E.; Hunter, R.G. Early life exposures, neurodevelopmental disorders, and transposable elements. Neurobiol. Stress 2019, 11, 100174. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Terrones, D.; Torres-Padilla, M.E. Nimble and Ready to Mingle: Transposon Outbursts of Early Development. Trends Genet. 2018, 34, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Lin, C.-J.; Yin, Y.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.; Ramalho-Santos, M. A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 2018, 174, 391–405.e319. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, G.J.; Billon, V. L1 retrotransposition in the soma: A field jumping ahead. Mob. DNA 2018, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Simons, C.; Pheasant, M.; Makunin, I.V.; Mattick, J.S. Transposon-free regions in mammalian genomes. Genome Res. 2006, 16, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Simons, C.; Makunin, I.V.; Pheasant, M.; Mattick, J.S. Maintenance of transposon-free regions throughout vertebrate evolution. BMC Genom. 2007, 8, 470. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.S.; Estécio, M.R.H.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.-P.J. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef]
- Reilly, M.T.; Faulkner, G.J.; Dubnau, J.; Ponomarev, I.; Gage, F.H. The role of transposable elements in health and diseases of the central nervous system. J. Neurosci. 2013, 33, 17577–17586. [Google Scholar] [CrossRef] [Green Version]
- Haggerty, C.; Kretzmer, H.; Riemenschneider, C.; Kumar, A.S.; Mattei, A.L.; Bailly, N.; Gottfreund, J.; Giesselmann, P.; Weigert, R.; Brändl, B.; et al. Dnmt1 has de novo activity targeted to transposable elements. Nat. Struct. Mol. Biol. 2021, 28, 594–603. [Google Scholar] [CrossRef]
- Bourc’his, D.; Bestor, T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004, 431, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A.; Steinhoff, C.; Florl, A.R. Methylation of endogenous human retroelements in health and disease. Curr. Top. Microbiol. Immunol. 2006, 310, 211–250. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.M.; Motta, V.; Panni, T.; Bertazzi, P.A.; Apostoli, P.; Hou, L.; Baccarelli, A.A. Evolutionary age of repetitive element subfamilies and sensitivity of DNA methylation to airborne pollutants. Part. Fibre Toxicol. 2013, 10, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miousse, I.R.; Chalbot, M.-C.G.; Lumen, A.; Ferguson, A.; Kavouras, I.G.; Koturbash, I. Response of transposable elements to environmental stressors. Mutat. Res. Rev. Mut. Res. 2015, 765, 19–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jintaridth, P.; Mutirangura, A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol. Genom. 2010, 41, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Hong, C.; Zhang, B.; Lowdon, R.F.; Xing, X.; Li, D.; Zhou, X.; Lee, H.J.; Maire, C.L.; Ligon, K.L.; et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat. Genet. 2013, 45, 836–841. [Google Scholar] [CrossRef]
- Philippe, C.; Vargas-Landin, D.B.; Doucet, A.J.; van Essen, D.; Vera-Otarola, J.; Kuciak, M.; Corbin, A.; Nigumann, P.; Cristofari, G. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. Elife 2016, 5, e13926. [Google Scholar] [CrossRef]
- Muotri, A.R.; Marchetto, M.C.N.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Almeida, M.V.; Vernaz, G.; Putman, A.L.K.; Miska, E.A. Taming transposable elements in vertebrates: From epigenetic silencing to domestication. Trends Genet. 2022, 38, 529–553. [Google Scholar] [CrossRef] [PubMed]
- Déléris, A.; Berger, F.; Duharcourt, S. Role of Polycomb in the control of transposable elements. Trends Genet. 2021, 37, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Yan, K.; Zheng, Q.; Ke, H.; Cheng, J.; Xiong, W.; Shi, X.; Wei, L.; Zhao, M.; Yang, F.; et al. Histone Demethylase KDM4B Promotes DNA Damage by Activating Long Interspersed Nuclear Element-1. Cancer Res. 2019, 79, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, D.; Vavrova-Anderson, J.; Oler, A.J.; Cowling, V.H.; Cairns, B.R.; White, R.J. SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation. Nat. Commun. 2015, 6, 6569. [Google Scholar] [CrossRef] [Green Version]
- Bruno, M.; Mahgoub, M.; Macfarlan, T.S. The Arms Race Between KRAB-Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annu. Rev. Genet. 2019, 53, 393–416. [Google Scholar] [CrossRef]
- Wolf, G.; Greenberg, D.; Macfarlan, T.S. Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Krüppel-associated box zinc finger protein family. Mob. DNA 2015, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Jacobs, F.M.J.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y. Diverse small non-coding RNAs in RNA interference pathways. Methods Mol. Biol. 2011, 764, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, R.; Gerstein, M. Protein fossils live on as RNA. Nature 2008, 453, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Seitz, H.; Horwich, M.D.; Li, C.; Du, T.; Lee, S.; Xu, J.; Kittler, E.L.; Zapp, M.L.; Weng, Z.; et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science 2008, 320, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Siomi, M.C. Small RNA-Mediated Quiescence of Transposable Elements in Animals. Dev. Cell 2010, 19, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Rajasethupathy, P.; Antonov, I.; Sheridan, R.; Frey, S.; Sander, C.; Tuschl, T.; Kandel, E.R. A Role for Neuronal piRNAs in the Epigenetic Control of Memory-Related Synaptic Plasticity. Cell 2012, 149, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Banerjee, S.; Zhou, H.; Jammalamadaka, A.; Arcila, M.; Manjunath, B.S.; Kosik, K.S. Identification of piRNAs in the central nervous system. RNA 2011, 17, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W. PIWI Proteins and piRNAs in the Nervous System. Mol. Cells 2019, 42, 828–835. [Google Scholar] [CrossRef]
- Watanabe, T.; Totoki, Y.; Toyoda, A.; Kaneda, M.; Kuramochi-Miyagawa, S.; Obata, Y.; Chiba, H.; Kohara, Y.; Kono, T.; Nakano, T.; et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 2008, 453, 539–543. [Google Scholar] [CrossRef]
- Tam, O.H.; Aravin, A.A.; Stein, P.; Girard, A.; Murchison, E.P.; Cheloufi, S.; Hodges, E.; Anger, M.; Sachidanandam, R.; Schultz, R.M.; et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 2008, 453, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Aravin, A.A.; Hannon, G.J.; Brennecke, J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007, 318, 761–764. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Yoshitake, K.; Asakawa, S. A Review of Discovery Profiling of PIWI-Interacting RNAs and Their Diverse Functions in Metazoans. Int. J. Mol. Sci. 2021, 22, 11166. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Siomi, M.C. The piRNA pathway in Drosophila ovarian germ and somatic cells. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2020, 96, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramochi-Miyagawa, S.; Watanabe, T.; Gotoh, K.; Totoki, Y.; Toyoda, A.; Ikawa, M.; Asada, N.; Kojima, K.; Yamaguchi, Y.; Ijiri, T.W.; et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 2008, 22, 908–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, B.; Munafò, M.; Ciabrelli, F.; Eastwood, E.L.; Fabry, M.H.; Kneuss, E.; Hannon, G.J. piRNA-Guided Genome Defense: From Biogenesis to Silencing. Annu. Rev. Genet. 2018, 52, 131–157. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Chandramohan, D.; Fioriti, L.; Melnick, A.M.; Hébert, J.M.; Mason, C.E.; Rajasethupathy, P.; Kandel, E.R. Roles for small noncoding RNAs in silencing of retrotransposons in the mammalian brain. Proc. Natl. Acad. Sci. USA 2016, 113, 12697–12702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, Y.; Saito, K.; Kin, T.; Ono, Y.; Asai, K.; Sunohara, T.; Okada, T.N.; Siomi, M.C.; Siomi, H. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature 2008, 453, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Soares, Z.G.; Gonçalves, A.N.A.; de Oliveira, K.P.V.; Marques, J.T. Viral RNA recognition by the Drosophila small interfering RNA pathway. Microb. Infect. 2014, 16, 1013–1021. [Google Scholar] [CrossRef]
- Okamura, K. Diversity of animal small RNA pathways and their biological utility. Wiley Interdiscip. Rev. RNA 2012, 3, 351–368. [Google Scholar] [CrossRef]
- Aliyari, R.; Wu, Q.; Li, H.-W.; Wang, X.-H.; Li, F.; Green, L.D.; Han, C.S.; Li, W.-X.; Ding, S.-W. Mechanism of Induction and Suppression of Antiviral Immunity Directed by Virus-Derived Small RNAs in Drosophila. Cell Host Microbe 2008, 4, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Kazazian, H.H., Jr. L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat. Struct. Mol. Biol. 2006, 13, 763–771. [Google Scholar] [CrossRef]
- Berrens, R.V.; Andrews, S.; Spensberger, D.; Santos, F.; Dean, W.; Gould, P.; Sharif, J.; Olova, N.; Chandra, T.; Koseki, H.; et al. An endosiRNA-Based Repression Mechanism Counteracts Transposon Activation during Global DNA Demethylation in Embryonic Stem Cells. Cell Stem Cell 2017, 21, 694–703.e697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Choudhry, H.; Cao, W. Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like family genes activation and regulation during tumorigenesis. Cancer Sci. 2018, 109, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Goubran, M.H.; Follack, T.B.; Chelico, L. Deamination-independent restriction of LINE-1 retrotransposition by APOBEC3H. Sci. Rep. 2017, 7, 10881. [Google Scholar] [CrossRef] [Green Version]
- Kinomoto, M.; Kanno, T.; Shimura, M.; Ishizaka, Y.; Kojima, A.; Kurata, T.; Sata, T.; Tokunaga, K. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007, 35, 2955–2964. [Google Scholar] [CrossRef]
- Koito, A.; Ikeda, T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 2013, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.A.; Tejwani, L.; Trujillo, C.A.; Negraes, P.D.; Herai, R.H.; Mesci, P.; Macia, A.; Crow, Y.J.; Muotri, A.R. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017, 21, 319–331.e318. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Du, J.; Han, X.; Goodier, J.L.; Li, P.; Zhou, X.; Wei, W.; Evans, S.L.; Li, L.; Zhang, W.; et al. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutières syndrome-related SAMHD1. Cell Rep. 2013, 4, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Aoto, S.; Katagiri, S.; Wang, Y.; Pagnamenta, A.T.; Sakamoto-Abutani, R.; Toyoda, M.; Umezawa, A.; Okamura, K. Frequent retrotransposition of endogenous genes in ERCC2-deficient cells derived from a patient with xeroderma pigmentosum. Stem Cell. Res. Ther. 2019, 10, 273. [Google Scholar] [CrossRef] [Green Version]
- Skariah, G.; Seimetz, J.; Norsworthy, M.; Lannom, M.C.; Kenny, P.J.; Elrakhawy, M.; Forsthoefel, C.; Drnevich, J.; Kalsotra, A.; Ceman, S. Mov10 suppresses retroelements and regulates neuronal development and function in the developing brain. BMC Biol. 2017, 15, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissinot, S.; Entezam, A.; Furano, A.V. Selection against deleterious LINE-1-containing loci in the human lineage. Mol. Biol. Evol. 2001, 18, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurnosov, A.A.; Ustyugova, S.V.; Nazarov, V.I.; Minervina, A.A.; Komkov, A.Y.; Shugay, M.; Pogorelyy, M.V.; Khodosevich, K.V.; Mamedov, I.Z.; Lebedev, Y.B. The Evidence for Increased L1 Activity in the Site of Human Adult Brain Neurogenesis. PLoS ONE 2015, 10, e0117854. [Google Scholar] [CrossRef] [PubMed]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; De Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479, 534–537. [Google Scholar] [CrossRef] [Green Version]
- Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.; James, S.J. Overexpression of LINE-1 Retrotransposons in Autism Brain. Mol. Neurobiol. 2018, 55, 1740–1749. [Google Scholar] [CrossRef]
- Kimura, R.H.; Choudary, P.V.; Schmid, C.W. Silk worm Bm1 SINE RNA increases following cellular insults. Nucleic Acids Res. 1999, 27, 3380–3387. [Google Scholar] [CrossRef] [Green Version]
- Capomaccio, S.; Verini-Supplizi, A.; Galla, G.; Vitulo, N.; Barcaccia, G.; Felicetti, M.; Silvestrelli, M.; Cappelli, K. Transcription of LINE-derived sequences in exercise-induced stress in horses. Anim. Genet. 2010, 41, 23–27. [Google Scholar] [CrossRef]
- Li, T.; Spearow, J.; Rubin, C.M.; Schmid, C.W. Physiological stresses increase mouse short interspersed element (SINE) RNA expression in vivo. Gene 1999, 239, 367–372. [Google Scholar] [CrossRef]
- Dubin, M.J.; Scheid, O.M.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef]
- Lanciano, S.; Mirouze, M. Transposable elements: All mobile, all different, some stress responsive, some adaptive? Curr. Opin. Genet. Dev. 2018, 49, 106–114. [Google Scholar] [CrossRef]
- Dai, J.; Xie, W.; Brady, T.L.; Gao, J.; Voytas, D.F. Phosphorylation Regulates Integration of the Yeast Ty5 Retrotransposon into Heterochromatin. Mol. Cell 2007, 27, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, G.; Marcantonio, P.; Del Re, B. LINE-1 retrotransposition in human neuroblastoma cells is affected by oxidative stress. Cell Tissue Res. 2011, 346, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.P.; Moore, L.; Deininger, P.L.; Roy-Engel, A.M. Heavy metals stimulate human LINE-1 retrotransposition. Int. J. Environ. Res. Public Health 2005, 2, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sawy, M.; Kale, S.P.; Dugan, C.; Nguyen, T.Q.; Belancio, V.; Bruch, H.; Roy-Engel, A.M.; Deininger, P.L. Nickel stimulates L1 retrotransposition by a post-transcriptional mechanism. J. Mol. Biol. 2005, 354, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Farkash, E.A.; Kao, G.D.; Horman, S.R.; Prak, E.T.L. Gamma radiation increases endonuclease-dependent L1 retrotransposition in a cultured cell assay. Nucleic Acids Res. 2006, 34, 1196–1204. [Google Scholar] [CrossRef]
- Okudaira, N.; Ishizaka, Y.; Nishio, H. Retrotransposition of long interspersed element 1 induced by methamphetamine or cocaine. J. Biol. Chem. 2014, 289, 25476–25485. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.M.; Chu, W.M.; Choudary, P.V.; Schmid, C.W. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res. 1995, 23, 1758–1765. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Thompson, C.B. Transcriptional activation of short interspersed elements by DNA-damaging agents. Genes Chromosomes Cancer 2001, 30, 64–71. [Google Scholar] [CrossRef]
- Mariner, P.D.; Walters, R.D.; Espinoza, C.A.; Drullinger, L.F.; Wagner, S.D.; Kugel, J.F.; Goodrich, J.A. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol. Cell 2008, 29, 499–509. [Google Scholar] [CrossRef]
- Espinoza, C.A.; Goodrich, J.A.; Kugel, J.F. Characterization of the structure, function, and mechanism of B2 RNA, an ncRNA repressor of RNA polymerase II transcription. RNA 2007, 13, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Chu, W.M.; Ballard, R.; Carpick, B.W.; Williams, B.R.; Schmid, C.W. Potential Alu function: Regulation of the activity of double-stranded RNA-activated kinase PKR. Mol. Cell. Biol. 1998, 18, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, V.; Merenciano, M.; González, J. Revisiting the Relationship between Transposable Elements and the Eukaryotic Stress Response. Trends Genet. 2017, 33, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Goodier, J.L.; Zhang, L.; Vetter, M.R.; Kazazian, H.H., Jr. LINE-1 ORF1 protein localizes in stress granules with other RNA-binding proteins, including components of RNA interference RNA-induced silencing complex. Mol. Cell. Biol. 2007, 27, 6469–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Li, J.; Xu, F.; Mei, S.; Le Duff, Y.; Yin, L.; Pang, X.; Cen, S.; Jin, Q.; Liang, C.; et al. SAMHD1 Inhibits LINE-1 Retrotransposition by Promoting Stress Granule Formation. PLoS Genet. 2015, 11, e1005367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.G.; Murakami, G.; Dewell, S.; Seligsohn, M.; Baker, M.E.R.; Datson, N.A.; McEwen, B.S.; Pfaff, D.W. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc. Natl. Acad. Sci. USA 2012, 109, 17657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Han, K.; Liang, P. Role of Transposable Elements in Gene Regulation in the Human Genome. Life 2021, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Naville, M.; Warren, I.A.; Haftek-Terreau, Z.; Chalopin, D.; Brunet, F.; Levin, P.; Galiana, D.; Volff, J.N. Not so bad after all: Retroviruses and long terminal repeat retrotransposons as a source of new genes in vertebrates. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2016, 22, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Kaneko-Ishino, T. The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front. Microbiol. 2012, 3, 262. [Google Scholar] [CrossRef] [Green Version]
- Volff, J.N. Turning junk into gold: Domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays 2006, 28, 913–922. [Google Scholar] [CrossRef]
- Estécio, M.R.H.; Gallegos, J.; Dekmezian, M.; Lu, Y.; Liang, S.; Issa, J.-P.J. SINE Retrotransposons Cause Epigenetic Reprogramming of Adjacent Gene Promoters. Mol. Cancer Res. 2012, 10, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Jacques, P.-É.; Jeyakani, J.; Bourque, G. The Majority of Primate-Specific Regulatory Sequences Are Derived from Transposable Elements. PLoS Genet. 2013, 9, e1003504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, M.; Makałowski, W. Transposable elements significantly contributed to the core promoters in the human genome. Sci. China Life Sci. 2019, 62, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Mätlik, K.; Redik, K.; Speek, M. L1 antisense promoter drives tissue-specific transcription of human genes. J. Biomed. Biotechnol. 2006, 2006, 71753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roller, M.; Stamper, E.; Villar, D.; Izuogu, O.; Martin, F.; Redmond, A.M.; Ramachanderan, R.; Harewood, L.; Odom, D.T.; Flicek, P. LINE retrotransposons characterize mammalian tissue-specific and evolutionarily dynamic regulatory regions. Genome Biol. 2021, 22, 62. [Google Scholar] [CrossRef]
- van de Lagemaat, L.N.; Landry, J.R.; Mager, D.L.; Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003, 19, 530–536. [Google Scholar] [CrossRef]
- Schoorlemmer, J.; Pérez-Palacios, R.; Climent, M.; Guallar, D.; Muniesa, P. Regulation of Mouse Retroelement MuERV-L/MERVL Expression by REX1 and Epigenetic Control of Stem Cell Potency. Front. Oncol. 2014, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Jjingo, D.; Conley, A.B.; Wang, J.; Mariño-Ramírez, L.; Lunyak, V.V.; Jordan, I.K. Mammalian-wide interspersed repeat (MIR)-derived enhancers and the regulation of human gene expression. Mob. DNA 2014, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Han, D.; Boyd-Kirkup, J.; Yu, X.; Han, J.J. Evolution of Alu elements toward enhancers. Cell Rep. 2014, 7, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.O.; Gingeras, T.R.; Weng, Z. Genome-wide analysis of polymerase III-transcribed Alu elements suggests cell-type-specific enhancer function. Genome Res. 2019, 29, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L.; Yang, L. ALUternative Regulation for Gene Expression. Trends Cell Biol. 2017, 27, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Vicente-García, C.; Seruggia, D.; Moltó, E.; Fernandez-Miñán, A.; Neto, A.; Lee, E.; Gómez-Skarmeta, J.L.; Montoliu, L.; Lunyak, V.V.; et al. MIR retrotransposon sequences provide insulators to the human genome. Proc. Natl. Acad. Sci. USA 2015, 112, E4428–E4437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, A.G.; Ouyang, N.; Boyle, A.P. Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes. Nat. Commun. 2020, 11, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Román, A.C.; González-Rico, F.J.; Moltó, E.; Hernando, H.; Neto, A.; Vicente-Garcia, C.; Ballestar, E.; Gómez-Skarmeta, J.L.; Vavrova-Anderson, J.; White, R.J.; et al. Dioxin receptor and SLUG transcription factors regulate the insulator activity of B1 SINE retrotransposons via an RNA polymerase switch. Genome Res. 2011, 21, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunyak, V.V.; Prefontaine, G.G.; Núñez, E.; Cramer, T.; Ju, B.G.; Ohgi, K.A.; Hutt, K.; Roy, R.; García-Díaz, A.; Zhu, X.; et al. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science 2007, 317, 248–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, V.J.; Leclerc, R.D.; May, G.; Wagner, G.P. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet. 2011, 43, 1154–1159. [Google Scholar] [CrossRef]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Lynch, V.J.; Nnamani, M.C.; Kapusta, A.; Brayer, K.; Plaza, S.L.; Mazur, E.C.; Emera, D.; Sheikh, S.Z.; Grützner, F.; Bauersachs, S.; et al. Ancient Transposable Elements Transformed the Uterine Regulatory Landscape and Transcriptome during the Evolution of Mammalian Pregnancy. Cell Rep. 2015, 10, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Chuong, E.B. Retroviruses facilitate the rapid evolution of the mammalian placenta. Bioessays 2013, 35, 853–861. [Google Scholar] [CrossRef]
- Chuong, E.B.; Rumi, M.A.K.; Soares, M.J.; Baker, J.C. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet. 2013, 45, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, A.; Marsano, R.M. Transposable elements: A jump toward the future of expression vectors. Crit. Rev. Biotechnol. 2021, 41, 792–808. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, X.; Tang, Z.; Grivainis, M.; Kahler, D.; Yun, C.; Mita, P.; Fenyö, D.; Boeke, J.D. Transcription factor profiling reveals molecular choreography and key regulators of human retrotransposon expression. Proc. Natl. Acad. Sci. USA 2018, 115, E5526–E5535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.R.; Dewan, A.; Zupnick, A.; Normart, R.; Gabriel, A.; Prives, C.; Levine, A.J.; Hoh, J. p53 responsive elements in human retrotransposons. Oncogene 2009, 28, 3857–3865. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Erwin, J.A.; Marchetto, M.C.; Gage, F.H. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 2014, 15, 497–506. [Google Scholar] [CrossRef]
- Sanchez-Luque, F.J.; Kempen, M.H.C.; Gerdes, P.; Vargas-Landin, D.B.; Richardson, S.R.; Troskie, R.L.; Jesuadian, J.S.; Cheetham, S.W.; Carreira, P.E.; Salvador-Palomeque, C.; et al. LINE-1 Evasion of Epigenetic Repression in Humans. Mol. Cell 2019, 75, 590–604.e512. [Google Scholar] [CrossRef]
- Athanikar, J.N.; Badge, R.M.; Moran, J.V. A YY1-binding site is required for accurate human LINE-1 transcription initiation. Nucleic Acids Res. 2004, 32, 3846–3855. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Zhang, L.; Zhang, Y.; Kazazian, H.H., Jr. An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Res. 2003, 31, 4929–4940. [Google Scholar] [CrossRef]
- Tchénio, T.; Casella, J.F.; Heidmann, T. Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 2000, 28, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.-J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. p53 genes function to restrain mobile elements. Genes Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, B.; Jones, A.E.; Caillet, C.J.; Das, S.; Royer, S.K.; Abrams, J.M. p53 directly represses human LINE1 transposons. Genes Dev. 2020, 34, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Domany, E. Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genom. 2006, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Piedrafita, F.J.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 1996, 271, 14412–14420. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bolten, C.; Bhat, B.G.; Woodring-Dietz, J.; Li, S.; Prayaga, S.K.; Xia, C.; Lala, D.S. Induction of Human Liver X Receptor α Gene Expression Via an Autoregulatory Loop Mechanism. Mol. Endocrinol. 2002, 16, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Norris, J.; Fan, D.; Aleman, C.; Marks, J.R.; Futreal, P.A.; Wiseman, R.W.; Iglehart, J.D.; Deininger, P.L.; McDonnell, D.P. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J. Biol. Chem. 1995, 270, 22777–22782. [Google Scholar] [CrossRef] [Green Version]
- Vansant, G.; Reynolds, W.F. The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc. Natl. Acad. Sci. USA 1995, 92, 8229–8233. [Google Scholar] [CrossRef] [Green Version]
- Antonaki, A.; Demetriades, C.; Polyzos, A.; Banos, A.; Vatsellas, G.; Lavigne, M.D.; Apostolou, E.; Mantouvalou, E.; Papadopoulou, D.; Mosialos, G.; et al. Genomic analysis reveals a novel nuclear factor-κB (NF-κB)-binding site in Alu-repetitive elements. J. Biol. Chem. 2011, 286, 38768–38782. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; de Llobet Cucalon, L.I.; Di Vona, C.; Le Dilly, F.; Vidal, E.; Lioutas, A.; Oliete, J.Q.; Jochem, L.; Cutts, E.; Dieci, G.; et al. TFIIIC Binding to Alu Elements Controls Gene Expression via Chromatin Looping and Histone Acetylation. Mol. Cell 2020, 77, 475–487.e411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crepaldi, L.; Policarpi, C.; Coatti, A.; Sherlock, W.T.; Jongbloets, B.C.; Down, T.A.; Riccio, A. Binding of TFIIIC to sine elements controls the relocation of activity-dependent neuronal genes to transcription factories. PLoS Genet. 2013, 9, e1003699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnada, S.M.; Isopi, A.; Tejada-Martinez, D.; Goubert, C.; Patoori, S.; Pagliaroli, L.; Tracewell, M.; Trizzino, M. Genomic features underlie the co-option of SVA transposons as cis-regulatory elements in human pluripotent stem cells. bioRxiv 2022, 2022.2001.2010.475682. [Google Scholar] [CrossRef]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, N.V.; Kraft, M.; Tondera, C.; Hanschmann, K.-M.; Löwer, J.; Löwer, R. Expression of the human endogenous retrovirus (HERV) group HML-2/HERV-K does not depend on canonical promoter elements but is regulated by transcription factors Sp1 and Sp3. J. Virol. 2011, 85, 3436–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manghera, M.; Douville, R.N. Endogenous retrovirus-K promoter: A landing strip for inflammatory transcription factors? Retrovirology 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-κB and IRF1 Induce Endogenous Retrovirus K Expression via Interferon-Stimulated Response Elements in Its 5′ Long Terminal Repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotea, V.; Makałowski, W. Do transposable elements really contribute to proteomes? Trends Genet. 2006, 22, 260–267. [Google Scholar] [CrossRef]
- Toll-Riera, M.; Bosch, N.; Bellora, N.; Castelo, R.; Armengol, L.; Estivill, X.; Mar Albà, M. Origin of Primate Orphan Genes: A Comparative Genomics Approach. Mol. Biol. Evol. 2009, 26, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Moran, J.V.; DeBerardinis, R.J.; Kazazian, H.H. Exon Shuffling by L1 Retrotransposition. Science 1999, 283, 1530. [Google Scholar] [CrossRef]
- Pickeral, O.K.; Makałowski, W.; Boguski, M.S.; Boeke, J.D. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 2000, 10, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancks, D.C.; Ewing, A.D.; Chen, J.E.; Tokunaga, K.; Kazazian, H.H., Jr. Exon-trapping mediated by the human retrotransposon SVA. Genome Res. 2009, 19, 1983–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev-Maor, G.; Sorek, R.; Shomron, N.; Ast, G. The birth of an alternatively spliced exon: 3’ splice-site selection in Alu exons. Science 2003, 300, 1288–1291. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Ast, G.; Graur, D. Alu-containing exons are alternatively spliced. Genome Res. 2002, 12, 1060–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorek, R. The birth of new exons: Mechanisms and evolutionary consequences. RNA 2007, 13, 1603–1608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.F.; Chasin, L.A. Comparison of multiple vertebrate genomes reveals the birth and evolution of human exons. Proc. Natl. Acad. Sci. USA 2006, 103, 13427–13432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal-Mark, N.; Schwartz, S.; Ast, G. Alternative splicing of Alu exons—Two arms are better than one. Nucleic Acids Res. 2008, 36, 2012–2023. [Google Scholar] [CrossRef] [Green Version]
- Sela, N.; Mersch, B.; Gal-Mark, N.; Lev-Maor, G.; Hotz-Wagenblatt, A.; Ast, G. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu’s unique role in shaping the human transcriptome. Genome Biol. 2007, 8, R127. [Google Scholar] [CrossRef]
- Mandal, A.K.; Pandey, R.; Jha, V.; Mukerji, M. Transcriptome-wide expansion of non-coding regulatory switches: Evidence from co-occurrence of Alu exonization, antisense and editing. Nucleic Acids Res. 2013, 41, 2121–2137. [Google Scholar] [CrossRef] [Green Version]
- Belancio, V.P.; Roy-Engel, A.M.; Deininger, P. The impact of multiple splice sites in human L1 elements. Gene 2008, 411, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Zemojtel, T.; Penzkofer, T.; Schultz, J.; Dandekar, T.; Badge, R.; Vingron, M. Exonization of active mouse L1s: A driver of transcriptome evolution? BMC Genom. 2007, 8, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaer, K.; Branovets, J.; Hallikma, A.; Nigumann, P.; Speek, M. Intronic L1 retrotransposons and nested genes cause transcriptional interference by inducing intron retention, exonization and cryptic polyadenylation. PLoS ONE 2011, 6, e26099. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, G.; Lowe, C.B.; Ahituv, N.; King, B.; Siepel, A.; Salama, S.R.; Rubin, E.M.; Kent, W.J.; Haussler, D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 2006, 441, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Roy-Engel, A.M.; El-Sawy, M.; Farooq, L.; Odom, G.L.; Perepelitsa-Belancio, V.; Bruch, H.; Oyeniran, O.O.; Deininger, P.L. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet. Genome Res. 2005, 110, 365–371. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ji, Z.; Tian, B. Phylogenetic analysis of mRNA polyadenylation sites reveals a role of transposable elements in evolution of the 3′-end of genes. Nucleic Acids Res. 2008, 36, 5581–5590. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Ara, T.; Gautheret, D. Using Alu elements as polyadenylation sites: A case of retroposon exaptation. Mol. Biol. Evol. 2009, 26, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-L.; DeCerbo, J.N.; Carmichael, G.G. Alu element-mediated gene silencing. EMBO J. 2008, 27, 1694–1705. [Google Scholar] [CrossRef]
- Chen, L.-L.; Carmichael, G.G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: Functional role of a nuclear noncoding RNA. Mol. Cell 2009, 35, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Daniel, C.; Silberberg, G.; Behm, M.; Öhman, M. Alu elements shape the primate transcriptome by cis-regulation of RNA editing. Genome Biol. 2014, 15, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswami, G.; Lin, W.; Piskol, R.; Tan, M.H.; Davis, C.; Li, J.B. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods 2012, 9, 579–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeman, Y.; Levanon, E.Y.; Jantsch, M.F.; Eisenberg, E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 2006, 12, 1802–1809. [Google Scholar] [CrossRef] [Green Version]
- Muotri, A.R.; Gage, F.H. Generation of neuronal variability and complexity. Nature 2006, 441, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef]
- Feng, Y.; Sansam, C.L.; Singh, M.; Emeson, R.B. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol. Cell. Biol. 2006, 26, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.-H.; Bindereif, A. Exon Circularization Requires Canonical Splice Signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Clayton, E.A.; Rishishwar, L.; Huang, T.-C.; Gulati, S.; Ban, D.; McDonald, J.F.; Jordan, I.K. An atlas of transposable element-derived alternative splicing in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190342. [Google Scholar] [CrossRef]
- Lehnert, S.; Van Loo, P.; Thilakarathne, P.J.; Marynen, P.; Verbeke, G.; Schuit, F.C. Evidence for co-evolution between human microRNAs and Alu-repeats. PLoS ONE 2009, 4, e4456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Jha, V.; Singhal, K.; Fatima, M.; Singh, D.; Chaturvedi, G.; Dholakia, D.; Kutum, R.; Pandey, R.; Bakken, T.E.; et al. Multiple Alu Exonization in 3’UTR of a Primate-Specific Isoform of CYP20A1 Creates a Potential miRNA Sponge. Genome Biol. Evol. 2021, 13, evaa233. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Nakabeppu, Y.; Furuichi, M.; Sekiguchi, M. Regulation of expression of the human MTH1 gene encoding 8-oxo-dGTPase. Alternative splicing of transcription products. J. Biol. Chem. 1997, 272, 17843–17850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henssen, A.G.; Henaff, E.; Jiang, E.; Eisenberg, A.R.; Carson, J.R.; Villasante, C.M.; Ray, M.; Still, E.; Burns, M.; Gandara, J.; et al. Genomic DNA transposition induced by human PGBD5. Elife 2015, 4, e10565. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005, 3, e181. [Google Scholar] [CrossRef] [Green Version]
- Carmona, L.M.; Fugmann, S.D.; Schatz, D.G. Collaboration of RAG2 with RAG1-like proteins during the evolution of V(D)J recombination. Genes Dev. 2016, 30, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Carmona, L.M.; Schatz, D.G. New insights into the evolutionary origins of the recombination-activating gene proteins and V(D)J recombination. FEBS J. 2017, 284, 1590–1605. [Google Scholar] [CrossRef]
- Chun, J.J.; Schatz, D.G.; Oettinger, M.A.; Jaenisch, R.; Baltimore, D. The recombination activating gene-1 (RAG-1) transcript is present in the murine central nervous system. Cell 1991, 64, 189–200. [Google Scholar] [CrossRef]
- Jessen, J.R.; Jessen, T.N.; Vogel, S.S.; Lin, S. Concurrent expression of recombination activating genes 1 and 2 in zebrafish olfactory sensory neurons. Genesis 2001, 29, 156–162. [Google Scholar] [CrossRef]
- Álvarez-Lindo, N.; Baleriola, J.; de Los Ríos, V.; Suárez, T.; de la Rosa, E.J. RAG-2 deficiency results in fewer phosphorylated histone H2AX foci, but increased retinal ganglion cell death and altered axonal growth. Sci. Rep. 2019, 9, 18486. [Google Scholar] [CrossRef]
- Mateo, L.; González, J. Pogo-like transposases have been repeatedly domesticated into CENP-B-related proteins. Genome Biol. Evol. 2014, 6, 2008–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Wang, Y.; Diaby, M.; Zong, W.; Shen, D.; Wang, S.; Chen, C.; Wang, X.; Song, C. Evolution of pogo, a separate superfamily of IS630-Tc1-mariner transposons, revealing recurrent domestication events in vertebrates. Mob. DNA 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Fachinetti, D. DNA Sequences in Centromere Formation and Function. In Centromeres and Kinetochores: Discovering the Molecular Mechanisms Underlying Chromosome Inheritance; Black, B.E., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 305–336. [Google Scholar]
- Bannert, N.; Kurth, R. Retroelements and the human genome: New perspectives on an old relation. Proc. Natl. Acad. Sci. USA 2004, 101, 14572–14579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupressoir, A.; Lavialle, C.; Heidmann, T. From ancestral infectious retroviruses to bona fide cellular genes: Role of the captured syncytins in placentation. Placenta 2012, 33, 663–671. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, Y.; Koshi, K.; Nakagawa, S.; Hashizume, K.; Miyazawa, T. Fematrin-1 is involved in fetomaternal cell-to-cell fusion in Bovinae placenta and has contributed to diversity of ruminant placentation. J. Virol. 2013, 87, 10563–10572. [Google Scholar] [CrossRef] [Green Version]
- Brandt, J.; Schrauth, S.; Veith, A.-M.; Froschauer, A.; Haneke, T.; Schultheis, C.; Gessler, M.; Leimeister, C.; Volff, J.-N. Transposable elements as a source of genetic innovation: Expression and evolution of a family of retrotransposon-derived neogenes in mammals. Gene 2005, 345, 101–111. [Google Scholar] [CrossRef]
- Henke, C.; Strissel, P.L.; Schubert, M.-T.; Mitchell, M.; Stolt, C.C.; Faschingbauer, F.; Beckmann, M.W.; Strick, R. Selective expression of sense and antisense transcripts of the sushi-ichi-related retrotransposon--derived family during mouse placentogenesis. Retrovirology 2015, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Rawn, S.M.; Cross, J.C. The Evolution, Regulation, and Function of Placenta-Specific Genes. Annu. Rev. Cell. Dev. Biol. 2008, 24, 159–181. [Google Scholar] [CrossRef]
- Naruse, M.; Ono, R.; Irie, M.; Nakamura, K.; Furuse, T.; Hino, T.; Oda, K.; Kashimura, M.; Yamada, I.; Wakana, S.; et al. Sirh7/Ldoc1 knockout mice exhibit placental P4 overproduction and delayed parturition. Development 2014, 141, 4763–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, M.; Yoshikawa, M.; Ono, R.; Iwafune, H.; Furuse, T.; Yamada, I.; Wakana, S.; Yamashita, Y.; Abe, T.; Ishino, F.; et al. Cognitive Function Related to the Sirh11/Zcchc16 Gene Acquired from an LTR Retrotransposon in Eutherians. PLoS Genet. 2015, 11, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Oshige, M.; Durant, S.T.; Rasila, K.K.; Williamson, E.A.; Ramsey, H.; Kwan, L.; Nickoloff, J.A.; Hromas, R. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaux, R.; Udit, S.; Batzer, M.A.; Feschotte, C. Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc. Natl. Acad. Sci. USA 2006, 103, 8101–8106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [Green Version]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [Green Version]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and evolution of human microRNAs from transposable elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef] [Green Version]
- Greenig, M. HERVs, immunity, and autoimmunity: Understanding the connection. PeerJ 2019, 7, e6711. [Google Scholar] [CrossRef] [Green Version]
- Hurst, T.P.; Magiorkinis, G. Activation of the innate immune response by endogenous retroviruses. J. Gen. Virol. 2015, 96, 1207–1218. [Google Scholar] [CrossRef]
- Perron, H.; Dougier-Reynaud, H.-L.; Lomparski, C.; Popa, I.; Firouzi, R.; Bertrand, J.-B.; Marusic, S.; Portoukalian, J.; Jouvin-Marche, E.; Villiers, C.L.; et al. Human endogenous retrovirus protein activates innate immunity and promotes experimental allergic encephalomyelitis in mice. PLoS ONE 2013, 8, e80128. [Google Scholar] [CrossRef]
- Curcio, M.J.; Belfort, M. The beginning of the end: Links between ancient retroelements and modern telomerases. Proc. Natl. Acad. Sci. USA 2007, 104, 9107–9108. [Google Scholar] [CrossRef] [Green Version]
- Belfort, M.; Curcio, M.J.; Lue, N.F. Telomerase and retrotransposons: Reverse transcriptases that shaped genomes. Proc. Natl. Acad. Sci. USA 2011, 108, 20304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casacuberta, E. Drosophila: Retrotransposons Making up Telomeres. Viruses 2017, 9, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podlevsky, J.D.; Chen, J.J.L. It all comes together at the ends: Telomerase structure, function, and biogenesis. Mutat. Res. 2012, 730, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelini, F.; Jalihal, A.P.; Francia, S.; Meers, C.; Neeb, Z.T.; Rossiello, F.; Gioia, U.; Aguado, J.; Jones-Weinert, C.; Luke, B.; et al. From “Cellular” RNA to “Smart” RNA: Multiple Roles of RNA in Genome Stability and Beyond. Chem. Rev. 2018, 118, 4365–4403. [Google Scholar] [CrossRef] [PubMed]
- Morrish, T.A.; Garcia-Perez, J.L.; Stamato, T.D.; Taccioli, G.E.; Sekiguchi, J.; Moran, J.V. Endonuclease-independent LINE-1 retrotransposition at mammalian telomeres. Nature 2007, 446, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Zvereva, M.I.; Shcherbakova, D.M.; Dontsova, O.A. Telomerase: Structure, functions, and activity regulation. Biochem. Biokhimiia 2010, 75, 1563–1583. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef]
- Mason, J.M.; Randall, T.A.; Frydrychova, R.C. Telomerase lost? Chromosoma 2016, 125, 65–73. [Google Scholar] [CrossRef]
- Fujiwara, H.; Osanai, M.; Matsumoto, T.; Kojima, K.K. Telomere-specific non-LTR retrotransposons and telomere maintenance in the silkworm, Bombyx mori. Chromosome Res. 2005, 13, 455–467. [Google Scholar] [CrossRef]
- Gladyshev, E.A.; Arkhipova, I.R. Telomere-associated endonuclease-deficient Penelope-like retroelements in diverse eukaryotes. Proc. Natl. Acad. Sci. USA 2007, 104, 9352–9357. [Google Scholar] [CrossRef] [Green Version]
- Saint-Leandre, B.; Nguyen, S.C.; Levine, M.T. Diversification and collapse of a telomere elongation mechanism. Genome Res. 2019, 29, 920–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meers, C.; Keskin, H.; Storici, F. DNA repair by RNA: Templated, or not templated, that is the question. DNA Repair 2016, 44, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Nakajima, S.; Böhm, S.; Bernstein, K.A.; Shen, Z.; Tsang, M.; Levine, A.S.; Lan, L. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc. Natl. Acad. Sci. USA 2015, 112, E3495–E3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puget, N.; Miller, K.M.; Legube, G. Non-canonical DNA/RNA structures during Transcription-Coupled Double-Strand Break Repair: Roadblocks or Bona fide repair intermediates? DNA Repair 2019, 81, 102661. [Google Scholar] [CrossRef]
- Su, Y.; Ghodke, P.P.; Egli, M.; Li, L.; Wang, Y.; Guengerich, F.P. Human DNA polymerase η has reverse transcriptase activity in cellular environments. J. Biol. Chem. 2019, 294, 6073–6081. [Google Scholar] [CrossRef] [Green Version]
- Meers, C.; Keskin, H.; Banyai, G.; Mazina, O.; Yang, T.; Gombolay, A.L.; Mukherjee, K.; Kaparos, E.I.; Newnam, G.; Mazin, A.; et al. Genetic Characterization of Three Distinct Mechanisms Supporting RNA-Driven DNA Repair and Modification Reveals Major Role of DNA Polymerase ζ. Mol. Cell 2020, 79, 1037–1050.e1035. [Google Scholar] [CrossRef]
- Teng, S.C.; Kim, B.; Gabriel, A. Retrotransposon reverse-transcriptase-mediated repair of chromosomal breaks. Nature 1996, 383, 641–644. [Google Scholar] [CrossRef]
- Keskin, H.; Shen, Y.; Huang, F.; Patel, M.; Yang, T.; Ashley, K.; Mazin, A.V.; Storici, F. Transcript-RNA-templated DNA recombination and repair. Nature 2014, 515, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Ono, R.; Ishii, M.; Fujihara, Y.; Kitazawa, M.; Usami, T.; Kaneko-Ishino, T.; Kanno, J.; Ikawa, M.; Ishino, F. Double strand break repair by capture of retrotransposon sequences and reverse-transcribed spliced mRNA sequences in mouse zygotes. Sci. Rep. 2015, 5, 12281. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef]
- Welty, S.; Teng, Y.; Liang, Z.; Zhao, W.; Sanders, L.H.; Greenamyre, J.T.; Rubio, M.E.; Thathiah, A.; Kodali, R.; Wetzel, R.; et al. RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 2018, 293, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Srikanta, D.; Sen, S.K.; Huang, C.T.; Conlin, E.M.; Rhodes, R.M.; Batzer, M.A. An alternative pathway for Alu retrotransposition suggests a role in DNA double-strand break repair. Genomics 2009, 93, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushman, D.M.; Chun, J. The genomically mosaic brain: Aneuploidy and more in neural diversity and disease. Semin. Cell Dev. Biol. 2013, 24, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehen, S.K.; Yung, Y.C.; McCreight, M.P.; Kaushal, D.; Yang, A.H.; Almeida, B.S.V.; Kingsbury, M.A.; Cabral, K.M.S.; McConnell, M.J.; Anliker, B.; et al. Constitutional aneuploidy in the normal human brain. J. Neurosci. 2005, 25, 2176–2180. [Google Scholar] [CrossRef]
- Cai, X.; Evrony, G.D.; Lehmann, H.S.; Elhosary, P.C.; Mehta, B.K.; Poduri, A.; Walsh, C.A. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2014, 8, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poduri, A.; Evrony, G.D.; Cai, X.; Walsh, C.A. Somatic Mutation, Genomic Variation, and Neurological Disease. Science 2013, 341, 1237758. [Google Scholar] [CrossRef] [Green Version]
- Kingsbury, M.A.; Friedman, B.; McConnell, M.J.; Rehen, S.K.; Yang, A.H.; Kaushal, D.; Chun, J. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc. Natl. Acad. Sci. USA 2005, 102, 6143–6147. [Google Scholar] [CrossRef] [Green Version]
- Westra, J.W.; Rivera, R.R.; Bushman, D.M.; Yung, Y.C.; Peterson, S.E.; Barral, S.; Chun, J. Neuronal DNA content variation (DCV) with regional and individual differences in the human brain. J. Comp. Neurol. 2010, 518, 3981–4000. [Google Scholar] [CrossRef] [Green Version]
- Lodato, M.A.; Woodworth, M.B.; Lee, S.; Evrony, G.D.; Mehta, B.K.; Karger, A.; Lee, S.; Chittenden, T.W.; D’Gama, A.M.; Cai, X.; et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015, 350, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wu, Q.; Ye, A.Y.; Guo, J.; Zheng, X.; Yang, X.; Yan, L.; Liu, Q.-R.; Hyde, T.M.; Wei, L.; et al. Somatic LINE-1 retrotransposition in cortical neurons and non-brain tissues of Rett patients and healthy individuals. PLoS Genet. 2019, 15, e1008043. [Google Scholar] [CrossRef] [Green Version]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehen, S.K.; McConnell, M.J.; Kaushal, D.; Kingsbury, M.A.; Yang, A.H.; Chun, J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl. Acad. Sci. USA 2001, 98, 13361–13366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kole, A.J.; Annis, R.P.; Deshmukh, M. Mature neurons: Equipped for survival. Cell Death Dis. 2013, 4, e689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.-C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Schwer, B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 2018, 71, 158–163. [Google Scholar] [CrossRef]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [Green Version]
- Terry, D.M.; Devine, S.E. Aberrantly High Levels of Somatic LINE-1 Expression and Retrotransposition in Human Neurological Disorders. Front. Genet. 2020, 10, 1244. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, G.J.; Garcia-Perez, J.L. L1 Mosaicism in Mammals: Extent, Effects, and Evolution. Trends Genet. 2017, 33, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O’Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460, 1127–1131. [Google Scholar] [CrossRef] [Green Version]
- Upton, K.R.; Gerhardt, D.J.; Jesuadian, J.S.; Richardson, S.R.; Sánchez-Luque, F.J.; Bodea, G.O.; Ewing, A.D.; Salvador-Palomeque, C.; van der Knaap, M.S.; Brennan, P.M.; et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 2015, 161, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macia, Á.; Widmann, T.J.; Heras, S.R.; Ayllón, V.; Sanchez, L.P.; Benkaddour-Boumzaouad, M.; Muñoz-López, M.; Rubio, A.M.; Amador-Cubero, S.; Blanco-Jimenez, E.; et al. Engineered LINE-1 retrotransposition in nondividing human neurons. Genome Res. 2017, 27, 335–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador-Palomeque, C.; Francisco, J.S.L.; Patrick, R.J.F.; Adam, D.E.; Ernst, J.W.; Sandra, R.R.; Geoffrey, J.F. Dynamic Methylation of an L1 Transduction Family during Reprogramming and Neurodifferentiation. Mol. Cell. Biol. 2019, 39, e00499-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, E.C.; Devine, S.E. The Role of Somatic L1 Retrotransposition in Human Cancers. Viruses 2017, 9, 131. [Google Scholar] [CrossRef]
- Naumova, O.Y.; Lee, M.; Rychkov, S.Y.; Vlasova, N.V.; Grigorenko, E.L. Gene expression in the human brain: The current state of the study of specificity and spatiotemporal dynamics. Child Dev. 2013, 84, 76–88. [Google Scholar] [CrossRef]
- Bae, B.I.; Jayaraman, D.; Walsh, C.A. Genetic changes shaping the human brain. Dev. Cell 2015, 32, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Negi, S.K.; Guda, C. Global gene expression profiling of healthy human brain and its application in studying neurological disorders. Sci. Rep. 2017, 7, 897. [Google Scholar] [CrossRef]
- Zylka, M.J.; Simon, J.M.; Philpot, B.D. Gene length matters in neurons. Neuron 2015, 86, 353–355. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.J.; Fire, A.Z. Intron and gene size expansion during nervous system evolution. BMC Genom. 2020, 21, 360. [Google Scholar] [CrossRef]
- McCoy, M.J.; Paul, A.J.; Victor, M.B.; Richner, M.; Gabel, H.W.; Gong, H.; Yoo, A.S.; Ahn, T.H. LONGO: An R package for interactive gene length dependent analysis for neuronal identity. Bioinformatics 2018, 34, i422–i428. [Google Scholar] [CrossRef]
- Wimmer, K.; Callens, T.; Wernstedt, A.; Messiaen, L. The NF1 gene contains hotspots for L1 endonuclease-dependent de novo insertion. PLoS Genet. 2011, 7, e1002371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, Y.P. Neurofibromin signaling and synapses. J. Biomed. Sci. 2007, 14, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Sur, D.; Kustwar, R.K.; Budania, S.; Mahadevan, A.; Hancks, D.C.; Yadav, V.; Shankar, S.K.; Mandal, P.K. Detection of the LINE-1 retrotransposon RNA-binding protein ORF1p in different anatomical regions of the human brain. Mob. DNA 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, J.J.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 151, 483–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrat, P.N.; DasGupta, S.; Wang, J.; Theurkauf, W.; Weng, Z.; Rosbash, M.; Waddell, S. Transposition-driven genomic heterogeneity in the Drosophila brain. Science 2013, 340, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Treiber, C.D.; Waddell, S. Resolving the prevalence of somatic transposition in Drosophila. Elife 2017, 6, e28297. [Google Scholar] [CrossRef] [Green Version]
- Li, J.B.; Church, G.M. Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat. Neurosci. 2013, 16, 1518–1522. [Google Scholar] [CrossRef] [Green Version]
- Maas, S.; Kawahara, Y.; Tamburro, K.M.; Nishikura, K. A-to-I RNA editing and human disease. RNA Biol. 2006, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S.; Mehler, M.F. RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci. 2008, 31, 227–233. [Google Scholar] [CrossRef]
- Yablonovitch, A.L.; Deng, P.; Jacobson, D.; Li, J.B. The evolution and adaptation of A-to-I RNA editing. PLoS Genet. 2017, 13, e1007064. [Google Scholar] [CrossRef] [Green Version]
- Mehler, M.F.; Mattick, J.S. Noncoding RNAs and RNA Editing in Brain Development, Functional Diversification, and Neurological Disease. Physiol. Rev. 2007, 87, 799–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlstedt, H.; Daniel, C.; Ensterö, M.; Ohman, M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009, 19, 978–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekdahl, Y.; Farahani, H.S.; Behm, M.; Lagergren, J.; Öhman, M. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res. 2012, 22, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, J.J.C.; Seeburg, P.H. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron 2012, 74, 432–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, S.; Kawahara, Y. Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J. Mol. Med. 2005, 83, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, M.E.; Brattås, P.L.; Gustafsson, C.; Petri, R.; Yudovich, D.; Pircs, K.; Verschuere, S.; Madsen, S.; Hansson, J.; Larsson, J.; et al. Activation of neuronal genes via LINE-1 elements upon global DNA demethylation in human neural progenitors. Nat. Commun. 2019, 10, 3182. [Google Scholar] [CrossRef] [Green Version]
- Turelli, P.; Playfoot, C.; Grun, D.; Raclot, C.; Pontis, J.; Coudray, A.; Thorball, C.; Duc, J.; Eugenia, V.P.; Deplancke, B.; et al. Primate-restricted KRAB zinc finger proteins and target retrotransposons control gene expression in human neurons. Sci. Adv. 2020, 6, eaba3200. [Google Scholar] [CrossRef]
- Wang, T.; Medynets, M.; Johnson, K.R.; Doucet-O’Hare, T.T.; DiSanza, B.; Li, W.; Xu, Y.; Bagnell, A.; Tyagi, R.; Sampson, K.; et al. Regulation of stem cell function and neuronal differentiation by HERV-K via mTOR pathway. Proc. Natl. Acad. Sci. USA 2020, 117, 17842–17853. [Google Scholar] [CrossRef]
- Nair, V.P.; Liu, H.; Ciceri, G.; Jungverdorben, J.; Frishman, G.; Tchieu, J.; Cederquist, G.Y.; Rothenaigner, I.; Schorpp, K.; Klepper, L.; et al. Activation of HERV-K(HML-2) disrupts cortical patterning and neuronal differentiation by increasing NTRK3. Cell Stem Cell 2021, 28, 1566–1581.e1568. [Google Scholar] [CrossRef]
- Vago, D.R.; Wallenstein, G.V.; Morris, L.S. Hippocampus. In Encyclopedia of the Neurological Sciences, 2nd ed.; Aminoff, M.J., Daroff, R.B., Eds.; Academic Press: Oxford, UK, 2014; pp. 566–570. [Google Scholar] [CrossRef]
- McEwen, B.S.; Nasca, C.; Gray, J.D. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.G.; Gagnidze, K.; McEwen, B.S.; Pfaff, D.W. Stress and the dynamic genome: Steroids, epigenetics, and the transposome. Proc. Natl. Acad. Sci. USA 2015, 112, 6828–6833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedrosian, T.A.; Quayle, C.; Novaresi, N.; Gage, F.H. Early life experience drives structural variation of neural genomes in mice. Science 2018, 359, 1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkina, O.I.; Zots, M.A.; Bezriadnov, D.V.; Anokhin, K.V. Erratum to: Increased 5-Bromo-2’-Deoxyuridine Incorporation in Various Brain Structures Following Passive Avoidance Training in Mice. Bull. Exp. Biol. Med. 2012, 154, 171–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salganik, R.I.; Parvez, H.; Tomson, V.P.; Shumskaya, I.A. Probable role of reverse transcription in learning: Correlation between hippocampal RNA-dependent DNA synthesis and learning ability in rats. Neurosci. Lett. 1983, 36, 317–322. [Google Scholar] [CrossRef]
- Kokaeva, F.F.; Den’mukhametova, S.V.; Kanapin, A.A.; Godukhin, O.V.; Il’in, Y.; Ivanov, V.A. Antisence oligodeoxyribonucleotides for fragments of the reverse transcriptase gene of the LINE-1 element of rats disturb the formation of long-term memory. Dokl. Biochem. Biophys. 2002, 383, 93–95. [Google Scholar] [CrossRef]
- Bachiller, S.; Del-Pozo-Martín, Y.; Carrión, Á.M. L1 retrotransposition alters the hippocampal genomic landscape enabling memory formation. Brain Behav. Immun. 2017, 64, 65–70. [Google Scholar] [CrossRef]
- Ponomarev, I.; Rau, V.; Eger, E.I.; Harris, R.A.; Fanselow, M.S. Amygdala transcriptome and cellular mechanisms underlying stress-enhanced fear learning in a rat model of posttraumatic stress disorder. Neuropsychopharmacology 2010, 35, 1402–1411. [Google Scholar] [CrossRef] [Green Version]
- Cappucci, U.; Torromino, G.; Casale, A.M.; Camon, J.; Capitano, F.; Berloco, M.; Mele, A.; Pimpinelli, S.; Rinaldi, A.; Piacentini, L. Stress-induced strain and brain region-specific activation of LINE-1 transposons in adult mice. Stress 2018, 21, 575–579. [Google Scholar] [CrossRef]
- Muotri, A.R.; Zhao, C.; Marchetto, M.C.; Gage, F.H. Environmental influence on L1 retrotransposons in the adult hippocampus. Hippocampus 2009, 19, 1002–1007. [Google Scholar] [CrossRef] [Green Version]
- Navabpour, S.; Rogers, J.; McFadden, T.; Jarome, T.J. DNA Double-Strand Breaks Are a Critical Regulator of Fear Memory Reconsolidation. Int. J. Mol. Sci. 2020, 21, 8995. [Google Scholar] [CrossRef]
- McConnell, M.J.; Moran, J.V.; Abyzov, A.; Akbarian, S.; Bae, T.; Cortes-Ciriano, I.; Erwin, J.A.; Fasching, L.; Flasch, D.A.; Freed, D.; et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leija-Salazar, M.; Piette, C.; Proukakis, C. Review: Somatic mutations in neurodegeneration. Neuropathol. Appl. Neurobiol. 2018, 44, 267–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, P.J. Genome integrity and disease prevention in the nervous system. Genes Dev. 2017, 31, 1180–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciaffardini, F.; Nicolai, S.; Caputo, M.; Canu, G.; Paccosi, E.; Costantino, M.; Frontini, M.; Balajee, A.S.; Proietti-De-Santis, L. The cockayne syndrome B protein is essential for neuronal differentiation and neuritogenesis. Cell Death Dis. 2014, 5, e1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitsera, N.; Gasteiger, K.; Lühnsdorf, B.; Allgayer, J.; Epe, B.; Carell, T.; Khobta, A. Cockayne syndrome: Varied requirement of transcription-coupled nucleotide excision repair for the removal of three structurally different adducts from transcribed DNA. PLoS ONE 2014, 9, e94405. [Google Scholar] [CrossRef] [Green Version]
- Emera, D.; Yin, J.; Reilly, S.K.; Gockley, J.; Noonan, J.P. Origin and evolution of developmental enhancers in the mammalian neocortex. Proc. Natl. Acad. Sci. USA 2016, 113, E2617–E2626. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Nishihara, H.; Hirakawa, M.; Fujimura, K.; Tanaka, M.; Kokubo, N.; Kimura-Yoshida, C.; Matsuo, I.; Sumiyama, K.; Saitou, N.; et al. Possible involvement of SINEs in mammalian-specific brain formation. Proc. Natl. Acad. Sci. USA 2008, 105, 4220–4225. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, A.; Kobayashi, N.; Suzuki-Hirano, A.; Nishihara, H.; Sasaki, T.; Hirakawa, M.; Sumiyama, K.; Shimogori, T.; Okada, N. A SINE-Derived Element Constitutes a Unique Modular Enhancer for Mammalian Diencephalic Fgf8. PLoS ONE 2012, 7, e43785. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, K.; Teissier, A.; Kobayashi, N.; Nakanishi, A.; Sasaki, T.; Yan, K.; Tarabykin, V.; Vigier, L.; Sumiyama, K.; Hirakawa, M.; et al. A Mammalian Conserved Element Derived from SINE Displays Enhancer Properties Recapitulating Satb2 Expression in Early-Born Callosal Projection Neurons. PLoS ONE 2011, 6, e28497. [Google Scholar] [CrossRef] [Green Version]
- Notwell, J.H.; Chung, T.; Heavner, W.; Bejerano, G. A family of transposable elements co-opted into developmental enhancers in the mouse neocortex. Nat. Commun. 2015, 6, 6644. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, A.M.; de Souza, F.S.J.; Franchini, L.F.; Bumaschny, V.F.; Low, M.J.; Rubinstein, M. Ancient Exaptation of a CORE-SINE Retroposon into a Highly Conserved Mammalian Neuronal Enhancer of the Proopiomelanocortin Gene. PLoS Genet. 2007, 3, e166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchini, L.F.; López-Leal, R.; Nasif, S.; Beati, P.; Gelman, D.M.; Low, M.J.; de Souza, F.J.; Rubinstein, M. Convergent evolution of two mammalian neuronal enhancers by sequential exaptation of unrelated retroposons. Proc. Natl. Acad. Sci. USA 2011, 108, 15270–15275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Policarpi, C.; Crepaldi, L.; Brookes, E.; Nitarska, J.; French, S.M.; Coatti, A.; Riccio, A. Enhancer SINEs Link Pol III to Pol II Transcription in Neurons. Cell Rep. 2017, 21, 2879–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The Neuronal Gene Arc Encodes a Repurposed Retrotransposon Gag Protein that Mediates Intercellular RNA Transfer. Cell 2018, 172, 275–288.e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, J.D. Arc—An endogenous neuronal retrovirus? Semin. Cell Dev. Biol. 2018, 77, 73–78. [Google Scholar] [CrossRef]
- Zhang, H.; Bramham, C.R. Arc/Arg3.1 function in long-term synaptic plasticity: Emerging mechanisms and unresolved issues. Eur. J. Neurosci. 2021, 54, 6696–6712. [Google Scholar] [CrossRef]
- Ashley, J.; Cordy, B.; Lucia, D.; Fradkin, L.G.; Budnik, V.; Thomson, T. Retrovirus-like Gag Protein Arc1 Binds RNA and Traffics across Synaptic Boutons. Cell 2018, 172, 262–274.e211. [Google Scholar] [CrossRef] [Green Version]
- Stessman, H.A.F.; Willemsen, M.H.; Fenckova, M.; Penn, O.; Hoischen, A.; Xiong, B.; Wang, T.; Hoekzema, K.; Vives, L.; Vogel, I.; et al. Disruption of POGZ is Associated with Intellectual Disability and Autism Spectrum Disorders. Am. J. Hum. Genet. 2016, 98, 541–552. [Google Scholar] [CrossRef] [Green Version]
- White, J.; Beck, C.R.; Harel, T.; Posey, J.E.; Jhangiani, S.N.; Tang, S.; Farwell, K.D.; Powis, Z.; Mendelsohn, N.J.; Baker, J.A.; et al. POGZ truncating alleles cause syndromic intellectual disability. Genome Med. 2016, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Seto, J.; Donovan, G.; Toth, M. Jerky, a Protein Deficient in a Mouse Epilepsy Model, Is Associated with Translationally Inactive mRNA in Neurons. J. Neurosci. 2002, 22, 176. [Google Scholar] [CrossRef]
- Edelstein, L.C.; Collins, T. The SCAN domain family of zinc finger transcription factors. Gene 2005, 359, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Korade, Z.; Kenchappa, R.S.; Mirnics, K.; Carter, B.D. NRIF is a Regulator of Neuronal Cholesterol Biosynthesis Genes. J. Mol. Neurosci. 2009, 38, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavelitz, T.; Gray, L.T.; Padilla, S.L.; Bailey, A.D.; Weiner, A.M. PGBD5: A neural-specific intron-containing piggyBac transposase domesticated over 500 million years ago and conserved from cephalochordates to humans. Mob. DNA 2013, 4, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer-Bensch, G. Emerging Roles of Long Non-Coding RNAs as Drivers of Brain Evolution. Cells 2019, 8, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafin, R.N. The Role of Transposable Elements in the Differentiation of Stem Cells. Mol. Gen. Microbiol. Virol. 2019, 34, 67–74. [Google Scholar] [CrossRef]
- Aprea, J.; Prenninger, S.; Dori, M.; Ghosh, T.; Monasor, L.S.; Wessendorf, E.; Zocher, S.; Massalini, S.; Alexopoulou, D.; Lesche, M.; et al. Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. EMBO J. 2013, 32, 3145–3160. [Google Scholar] [CrossRef] [PubMed]
- Skryabin, B.V.; Kremerskothen, J.; Vassilacopoulou, D.; Disotell, T.R.; Kapitonov, V.V.; Jurka, J.; Brosius, J. The BC200 RNA gene and its neural expression are conserved in Anthropoidea (Primates). J. Mol. Evol. 1998, 47, 677–685. [Google Scholar] [CrossRef]
- Kuryshev, V.Y.; Skryabin, B.V.; Kremerskothen, J.; Jurka, J.; Brosius, J. Birth of a gene: Locus of neuronal BC200 snmRNA in three prosimians and human BC200 pseudogenes as archives of change in the Anthropoidea lineage. J. Mol. Biol. 2001, 309, 1049–1066. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Martignetti, J.A.; Shen, M.R.; Brosius, J.; Deininger, P. Rodent BC1 RNA gene as a master gene for ID element amplification. Proc. Natl. Acad. Sci. USA 1994, 91, 3607–3611. [Google Scholar] [CrossRef] [Green Version]
- Martignetti, J.A.; Brosius, J. BC1 RNA: Transcriptional analysis of a neural cell-specific RNA polymerase III transcript. Mol. Cell. Biol. 1995, 15, 1642–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesnokova, E.; Kolosov, P. Local Protein Synthesis in Dendritic Terminals and Its Regulation in Normal Conditions and during Plastic Changes. Neurosci. Behav. Physiol. 2017, 47, 595–607. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, H.S.; Kim, M.; Shin, H. Brain Cytoplasmic RNAs in Neurons: From Biosynthesis to Function. Biomolecules 2020, 10, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Schein, A.; Zucchelli, S.; Kauppinen, S.; Gustincich, S.; Carninci, P. Identification of antisense long noncoding RNAs that function as SINEUPs in human cells. Sci. Rep. 2016, 6, 33605. [Google Scholar] [CrossRef] [Green Version]
- Cartault, F.; Munier, P.; Benko, E.; Desguerre, I.; Hanein, S.; Boddaert, N.; Bandiera, S.; Vellayoudom, J.; Krejbich-Trotot, P.; Bintner, M.; et al. Mutation in a primate-conserved retrotransposon reveals a noncoding RNA as a mediator of infantile encephalopathy. Proc. Natl. Acad. Sci. USA 2012, 109, 4980. [Google Scholar] [CrossRef] [Green Version]
- Smalheiser, N.R. The search for endogenous siRNAs in the mammalian brain. Exp. Neurol. 2012, 235, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.F.; Coffin, J.M. Evidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution. Nat. Genet. 2001, 29, 487–489. [Google Scholar] [CrossRef]
- Ricci, M.; Peona, V.; Guichard, E.; Taccioli, C.; Boattini, A. Transposable Elements Activity is Positively Related to Rate of Speciation in Mammals. J. Mol. Evol. 2018, 86, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.A.; Liu, G.; Eichler, E.E. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003, 73, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Luttig, C.T.; Tsui, C.; Pittard, W.S.; Devine, S.E. Recently mobilized transposons in the human and chimpanzee genomes. Am. J. Hum. Genet. 2006, 78, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Hormozdiari, F.; Miriam, K.K.; Prado-Martinez, J.; Chiatante, G.; Irene, H.H.; Jerilyn, A.W.; Nelson, B.; Alkan, C.; Peter, H.S.; Huddleston, J.; et al. Rates and patterns of great ape retrotransposition. Proc. Natl. Acad. Sci. USA 2013, 110, 13457–13462. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, K.; Hattori, M.; Yada, T.; Gojobori, T.; Sakaki, Y.; Okada, N. Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol. 2003, 4, R74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human Genomic Deletions Mediated by Recombination between Alu Elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, A.-L.; Wang, Y.-Q.; Zhang, H.; Liao, C.-H.; Wang, J.-K.; Zhang, R.; Che, J.; Su, B. Rapid evolution and copy number variation of primate RHOXF2, an X-linked homeobox gene involved in male reproduction and possibly brain function. BMC Evol. Biol. 2011, 11, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guffanti, G.; Bartlett, A.; Klengel, T.; Klengel, C.; Hunter, R.; Glinsky, G.; Macciardi, F. Novel Bioinformatics Approach Identifies Transcriptional Profiles of Lineage-Specific Transposable Elements at Distinct Loci in the Human Dorsolateral Prefrontal Cortex. Mol. Biol. Evol. 2018, 35, 2435–2453. [Google Scholar] [CrossRef]
- del Rosario, R.C.; Rayan, N.A.; Prabhakar, S. Noncoding origins of anthropoid traits and a new null model of transposon functionalization. Genome Res. 2014, 24, 1469–1484. [Google Scholar] [CrossRef] [Green Version]
- Belaghzal, H.; Dekker, J.; Gibcus, J.H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 2017, 123, 56–65. [Google Scholar] [CrossRef]
- Luo, X.; Liu, Y.; Dang, D.; Hu, T.; Hou, Y.; Meng, X.; Zhang, F.; Li, T.; Wang, C.; Li, M.; et al. 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 2021, 184, 723–740.e721. [Google Scholar] [CrossRef]
- Nataf, S.; Uriagereka, J.; Benitez-Burraco, A. The Promoter Regions of Intellectual Disability-Associated Genes Are Uniquely Enriched in LTR Sequences of the MER41 Primate-Specific Endogenous Retrovirus: An Evolutionary Connection Between Immunity and Cognition. Front. Genet. 2019, 10, 321. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Pederson, S.M.; Cao, D.; Qu, Z.; Hu, Z.; Adelson, D.L.; Wei, C. Genome-Wide Analysis of the Association of Transposable Elements with Gene Regulation Suggests that Alu Elements Have the Largest Overall Regulatory Impact. J. Comput. Biol. 2018, 25, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Lie, D.C.; DeCicco, K.L.; Shi, Y.; DeLuca, L.M.; Gage, F.H.; Evans, R.M. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2006, 103, 3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haushalter, C.; Asselin, L.; Fraulob, V.; Dollé, P.; Rhinn, M. Retinoic acid controls early neurogenesis in the developing mouse cerebral cortex. Dev. Biol. 2017, 430, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Laperriere, D.; Wang, T.T.; White, J.H.; Mader, S. Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genom. 2007, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, E.; Nemzer, S.; Kinar, Y.; Sorek, R.; Rechavi, G.; Levanon, E.Y. Is abundant A-to-I RNA editing primate-specific? Trends Genet. 2005, 21, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz-Yaacov, N.; Levanon, E.Y.; Nevo, E.; Kinar, Y.; Harmelin, A.; Jacob-Hirsch, J.; Amariglio, N.; Eisenberg, E.; Rechavi, G. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc. Natl. Acad. Sci. USA 2010, 107, 12174–12179. [Google Scholar] [CrossRef] [Green Version]
- Ramaswami, G.; Li, J.B. RADAR: A rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014, 42, D109–D113. [Google Scholar] [CrossRef] [Green Version]
- Larsen, P.A.; Hunnicutt, K.E.; Larsen, R.J.; Yoder, A.D.; Saunders, A.M. Warning SINEs: Alu elements, evolution of the human brain, and the spectrum of neurological disease. Chromosome Res. 2018, 26, 93–111. [Google Scholar] [CrossRef] [Green Version]
- Porath, H.T.; Schaffer, A.A.; Kaniewska, P.; Alon, S.; Eisenberg, E.; Rosenthal, J.; Levanon, E.Y.; Levy, O. A-to-I RNA Editing in the Earliest-Diverging Eumetazoan Phyla. Mol. Biol. Evol. 2017, 34, 1890–1901. [Google Scholar] [CrossRef] [Green Version]
- Behm, M.; Öhman, M. RNA Editing: A Contributor to Neuronal Dynamics in the Mammalian Brain. Trends Genet. 2016, 32, 165–175. [Google Scholar] [CrossRef]
- Lev-Maor, G.; Sorek, R.; Levanon, E.Y.; Paz, N.; Eisenberg, E.; Ast, G. RNA-editing-mediated exon evolution. Genome Biol. 2007, 8, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Lin, L.; Cai, J.J.; Jiang, P.; Kenkel, E.J.; Stroik, M.R.; Sato, S.; Davidson, B.L.; Xing, Y. Widespread establishment and regulatory impact of Alu exons in human genes. Proc. Natl. Acad. Sci. USA 2011, 108, 2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, B.-I.; Tietjen, I.; Atabay, K.D.; Evrony, G.D.; Johnson, M.B.; Asare, E.; Wang, P.P.; Murayama, A.Y.; Im, K.; Lisgo, S.N.; et al. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 2014, 343, 764–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrott, A.M.; Tsai, M.; Batchu, P.; Ryan, K.; Ozer, H.L.; Tian, B.; Mathews, M.B. The evolution and expression of the snaR family of small non-coding RNAs. Nucleic Acids Res. 2011, 39, 1485–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, H.-H.; Hayakawa, T.; Diaz, S.; Krings, M.; Indriati, E.; Leakey, M.; Paabo, S.; Satta, Y.; Takahata, N.; Varki, A. Inactivation of CMP-N-acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 11736–11741. [Google Scholar] [CrossRef] [Green Version]
- Hedlund, M.; Tangvoranuntakul, P.; Takematsu, H.; Long, J.M.; Housley, G.D.; Kozutsumi, Y.; Suzuki, A.; Wynshaw-Boris, A.; Ryan, A.F.; Gallo, R.L.; et al. N-glycolylneuraminic acid deficiency in mice: Implications for human biology and evolution. Mol. Cell. Biol. 2007, 27, 4340–4346. [Google Scholar] [CrossRef] [Green Version]
- Naito-Matsui, Y.; Davies, L.R.L.; Takematsu, H.; Chou, H.-H.; Tangvoranuntakul, P.; Carlin, A.F.; Verhagen, A.; Heyser, C.J.; Yoo, S.-W.; Choudhury, B.; et al. Physiological Exploration of the Long Term Evolutionary Selection against Expression of N-Glycolylneuraminic Acid in the Brain. J. Biol. Chem. 2017, 292, 2557–2570. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr.; Wong, C.; Youssoufian, H.; Scott, A.F.; Phillips, D.G.; Antonarakis, S.E. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 1988, 332, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guffanti, G.; Gaudi, S.; Fallon, J.H.; Sobell, J.; Potkin, S.G.; Pato, C.; Macciardi, F. Transposable elements and psychiatric disorders. Am. J. Med. Genet. B 2014, 165, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Nätt, D.; Thorsell, A. Stress-induced transposon reactivation: A mediator or an estimator of allostatic load? Environ. Epigenet. 2016, 2, dvw015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravel-Godreuil, C.; Znaidi, R.; Bonnifet, T.; Joshi, R.L.; Fuchs, J. Transposable elements as new players in neurodegenerative diseases. FEBS Lett. 2021, 595, 2733–2755. [Google Scholar] [CrossRef]
- Peze-Heidsieck, E.; Bonnifet, T.; Znaidi, R.; Ravel-Godreuil, C.; Massiani-Beaudoin, O.; Joshi, R.L.; Fuchs, J. Retrotransposons as a Source of DNA Damage in Neurodegeneration. Front. Aging Neurosci. 2022, 13, 786897. [Google Scholar] [CrossRef]
- Tan, H.; Qurashi, A.; Poidevin, M.; Nelson, D.L.; Li, H.; Jin, P. Retrotransposon activation contributes to fragile X premutation rCGG-mediated neurodegeneration. Hum. Mol. Genet. 2011, 21, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Bravo, P.; Darvish, H.; Tafakhori, A.; Azcona, L.J.; Johari, A.H.; Jamali, F.; Paisán-Ruiz, C. Molecular characterization of PRKN structural variations identified through whole-genome sequencing. Mol. Genet. Genomic Med. 2018, 6, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Guffanti, G.; Gaudi, S.; Klengel, T.; Fallon, J.H.; Mangalam, H.; Madduri, R.; Rodriguez, A.; DeCrescenzo, P.; Glovienka, E.; Sobell, J.; et al. LINE1 insertions as a genomic risk factor for schizophrenia: Preliminary evidence from an affected family. Am. J. Med. Genet. B 2016, 171, 534–545. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, B.; Pattni, R.; Gleason, K.; Tan, C.; Kalinowski, A.; Sloan, S.; Fiston-Lavier, A.-S.; Mariani, J.; Petrov, D.; et al. Machine learning reveals bilateral distribution of somatic L1 insertions in human neurons and glia. Nat. Neurosci. 2021, 24, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Abrusán, G. Somatic transposition in the brain has the potential to influence the biosynthesis of metabolites involved in Parkinson’s disease and schizophrenia. Biol. Direct. 2012, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaribeygi, H.; Panahi, Y.; Sahraei, H.; Johnston, T.P.; Sahebkar, A. The impact of stress on body function: A review. EXCLI J. 2017, 16, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Cuarenta, A.; Kigar, S.L.; Henion, I.C.; Karls, K.E.; Chang, L.; Bakshi, V.P.; Auger, A.P. Early life stress increases Line1 within the developing brain in a sex-dependent manner. Brain Res. 2020, 1748, 147123. [Google Scholar] [CrossRef] [PubMed]
- Cuarenta, A.; Kigar, S.L.; Henion, I.C.; Chang, L.; Bakshi, V.P.; Auger, A.P. Early life stress during the neonatal period alters social play and Line1 during the juvenile stage of development. Sci. Rep. 2021, 11, 3549. [Google Scholar] [CrossRef]
- Nätt, D.; Johansson, I.; Faresjö, T.; Ludvigsson, J.; Thorsell, A. High cortisol in 5-year-old children causes loss of DNA methylation in SINE retrotransposons: A possible role for ZNF263 in stress-related diseases. Clin. Epigenetics 2015, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 2013, 5, 867–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De Cecco, M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885.e875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meter, M.; Kashyap, M.; Rezazadeh, S.; Geneva, A.J.; Morello, T.D.; Seluanov, A.; Gorbunova, V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014, 5, 5011. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.; Zuniga, G.; Sun, W.; Beckmann, A.; Ochoa, E.; DeVos, S.L.; Hyman, B.; Chiu, G.; Roy, E.R.; Cao, W.; et al. Pathogenic tau accelerates aging-associated activation of transposable elements in the mouse central nervous system. Prog. Neurobiol. 2022, 208, 102181. [Google Scholar] [CrossRef]
- Li, W.; Prazak, L.; Chatterjee, N.; Grüninger, S.; Krug, L.; Theodorou, D.; Dubnau, J. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat. Neurosci. 2013, 16, 529–531. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Peferoen, L.A.N.; Vogel, D.Y.S.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Jeppesen, R.; Benros, M.E. Autoimmune Diseases and Psychotic Disorders. Front. Psychiatry 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef]
- Perron, H.; Hamdani, N.; Faucard, R.; Lajnef, M.; Jamain, S.; Daban-Huard, C.; Sarrazin, S.; LeGuen, E.; Houenou, J.; Delavest, M.; et al. Molecular characteristics of Human Endogenous Retrovirus type-W in schizophrenia and bipolar disorder. Transl. Psychiatry 2012, 2, e201. [Google Scholar] [CrossRef] [PubMed]
- Licastro, F.; Porcellini, E. Activation of Endogenous Retrovirus, Brain Infections and Environmental Insults in Neurodegeneration and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7263. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, P.A.; Lutz, M.W.; Hunnicutt, K.E.; Mihovilovic, M.; Saunders, A.M.; Yoder, A.D.; Roses, A.D. The Alu neurodegeneration hypothesis: A primate-specific mechanism for neuronal transcription noise, mitochondrial dysfunction, and manifestation of neurodegenerative disease. Alzheimers. Dement. 2017, 13, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Baeken, M.W.; Moosmann, B.; Hajieva, P. Retrotransposon activation by distressed mitochondria in neurons. Biochem. Biophys. Res. Commun. 2020, 525, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Jeong, H.H.; Hsieh, Y.C.; Klein, H.U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- Bollati, V.; Galimberti, D.; Pergoli, L.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Bertazzi, P.A.; Baccarelli, A. DNA methylation in repetitive elements and Alzheimer disease. Brain Behav. Immun. 2011, 25, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Protasova, M.S.; Gusev, F.E.; Grigorenko, A.P.; Kuznetsova, I.L.; Rogaev, E.I.; Andreeva, T.V. Quantitative Analysis of L1-Retrotransposons in Alzheimer’s Disease and Aging. Biochem. Biokhimiia 2017, 82, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.-O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, 1. [Google Scholar] [CrossRef] [PubMed]
- de Thé, F.X.B.; Rekaik, H.; Peze-Heidsieck, E.; Massiani-Beaudoin, O.; Joshi, R.L.; Fuchs, J.; Prochiantz, A. Engrailed homeoprotein blocks degeneration in adult dopaminergic neurons through LINE-1 repression. EMBO J. 2018, 37, e97374. [Google Scholar] [CrossRef]
- Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. An Increased Burden of Highly Active Retrotransposition Competent L1s is Associated with Parkinson’s Disease Risk and Progression in the PPMI Cohort. Int. J. Mol. Sci. 2020, 21, 6562. [Google Scholar] [CrossRef]
- Nielsen, S.S.; Checkoway, H.; Butler, R.A.; Nelson, H.H.; Farin, F.M.; Longstreth, W.T., Jr.; Franklin, G.M.; Swanson, P.D.; Kelsey, K.T. LINE-1 DNA methylation, smoking and risk of Parkinson’s disease. J. Parkinsons Dis. 2012, 2, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floreani, L.; Ansaloni, F.; Mangoni, D.; Agostoni, E.; Sanges, R.; Persichetti, F.; Gustincich, S. Analysis of LINE1 Retrotransposons in Huntington’s Disease. Front. Cell. Neurosci. 2022, 15, 743797. [Google Scholar] [CrossRef]
- Ruzo, A.; Croft, G.F.; Metzger, J.J.; Galgoczi, S.; Gerber, L.J.; Pellegrini, C.; Wang, H., Jr.; Fenner, M.; Tse, S.; Marks, A.; et al. Chromosomal instability during neurogenesis in Huntington’s disease. Development 2018, 145, dev156844. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Wu, C.; Jin, L. A Possible Role for Long Interspersed Nuclear Elements-1 (LINE-1) in Huntington’s Disease Progression. Med. Sci. Monit. 2018, 24, 3644–3652. [Google Scholar] [CrossRef]
- Casale, A.M.; Liguori, F.; Ansaloni, F.; Cappucci, U.; Finaurini, S.; Spirito, G.; Persichetti, F.; Sanges, R.; Gustincich, S.; Piacentini, L. Transposable element activation promotes neurodegeneration in a Drosophila model of Huntington’s disease. iScience 2022, 25, 103702. [Google Scholar] [CrossRef]
- Chaytow, H.; Faller, K.M.E.; Huang, Y.T.; Gillingwater, T.H. Spinal muscular atrophy: From approved therapies to future therapeutic targets for personalized medicine. Cell Rep. Med. 2021, 2, 100346. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Seo, J.; Singh, N.N.; Singh, R.N. A Multilayered Control of the Human Survival Motor Neuron Gene Expression by Alu Elements. Front. Microbiol. 2017, 8, 2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfahad, T.; Nath, A. Retroviruses and amyotrophic lateral sclerosis. Antiviral. Res. 2013, 99, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Pereira, G.C.; Sanchez, L.; Schaughency, P.M.; Rubio-Roldán, A.; Choi, J.A.; Planet, E.; Batra, R.; Turelli, P.; Trono, D.; Ostrow, L.W.; et al. Properties of LINE-1 proteins and repeat element expression in the context of amyotrophic lateral sclerosis. Mob. DNA 2018, 9, 35. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Miller, B.L. Frontotemporal lobar degeneration: Epidemiology, pathophysiology, diagnosis and management. CNS Drugs 2010, 24, 375–398. [Google Scholar] [CrossRef]
- Mohandas, E.; Rajmohan, V. Frontotemporal dementia: An updated overview. Indian J. Psychiatry 2009, 51, S65–S69. [Google Scholar]
- Cohen, T.; Lee, V.; Trojanowski, J. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Shan, X.; Chiang, P.-M.; Price, D.L.; Wong, P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.Y.; Russ, J.; Cali, C.P.; Phan, J.M.; Amlie-Wolf, A.; Lee, E.B. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 2019, 27, 1409–1421.e1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, L.; Chatterjee, N.; Borges-Monroy, R.; Hearn, S.; Liao, W.-W.; Morrill, K.; Prazak, L.; Rozhkov, N.; Theodorou, D.; Hammell, M.; et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017, 13, e1006635. [Google Scholar] [CrossRef] [Green Version]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Fagegaltier, D.; Harris, B.T.; Ostrow, L.W.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e1165. [Google Scholar] [CrossRef] [Green Version]
- Antony, J.M.; van Marle, G.; Opii, W.; Butterfield, D.A.; Mallet, F.; Yong, V.W.; Wallace, J.L.; Deacon, R.M.; Warren, K.; Power, C. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat. Neurosci. 2004, 7, 1088–1095. [Google Scholar] [CrossRef]
- Duperray, A.; Barbe, D.; Raguenez, G.; Weksler, B.B.; Romero, I.A.; Couraud, P.-O.; Perron, H.; Marche, P.N. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int. Immunol. 2015, 27, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Mameli, G.; Poddighe, L.; Astone, V.; Delogu, G.; Arru, G.; Sotgiu, S.; Serra, C.; Dolei, A. Novel reliable real-time PCR for differential detection of MSRVenv and syncytin-1 in RNA and DNA from patients with multiple sclerosis. J. Virol. Methods 2009, 161, 98–106. [Google Scholar] [CrossRef]
- Perron, H.; Bernard, C.; Bertrand, J.B.; Lang, A.B.; Popa, I.; Sanhadji, K.; Portoukalian, J. Endogenous retroviral genes, Herpesviruses and gender in Multiple Sclerosis. J. Neurol. Sci. 2009, 286, 65–72. [Google Scholar] [CrossRef]
- Evans, T.A.; Erwin, J.A. Retroelement-derived RNA and its role in the brain. Semin. Cell Dev. Biol. 2021, 114, 68–80. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.A.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Carreira, P.E.; Ewing, A.D.; Li, G.; Schauer, S.N.; Upton, K.R.; Fagg, A.C.; Morell, S.; Kindlova, M.; Gerdes, P.; Richardson, S.R.; et al. Evidence for L1-associated DNA rearrangements and negligible L1 retrotransposition in glioblastoma multiforme. Mob. DNA 2016, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodier, J.L. Retrotransposition in tumors and brains. Mob. DNA 2014, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef]
- Petri, R.; Brattås, P.L.; Sharma, Y.; Jönsson, M.E.; Pircs, K.; Bengzon, J.; Jakobsson, J. LINE-2 transposable elements are a source of functional human microRNAs and target sites. PLoS Genet. 2019, 15, e1008036. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Sharma, S.R.; Gonda, X.; Tarazi, F.I. Autism Spectrum Disorder: Classification, diagnosis and therapy. Pharmacol. Ther. 2018, 190, 91–104. [Google Scholar] [CrossRef]
- Hu, V.W.; Steinberg, M.E. Novel clustering of items from the Autism Diagnostic Interview-Revised to define phenotypes within autism spectrum disorders. Autism Res. 2009, 2, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Tangsuwansri, C.; Saeliw, T.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Investigation of epigenetic regulatory networks associated with autism spectrum disorder (ASD) by integrated global LINE-1 methylation and gene expression profiling analyses. PLoS ONE 2018, 13, e0201071. [Google Scholar] [CrossRef] [Green Version]
- Saeliw, T.; Tangsuwansri, C.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Integrated genome-wide Alu methylation and transcriptome profiling analyses reveal novel epigenetic regulatory networks associated with autism spectrum disorder. Mol. Autism 2018, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrieri, E.; Arpino, C.; Matteucci, C.; Sorrentino, R.; Pica, F.; Alessandrelli, R.; Coniglio, A.; Curatolo, P.; Rezza, G.; Macciardi, F.; et al. HERVs expression in Autism Spectrum Disorders. PLoS ONE 2012, 7, e48831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohodich, A.E.; Zoghbi, H.Y. Rett syndrome: Disruption of epigenetic control of postnatal neurological functions. Hum. Mol. Genet. 2015, 24, R10–R16. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- MacDonald, A.W., 3rd; Carter, C.S. Event-related FMRI study of context processing in dorsolateral prefrontal cortex of patients with schizophrenia. J. Abnorm. Psychol. 2003, 112, 689–697. [Google Scholar] [CrossRef]
- Guillozet-Bongaarts, A.L.; Hyde, T.M.; Dalley, R.A.; Hawrylycz, M.J.; Henry, A.; Hof, P.R.; Hohmann, J.; Jones, A.R.; Kuan, C.L.; Royall, J.; et al. Altered gene expression in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2014, 19, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Van Winkel, R.; Esquivel, G.; Kenis, G.; Wichers, M.; Collip, D.; Peerbooms, O.; Rutten, B.; Myin-Germeys, I.; Van Os, J. REVIEW: Genome-Wide Findings in Schizophrenia and the Role of Gene–Environment Interplay. CNS Neurosci. Ther. 2010, 16, e185–e192. [Google Scholar] [CrossRef]
- Doyle, G.A.; Crist, R.C.; Karatas, E.T.; Hammond, M.J.; Ewing, A.D.; Ferraro, T.N.; Hahn, C.-G.; Berrettini, W.H. Analysis of LINE-1 Elements in DNA from Postmortem Brains of Individuals with Schizophrenia. Neuropsychopharmacology 2017, 42, 2602–2611. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.L.; O’Donovan, M.C.; Owen, M.J. Recent genomic advances in schizophrenia. Clin. Genet. 2012, 81, 103–109. [Google Scholar] [CrossRef]
- Cardno, A.G.; Owen, M.J. Genetic Relationships Between Schizophrenia, Bipolar Disorder, and Schizoaffective Disorder. Schizophr. Bull. 2014, 40, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slokar, G.; Hasler, G. Human Endogenous Retroviruses as Pathogenic Factors in the Development of Schizophrenia. Front. Psychiatry 2016, 6, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bundo, M.; Toyoshima, M.; Okada, Y.; Akamatsu, W.; Ueda, J.; Nemoto-Miyauchi, T.; Sunaga, F.; Toritsuka, M.; Ikawa, D.; Kakita, A.; et al. Increased L1 Retrotransposition in the Neuronal Genome in Schizophrenia. Neuron 2014, 81, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelson, J.J.; Shin, M.K.; Koh, J.Y.; Brueggeman, L.; Zhang, A.; Katzman, A.; McDaniel, L.; Fang, M.; Pufall, M.; Pieper, A.A. Neuronal PAS Domain Proteins 1 and 3 Are Master Regulators of Neuropsychiatric Risk Genes. Biol. Psychiatry 2017, 82, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, H.; Schröder, J.; Bachmann, S.; Bottmer, C.; Yolken, R.H. HERV-W-related RNA detected in plasma from individuals with recent-onset schizophrenia or schizoaffective disorder. Mol. Psychiatry 2004, 9, 12–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Du, T.; Liu, Z.; Shen, Y.; Xiu, J.; Xu, Q. Inverse changes in L1 retrotransposons between blood and brain in major depressive disorder. Sci. Rep. 2016, 6, 37530. [Google Scholar] [CrossRef] [PubMed]
- Rusiecki, J.A.; Chen, L.; Srikantan, V.; Zhang, L.; Yan, L.; Polin, M.L.; Baccarelli, A. DNA methylation in repetitive elements and post-traumatic stress disorder: A case-control study of US military service members. Epigenomics 2012, 4, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mews, P.; Calipari, E.S. Cross-talk between the epigenome and neural circuits in drug addiction. Prog. Brain Res. 2017, 235, 19–63. [Google Scholar] [CrossRef]
- Volkow, N.D.; Michaelides, M.; Baler, R. The Neuroscience of Drug Reward and Addiction. Physiol. Rev. 2019, 99, 2115–2140. [Google Scholar] [CrossRef]
- Voskresenskiy, A.M.; Sun, L.S. The housekeeping gene (GA3PDH) and the long interspersed nuclear element (LINE) in the blood and organs of rats treated with cocaine. Ann. N. Y. Acad. Sci. 2008, 1137, 309–315. [Google Scholar] [CrossRef]
- Maze, I.; Feng, J.; Wilkinson, M.B.; Sun, H.; Shen, L.; Nestler, E.J. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc. Natl. Acad. Sci. USA 2011, 108, 3035–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, G.A.; Doucet-O’Hare, T.T.; Hammond, M.J.; Crist, R.C.; Ewing, A.D.; Ferraro, T.N.; Mash, D.C.; Kazazian Jr, H.H.; Berrettini, W.H. Reading LINE s within the cocaine addicted brain. Brain Behav. 2017, 7, e00678. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.; Shah, J.; Hodgson, N.; Byun, H.M.; Deth, R. Morphine induces redox-based changes in global DNA methylation and retrotransposon transcription by inhibition of excitatory amino acid transporter type 3-mediated cysteine uptake. Mol. Pharmacol. 2014, 85, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Maze, I.; Dietz, D.M.; Scobie, K.N.; Kennedy, P.J.; Damez-Werno, D.; Neve, R.L.; Zachariou, V.; Shen, L.; Nestler, E.J. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J. Neurosci. 2012, 32, 17454–17464. [Google Scholar] [CrossRef]
- Ponomarev, I.; Wang, S.; Zhang, L.; Harris, R.A.; Mayfield, R.D. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J. Neurosci. 2012, 32, 1884–1897. [Google Scholar] [CrossRef]
- Sitammagari, K.K.; Masood, W. Creutzfeldt Jakob Disease. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Manix, M.; Kalakoti, P.; Henry, M.; Thakur, J.; Menger, R.; Guthikonda, B.; Nanda, A. Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg. Focus 2015, 39, E2. [Google Scholar] [CrossRef] [Green Version]
- Jeong, B.-H.; Lee, Y.-J.; Carp, R.I.; Kim, Y.-S. The prevalence of human endogenous retroviruses in cerebrospinal fluids from patients with sporadic Creutzfeldt–Jakob disease. J. Clin. Virol. 2010, 47, 136–142. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Giovannini, M. Mouse models of neurofibromatosis 1 and 2. Neoplasia 2002, 4, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Vivarelli, R.; Grosso, S.; Calabrese, F.; Farnetani, M.; Di Bartolo, R.; Morgese, G.; Balestri, P. Epilepsy in neurofibromatosis 1. J. Child Neurol. 2003, 18, 338–342. [Google Scholar] [CrossRef]
- Garg, S.; Lehtonen, A.; Huson, S.M.; Emsley, R.; Trump, D.; Evans, D.G.; Green, J. Autism and other psychiatric comorbidity in neurofibromatosis type 1: Evidence from a population-based study. Dev. Med. Child Neurol. 2013, 55, 139–145. [Google Scholar] [CrossRef]
- Peltonen, S.; Kallionpää, R.A.; Peltonen, J. Neurofibromatosis type 1 (NF1) gene: Beyond café au lait spots and dermal neurofibromas. Exp. Dermatol. 2017, 26, 645–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.; Yoon, H.M.; Lee, B.H. Neurofibromatosis type I: Points to be considered by general pediatricians. Clin. Exp. Pediatr. 2021, 64, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.M.; Silva, A.J. Molecular and cellular mechanisms underlying the cognitive deficits associated with neurofibromatosis 1. J. Child Neurol. 2002, 17, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aneichyk, T.; Hendriks, W.T.; Yadav, R.; Shin, D.; Gao, D.; Vaine, C.A.; Collins, R.L.; Domingo, A.; Currall, B.; Stortchevoi, A.; et al. Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell 2018, 172, 897–909.e821. [Google Scholar] [CrossRef] [Green Version]


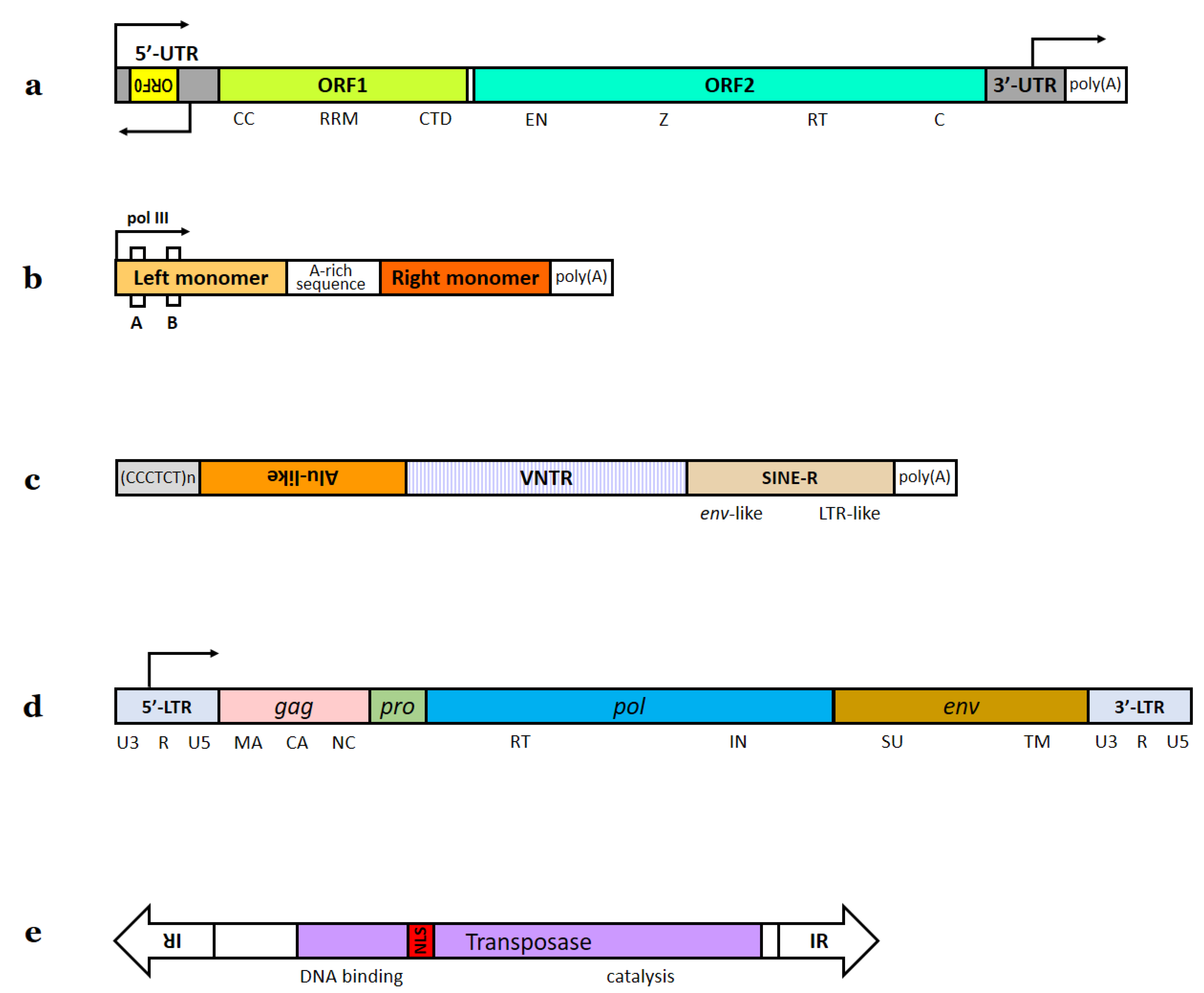{kind=link}
{kind=link}
| TE repression | Epigenetic | DNA methylation | 1.7.1 | |
| Histone methylation | 1.7.2 | |||
| KRAB-ZFP transcriptional repressors | 1.7.3 | |||
| Epigenetic and post-transcriptional | RNA interference | 1.7.4 | ||
| Other mechanisms | 1.7.5 | |||
| TE exaptation | TE derepression in cell stress conditions | 1.7.7 | ||
| Regulation of host gene expression by TE sequences | Gene expression rate tuning | 1.8.1 | ||
| Promoters, enhancers, insulators | 1.8.2 | |||
| TF binding sites | 1.8.3 | |||
| TE insertions affecting protein sequence | Insertions within exons, coding sequence mobilizations | 1.8.4 | ||
| TE insertions affecting the fate of RNA | Exonization (new splice isoforms) | 1.8.5 | ||
| Alternative polyadenylation sites | 1.8.6 | |||
| RNA A-I editing | 1.8.7 | |||
| Other mechanisms | 1.8.8 | |||
| TE-encoded proteins and non-coding RNAs used by the host cell | 1.8.9 | |||
| TEs and DNA repair | 1.8.10 | |||
| Function | Cell Type | Type of TE Insertions | Section Number |
|---|---|---|---|
| A source of mutagenesis contributing to neuronal mosaicism | Neurons and NPCs | Somatic | 2.2 |
| RNA A-I editing of neuronal transcripts | Neurons, possibly NPCs | Germline | 2.3 |
| Facilitating neuronal differentiation via host gene transcription regulation | NPCs | Germline | 2.4 |
| Possible TE participation in neuronal plasticity | Neurons, possibly NPCs | Somatic | 2.5 |
| Possible TE participation in DNA repair | Neurons | Somatic | 2.6 |
| TE-derived cis-regulatory sequences in neuronal gene expression | Neurons and NPCs | Germline | 2.7.1 |
| TE-encoded neuronal proteins | Neurons, possibly NPCs | Germline | 2.7.2 |
| TE-encoded neuronal ncRNAs | Neurons and NPCs | Germline | 2.7.3 |
| Disease | Type of Disease | Associated TE Activity | Suggested Mechanisms of TE Participating in Pathogenesis | Section Number |
|---|---|---|---|---|
| Alzheimer’s disease | Neurodegenerative | Transcriptional activation of different TE types | Neuroinflammation; generation of defective APP retrocopies; speculated Alu-mediated alterations of MAPT transcripts | 3.2.1 |
| Parkinson’s disease | Neurodegenerative | Increased L1 transcription; germline TE insertions | Aggravation of oxidative stress; germline PRKN mutations | 3.2.2 |
| Huntington’s disease | Neurodegenerative | Increased L1 mobilization and transcription | Dysregulation of cell survival signaling pathways | 3.2.3 |
| Ataxia telangiectasia | Neurodegenerative | Increased L1 mobilization; L1 insertions with increased average length | No specific mechanisms proposed | 3.2.4 |
| Spinal muscular atrophy | Neurodegenerative, neuromuscular | Alu-mediated recombinations | Germline SMN1 mutations | 3.2.5 |
| Amyotrophic lateral sclerosis | Neurodegenerative | Increased HERV-K transcription; increased L1 mobilization | Env protein toxicity; ORF1p aggregation with mutant host proteins | 3.2.6 |
| Fronto-temporal lobar degeneration | Neurodegenerative | Decreased binding between TDP-43 and its multiple TE RNA targets; increased L1 mobilization and transcription | No specific mechanisms proposed | 3.2.6 |
| Fragile X-associated tremor/ataxia syndrome | Neurodegenerative | Increased LTR transcription | The causal role of TEs in pathogenesis was confirmed, but the exact mechanisms of damage are unknown | 3.2.7 |
| Multiple sclerosis | Autoimmune, inflammatory | Increased HERV-W transcription | Env protein toxicity; Env-induced autoimmunity to myelin proteins | 3.2.8 |
| Aicardi–Goutières syndrome | Inflammatory, neurodelopmental | Increased L1 mobilization and transcription | Accumulation of L1 cDNA in the cytoplasm leading to IFN-I response and neuroinflammation | 3.2.9 |
| Glioblastoma | Malignant tumor | Increased HERV transcription; selective packaging of HERV RNA in microvesicles | No specific mechanisms proposed | 3.2.10 |
| Autism spectrum disorders | Neurodevelopmental | Increased L1 transcription; germline L1 and Alu insertions in autism-associated genes; increased HERV-H transcription | Increased L1 RNA level correlated with oxidative stress; germline TE insertions likely affect gene expression rate | 3.2.11 |
| Rett syndrome | Neurodevelopmental | Increased L1 mobilization; less frequent L1 insertions in exons | No specific mechanisms proposed | 3.2.12 |
| Schizophrenia | Mental (psychotic) | Increased L1 mobilization; germline and somatic L1 insertions in genes associated with SZ and similar pathologies; increased HERV-K10 and MSRV env transcription | L1 insertions affect gene expression rate (confirmed in vitro for somatic insertions). A link between ERV expression and myelin inflammation was proposed | 3.2.13 |
| Bipolar disorder | Mental (mood disorder) | Increased HERV-K10 and MSRV env transcription | A link between ERV expression and myelin inflammation was proposed | 3.2.13 |
| Major depressive disorder | Mental (mood disorder) | Increased L1 mobilization in blood cells; decreased L1 mobilization in prefrontal cortex | No specific mechanisms proposed | 3.2.14 |
| Post-traumatic stress disorder | Mental (anxiety) | Altered L1 and Alu methylation levels in blood cells; increased L1 transcription in the brain after stress exposure | No specific mechanisms proposed | 3.2.15 |
| Drug addiction | Substance use disorder | Increased L1 mobilization and transcription caused by metamphetamine, cocaine, morphine administration; L1 insertions in genes associated with cocaine addiction in addict brain samples; increased LTR and L1 transcription in alcoholic brain samples | Germline or early developmental stage somatic insertions in genes influencing predisposition to cocaine addiction | 3.2.16 |
| Creutzfeldt–Jakob disease | Neurodegenerative, prion | Increased HERV-W and HERV-L transcription | Neuroinflammation | 3.2.17 |
| Neurofibromatosis type I | Neurocutaneous | Germline TE insertions within NF1 with unexpectedly high rate | Germline NF1 mutations | 3.2.18 |
| Age-related macular degeneration | Neurodegenerative | Increased Alu transcription | Alu RNA accumulation in the cytoplasm causing innate immune response and apoptosis | 3.2.19 |
| X-linked dystonia-parkinsonism | Neurodegenerative | SVA insertion | Germline TAF1 mutation | 3.2.20 |
| Ravine encephalopathy | Neurodegenerative | Point mutation within a TE-derived region | Germline SLC7A2 mutation | 3.2.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chesnokova, E.; Beletskiy, A.; Kolosov, P. The Role of Transposable Elements of the Human Genome in Neuronal Function and Pathology. Int. J. Mol. Sci. 2022, 23, 5847. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105847
Chesnokova E, Beletskiy A, Kolosov P. The Role of Transposable Elements of the Human Genome in Neuronal Function and Pathology. International Journal of Molecular Sciences. 2022; 23(10):5847. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105847
Chicago/Turabian StyleChesnokova, Ekaterina, Alexander Beletskiy, and Peter Kolosov. 2022. "The Role of Transposable Elements of the Human Genome in Neuronal Function and Pathology" International Journal of Molecular Sciences 23, no. 10: 5847. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105847