
Challenges in Analyzing Functional Epigenetic Data in Perspective of Adolescent Psychiatric Health


Abstract
:1. Introduction
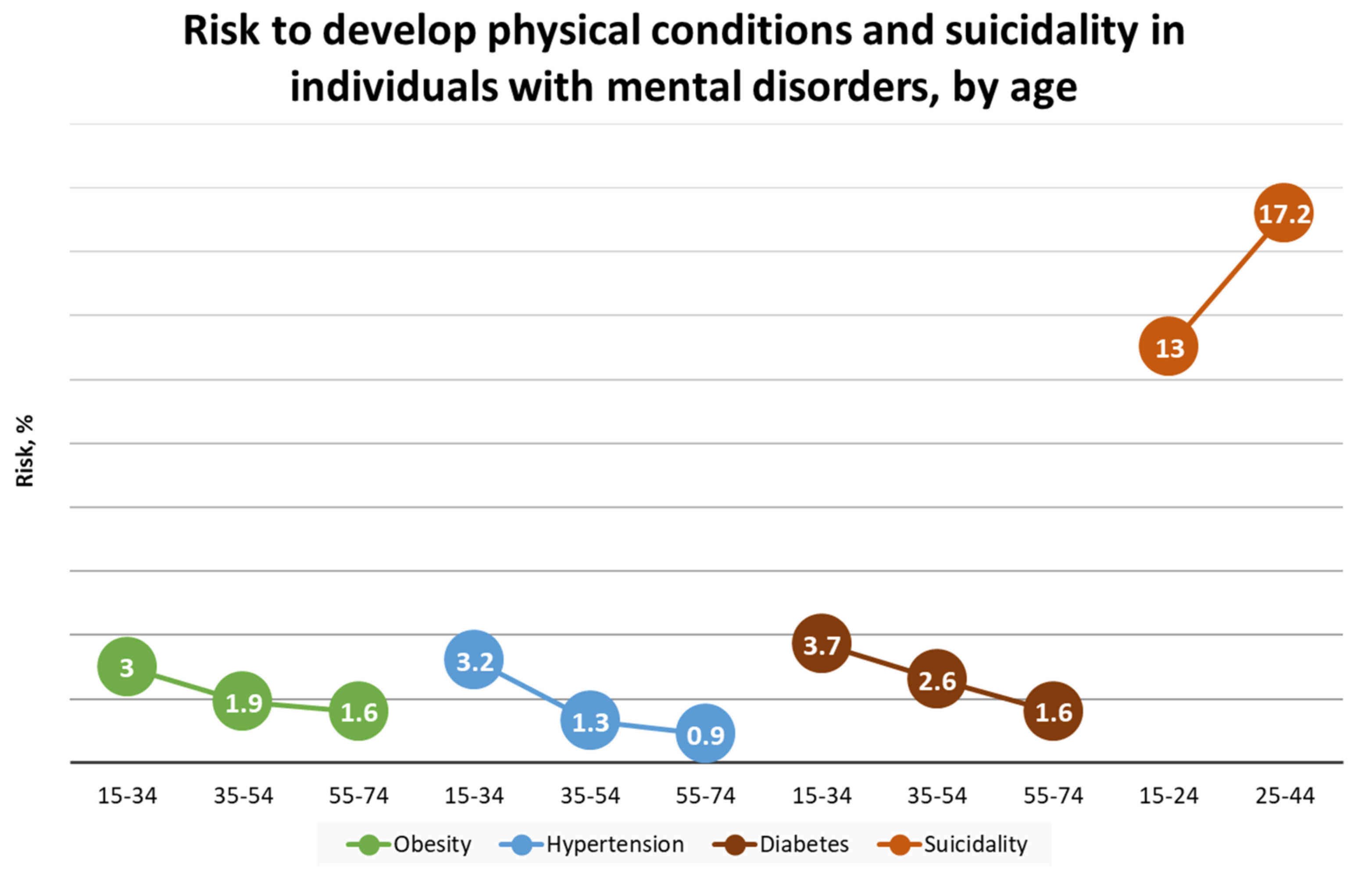
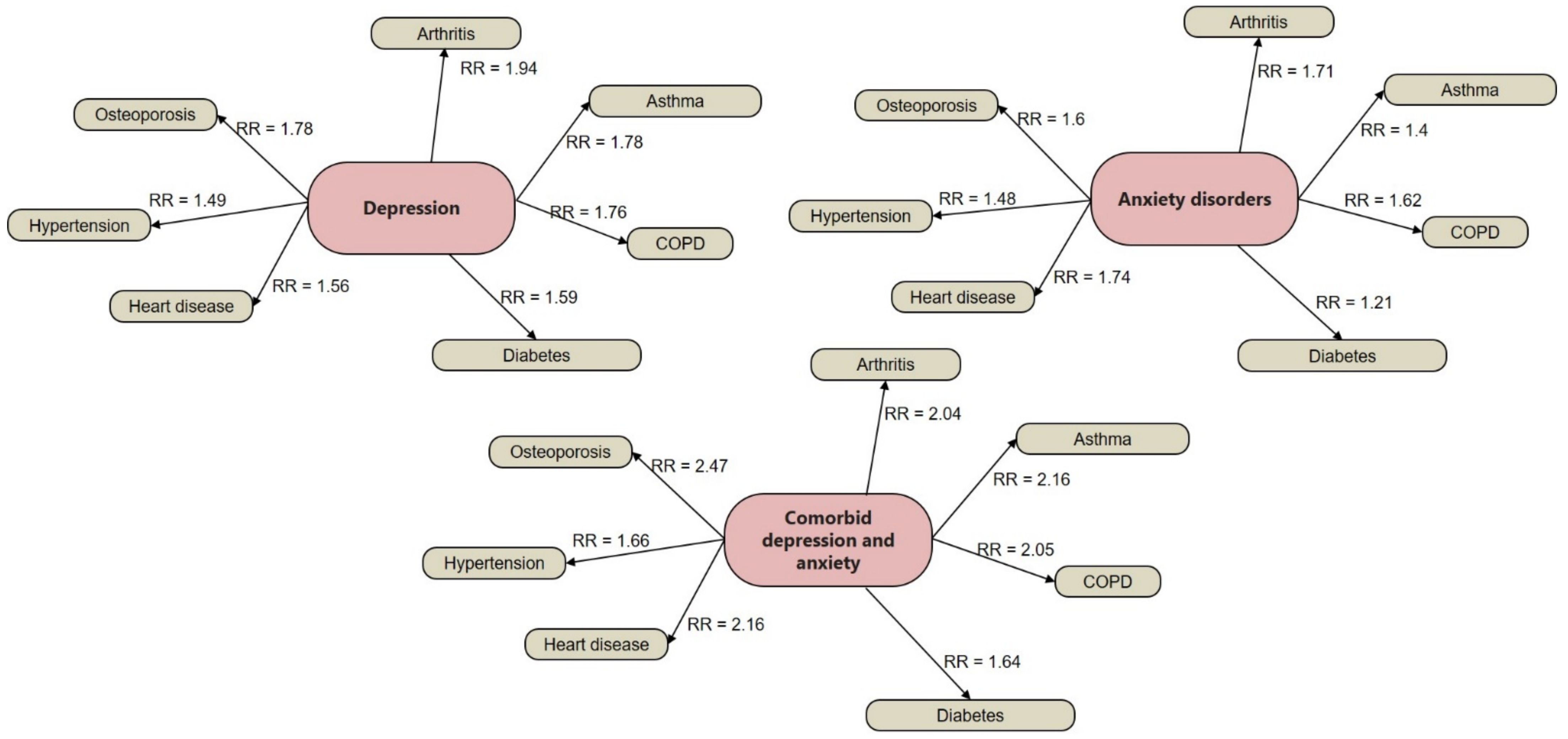
1.1. Prevalence and Risk Conditions of MDD and GAD
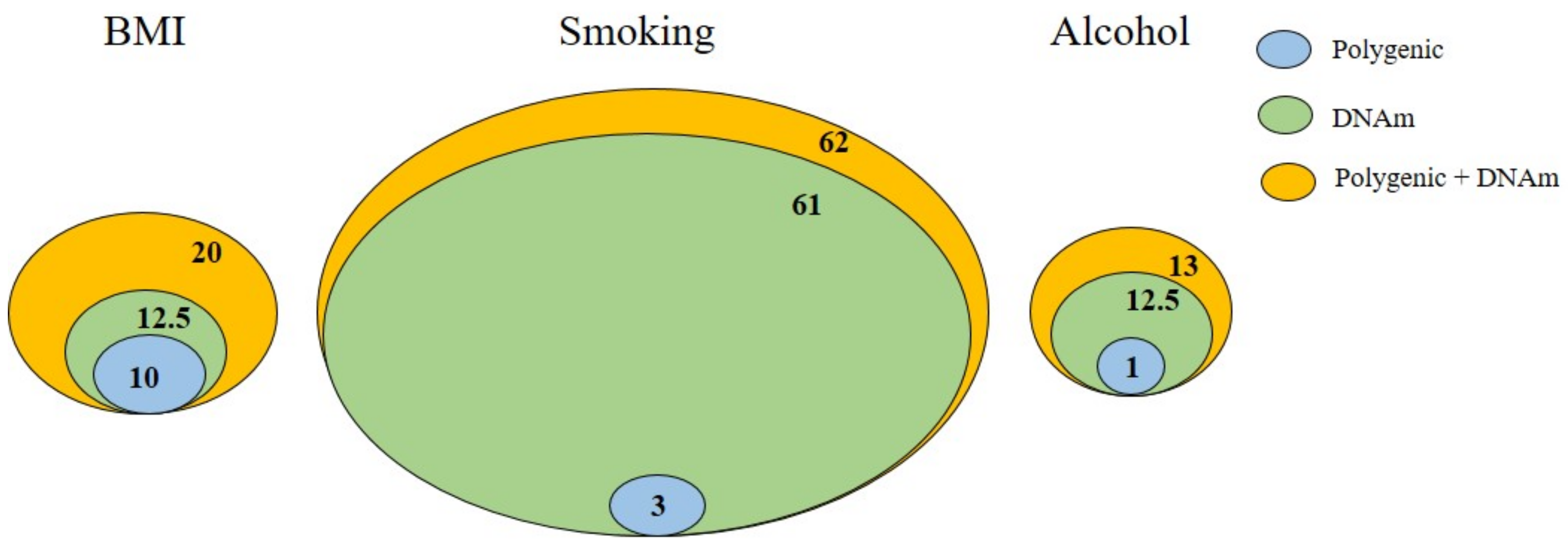
1.2. DNAm as Gene–Environment Interplay in Depression and GAD
1.3. Methodological Challenges in DNAm Analysis in Psychiatric Disorders
1.3.1. The Choice of the Investigated Tissue
1.3.2. Available Tools for the Biological Interpretation of DNAm Findings
1.3.3. Validation of the DNAm Findings by a Different Technique or in an Independent Cohort
1.3.4. Composition of the Statistical Models when Analyzing DNAm and Statistical Significance
1.3.5. Adjusting for Cell-Type Proportions in Whole Blood
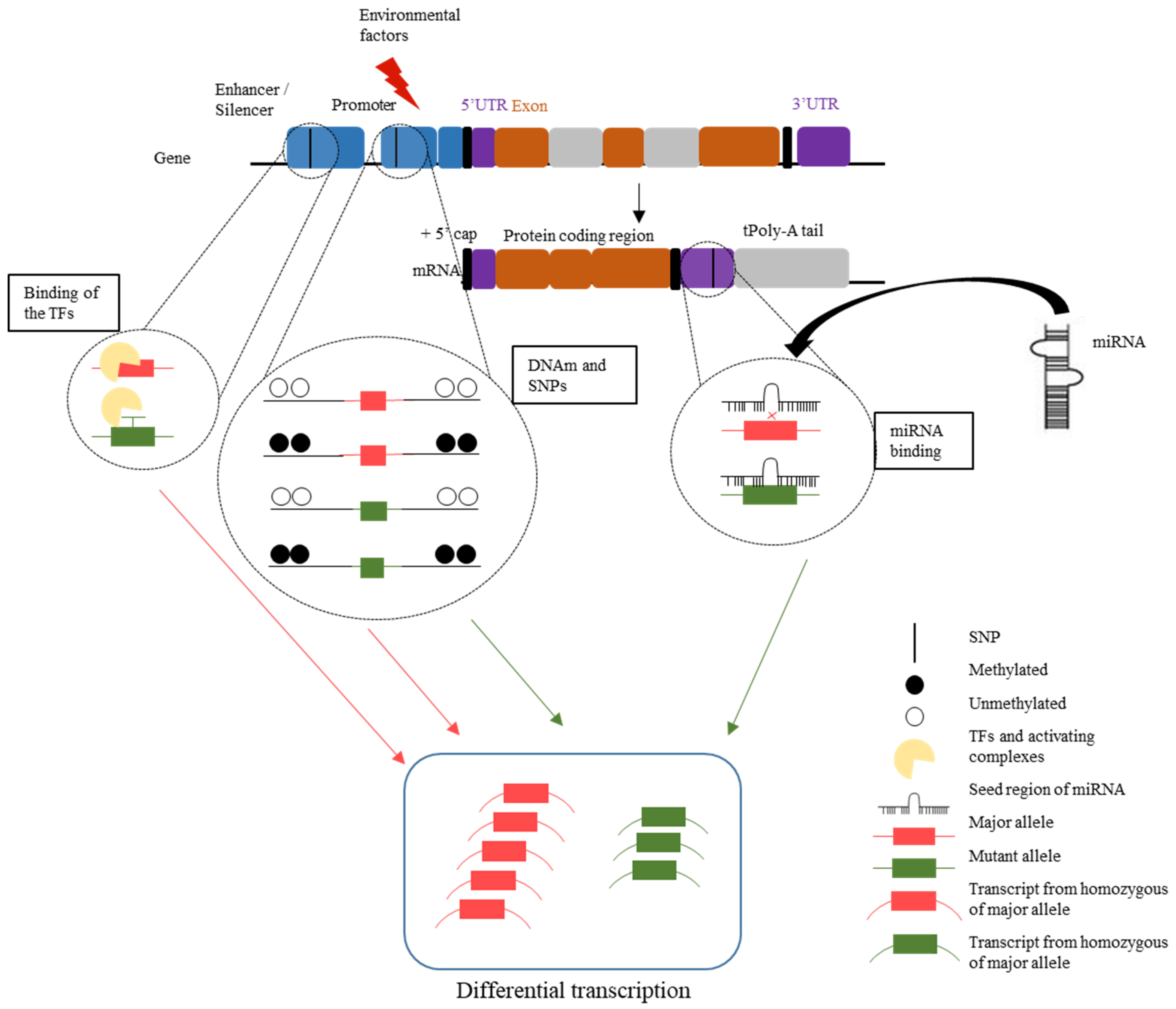
1.3.6. Interpretation of DNAm Variation, SNPs, and miRNA as a Complex Network
2. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2008.
- Sugden, K.; Hannon, E.J.; Arseneault, L.; Belsky, D.W.; Broadbent, J.M.; Corcoran, D.L.; Hancox, R.J.; Houts, R.M.; Moffitt, T.E.; Poulton, R.; et al. Establishing a generalized polyepigenetic biomarker for tobacco smoking. Transl. Psychiatry 2019, 9, 92. [Google Scholar] [CrossRef]
- Córdova-Palomera, A.; Fatjó-Vilas, M.; Gastó, C.; Navarro, V.; Krebs, M.O.; Fañanás, L. Genome-wide methylation study on depression: Differential methylation and variable methylation in monozygotic twins. Transl. Psychiatry 2015, 5, e557. [Google Scholar] [CrossRef]
- Ciuculete, D.M.; Voisin, S.; Kular, L.; Welihinda, N.; Jonsson, J.; Jagodic, M.; Mwinyi, J.; Schiöth, H.B. Longitudinal DNA methylation changes at MET may alter HGF/c-MET signalling in adolescents at risk for depression. Epigenetics 2020, 15, 646–663. [Google Scholar] [CrossRef] [Green Version]
- Ciuculete, D.M.; Bostrom, A.E.; Voisin, S.; Philipps, H.; Titova, O.E.; Bandstein, M.; Nikontovic, L.; Williams, M.J.; Mwinyi, J.; Schioth, H.B. A methylome-wide mQTL analysis reveals associations of methylation sites with GAD1 and HDAC3 SNPs and a general psychiatric risk score. Transl. Psychiatry 2017, 7, e1002. [Google Scholar] [CrossRef]
- Boström, A.E.; Ciuculete, D.M.; Attwood, M.; Krattinger, R.; Nikontovic, L.; Titova, O.E.; Kullak-Ublick, G.A.; Mwinyi, J.; Schiöth, H.B. A MIR4646 associated methylation locus is hypomethylated in adolescent depression. J. Affect Disord 2017, 220, 117–128. [Google Scholar] [CrossRef]
- Jokinen, J.; Boström, A.E.; Dadfar, A.; Ciuculete, D.M.; Chatzittofis, A.; Åsberg, M.; Schiöth, H.B. Epigenetic Changes in the CRH Gene are Related to Severity of Suicide Attempt and a General Psychiatric Risk Score in Adolescents. EBioMedicine 2018, 27, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Ciuculete, D.M.; Boström, A.E.; Tuunainen, A.K.; Sohrabi, F.; Kular, L.; Jagodic, M.; Voisin, S.; Mwinyi, J.; Schiöth, H.B. Changes in methylation within the STK32B promoter are associated with an increased risk for generalized anxiety disorder in adolescents. J. Psychiatr. Res. 2018, 102, 44–51. [Google Scholar] [CrossRef]
- Boström, A.E.; Chatzittofis, A.; Ciuculete, D.M.; Flanagan, J.N.; Krattinger, R.; Bandstein, M.; Mwinyi, J.; Kullak-Ublick, G.A.; Öberg, K.G.; Arver, S.; et al. Hypermethylation-associated downregulation of microRNA-4456 in hypersexual disorder with putative influence on oxytocin signalling: A DNA methylation analysis of miRNA genes. Epigenetics 2020, 15, 145–160. [Google Scholar] [CrossRef]
- Rasmusson, A.J.; Gallwitz, M.; Soltanabadi, B.; Ciuculete, D.M.; Mengel-From, J.; Christensen, K.; Nygaard, M.; Soerensen, M.; Boström, A.E.; Fredriksson, R.; et al. Toll-like receptor 4 methylation grade is linked to depressive symptom severity. Transl. Psychiatry 2021, 11, 371. [Google Scholar] [CrossRef]
- Chatzittofis, A.; Boström, A.D.E.; Ciuculete, D.M.; Öberg, K.G.; Arver, S.; Schiöth, H.B.; Jokinen, J. HPA axis dysregulation is associated with differential methylation of CpG-sites in related genes. Sci. Rep. 2021, 11, 20134. [Google Scholar] [CrossRef]
- England, Public Helath. Severe Mental Illness (SMI) and Physical Health Inequalities: Briefing; England, Public Helath: London, UK, 2018. [Google Scholar]
- Hoang, U.; Goldacre, M.; James, A. Mortality following hospital discharge with a diagnosis of eating disorder: National record linkage study, England, 2001–2009. Int. J. Eat. Disord. 2014, 47, 507–515. [Google Scholar] [CrossRef]
- Manu, D.-M. Functional Epigenetic Analyses in the Context of Psychiatric Health in Adolescence. Ph.D. Thesis, comprehensive summary. Acta Universitatis Upsaliensis, Uppsala, Sweden, 2022. [Google Scholar]
- Wittchen, H.U. Generalized anxiety disorder: Prevalence, burden, and cost to society. Depress. Anxiety 2002, 16, 162–171. [Google Scholar] [CrossRef]
- Bromet, E.; Andrade, L.H.; Hwang, I.; Sampson, N.A.; Alonso, J.; de Girolamo, G.; de Graaf, R.; Demyttenaere, K.; Hu, C.; Iwata, N.; et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med. 2011, 9, 90. [Google Scholar] [CrossRef]
- Moffitt, T.E.; Caspi, A.; Taylor, A.; Kokaua, J.; Milne, B.J.; Polanczyk, G.; Poulton, R. How common are common mental disorders? Evidence that lifetime prevalence rates are doubled by prospective versus retrospective ascertainment. Psychol. Med. 2010, 40, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Smith, K. Mental health: A world of depression. Nature 2014, 515, 180–181. [Google Scholar] [CrossRef]
- Suicide. 2016. Available online: http://www.who.int/topics/suicide/en/ (accessed on 13 April 2022).
- Chesney, E.; Goodwin, G.M.; Fazel, S. Risks of all-cause and suicide mortality in mental disorders: A meta-review. World Psychiatry Off. J. World Psychiatr. Assoc. (WPA) 2014, 13, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, R.; Shen, C.; Sambamoorthi, U. Excess risk of chronic physical conditions associated with depression and anxiety. BMC Psychiatry 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; West, J.; Beck, S. Age-associated epigenetic drift: Implications, and a case of epigenetic thrift? Hum. Mol. Genet. 2013, 22, R7–R15. [Google Scholar] [CrossRef]
- Takeuchi, N.; Nonen, S.; Kato, M.; Wakeno, M.; Takekita, Y.; Kinoshita, T.; Kugawa, F. Therapeutic Response to Paroxetine in Major Depressive Disorder Predicted by DNA Methylation. Neuropsychobiology 2017, 75, 81–88. [Google Scholar] [CrossRef]
- Voisin, S.; Eynon, N.; Yan, X.; Bishop, D.J. Exercise training and DNA methylation in humans. Acta Physiol. 2015, 213, 39–59. [Google Scholar] [CrossRef]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef] [Green Version]
- Nagy, C.; Vaillancourt, K.; Turecki, G. A role for activity-dependent epigenetics in the development and treatment of major depressive disorder. Genes Brain Behav. 2018, 17, e12446. [Google Scholar] [CrossRef] [Green Version]
- Habano, W.; Kawamura, K.; Iizuka, N.; Terashima, J.; Sugai, T.; Ozawa, S. Analysis of DNA methylation landscape reveals the roles of DNA methylation in the regulation of drug metabolizing enzymes. Clin. Epigenetics 2015, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Maschietto, M.; Bastos, L.C.; Tahira, A.C.; Bastos, E.P.; Euclydes, V.L.; Brentani, A.; Fink, G.; de Baumont, A.; Felipe-Silva, A.; Francisco, R.P.; et al. Sex differences in DNA methylation of the cord blood are related to sex-bias psychiatric diseases. Sci. Rep. 2017, 7, 44547. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, P.; Huen, K.; Davé, V.; Barcellos, L.; Eskenazi, B.; Holland, N. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genom. 2015, 16, 911. [Google Scholar] [CrossRef] [Green Version]
- Singmann, P.; Shem-Tov, D.; Wahl, S.; Grallert, H.; Fiorito, G.; Shin, S.-Y.; Schramm, K.; Wolf, P.; Kunze, S.; Baran, Y.; et al. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenetics Chromatin 2015, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wang, F.; Liu, Y.; Yu, Y.; Gelernter, J.; Zhang, H. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum. Mol. Genet. 2014, 23, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Spiers, H.; Hannon, E.; Schalkwyk, L.C.; Smith, R.; Wong, C.C.; O’Donovan, M.C.; Bray, N.J.; Mill, J. Methylomic trajectories across human fetal brain development. Genome Res. 2015, 25, 338–352. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.; Pickles, A.; Wright, N.; Quinn, J.P.; Murgatroyd, C.; Sharp, H. Mismatched Prenatal and Postnatal Maternal Depressive Symptoms and Child Behaviours: A Sex-Dependent Role for NR3C1 DNA Methylation in the Wirral Child Health and Development Study. Cells 2019, 8, 943. [Google Scholar] [CrossRef] [Green Version]
- Krol, K.M.; Grossmann, T. Psychological effects of breastfeeding on children and mothers. Bundesgesundheitsblatt Gesundh. Gesundh. 2018, 61, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.T.; Spector, T.D. DNA methylation studies using twins: What are they telling us? Genome Biol. 2012, 13, 172. [Google Scholar] [CrossRef]
- Gertz, J.; Varley, K.E.; Reddy, T.E.; Bowling, K.M.; Pauli, F.; Parker, S.L.; Kucera, K.S.; Willard, H.F.; Myers, R.M. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011, 7, e1002228. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.A.; Shao, X.; Morin, A.; Siroux, V.; Kwan, T.; Ge, B.; Aïssi, D.; Chen, L.; Vasquez, L.; Allum, F.; et al. Correction to: Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome. Genome Biol. 2019, 20, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dongen, J.; Nivard, M.G.; Willemsen, G.; Hottenga, J.-J.; Helmer, Q.; Dolan, C.V.; Ehli, E.A.; Davies, G.E.; van Iterson, M.; Breeze, C.E.; et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 2016, 7, 11115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Ge, B.; Casale, F.P.; Vasquez, L.; Kwan, T.; Garrido-Martín, D.; Watt, S.; Yan, Y.; Kundu, K.; Ecker, S.; et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 2016, 167, 1398–1414.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannon, E.; Weedon, M.; Bray, N.; O’Donovan, M.; Mill, J. Pleiotropic Effects of Trait-Associated Genetic Variation on DNA Methylation: Utility for Refining GWAS Loci. Am. J. Hum. Genet. 2017, 100, 954–959. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.L.; Tong, L.; Argos, M.; Demanelis, K.; Jasmine, F.; Rakibuz-Zaman, M.; Sarwar, G.; Islam, M.T.; Shahriar, H.; Islam, T.; et al. Co-occurring expression and methylation QTLs allow detection of common causal variants and shared biological mechanisms. Nat. Commun. 2018, 9, 804. [Google Scholar] [CrossRef]
- Gaunt, T.R.; Shihab, H.A.; Hemani, G.; Min, J.L.; Woodward, G.; Lyttleton, O.; Zheng, J.; Duggirala, A.; McArdle, W.L.; Ho, K.; et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.K.; Kilaru, V.; Kocak, M.; Almli, L.M.; Mercer, K.B.; Ressler, K.J.; Tylavsky, F.A.; Conneely, K.N. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genom. 2014, 15, 145. [Google Scholar] [CrossRef] [Green Version]
- Min, J.L.; Hemani, G.; Hannon, E.; Dekkers, K.F.; Castillo-Fernandez, J.; Luijk, R.; Carnero-Montoro, E.; Lawson, D.J.; Burrows, K.; Suderman, M.; et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat. Genet. 2021, 53, 1311–1321. [Google Scholar] [CrossRef]
- Thoenen, H. Neurotrophins and neuronal plasticity. Science 1995, 270, 593–598. [Google Scholar] [CrossRef]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The Role of BDNF on Neural Plasticity in Depression. Front. Cell Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Carlberg, L.; Scheibelreiter, J.; Hassler, M.R.; Schloegelhofer, M.; Schmoeger, M.; Ludwig, B.; Kasper, S.; Aschauer, H.; Egger, G.; Schosser, A. Brain-derived neurotrophic factor (BDNF)-epigenetic regulation in unipolar and bipolar affective disorder. J. Affect. Disord. 2014, 168, 399–406. [Google Scholar] [CrossRef]
- Tadić, A.; Müller-Engling, L.; Schlicht, K.F.; Kotsiari, A.; Dreimüller, N.; Kleimann, A.; Bleich, S.; Lieb, K.; Frieling, H. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol. Psychiatry 2014, 19, 281–283. [Google Scholar] [CrossRef]
- Dell’Osso, B.; D’Addario, C.; Carlotta Palazzo, M.; Benatti, B.; Camuri, G.; Galimberti, D.; Fenoglio, C.; Scarpini, E.; Di Francesco, A.; Maccarrone, M.; et al. Epigenetic modulation of BDNF gene: Differences in DNA methylation between unipolar and bipolar patients. J. Affect. Disord. 2014, 166, 330–333. [Google Scholar] [CrossRef]
- Schröter, K.; Brum, M.; Brunkhorst-Kanaan, N.; Tole, F.; Ziegler, C.; Domschke, K.; Reif, A.; Kittel-Schneider, S. Longitudinal multi-level biomarker analysis of BDNF in major depression and bipolar disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 169–181. [Google Scholar] [CrossRef]
- Lam, D.; Ancelin, M.-L.; Ritchie, K.; Freak-Poli, R.; Saffery, R.; Ryan, J. Genotype-dependent associations between serotonin transporter gene (SLC6A4) DNA methylation and late-life depression. BMC Psychiatry 2018, 18, 282. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Sun, H.; Xu, Y.; Wang, Z.; Cui, H.; Wang, C.; Liu, W.; An, G.; Hu, J. Methylation Status of the Serotonin Transporter Promoter CpG Island Is Associated With Major Depressive Disorder in Chinese Han Population: A Case-Control Study. J. Nerv. Ment. Dis. 2017, 205, 641–646. [Google Scholar] [CrossRef]
- Domschke, K.; Tidow, N.; Schwarte, K.; Deckert, J.; Lesch, K.P.; Arolt, V.; Zwanzger, P.; Baune, B.T. Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int. J. Neuropsychopharmacol. 2014, 17, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- Murgatroyd, C.; Quinn, J.P.; Sharp, H.M.; Pickles, A.; Hill, J. Effects of prenatal and postnatal depression, and maternal stroking, at the glucocorticoid receptor gene. Transl. Psychiatry 2015, 5, e560. [Google Scholar] [CrossRef] [Green Version]
- Na, K.S.; Chang, H.S.; Won, E.; Han, K.M.; Choi, S.; Tae, W.S.; Yoon, H.K.; Kim, Y.K.; Joe, S.H.; Jung, I.K.; et al. Association between glucocorticoid receptor methylation and hippocampal subfields in major depressive disorder. PLoS ONE 2014, 9, e85425. [Google Scholar]
- Efstathopoulos, P.; Andersson, F.; Melas, P.A.; Yang, L.L.; Villaescusa, J.C.; Rȕegg, J.; Ekström, T.J.; Forsell, Y.; Galanti, M.R.; Lavebratt, C. NR3C1 hypermethylation in depressed and bullied adolescents. Transl. Psychiatry 2018, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Starnawska, A.; Bukowski, L.; Chernomorchenko, A.; Elfving, B.; Müller, H.K.; van den Oord, E.; Aberg, K.; Guintivano, J.; Grove, J.; Mors, O.; et al. DNA methylation of the KLK8 gene in depression symptomatology. Clin. Epigenetics 2021, 13, 200. [Google Scholar] [CrossRef] [PubMed]
- Story Jovanova, O.; Nedeljkovic, I.; Spieler, D.; Walker, R.M.; Liu, C.; Luciano, M.; Bressler, J.; Brody, J.; Drake, A.J.; Evans, K.L.; et al. DNA Methylation Signatures of Depressive Symptoms in Middle-aged and Elderly Persons: Meta-analysis of Multiethnic Epigenome-wide Studies. JAMA Psychiatry 2018, 75, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson-Nay, R.; Lapato, D.M.; Wolen, A.R.; Lancaster, E.E.; Webb, B.T.; Verhulst, B.; Hettema, J.M.; York, T.P. An epigenome-wide association study of early-onset major depression in monozygotic twins. Transl. Psychiatry 2020, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Caramaschi, D.; Adams, M.J.; Walker, R.M.; Min, J.L.; Kwong, A.; Hemani, G.; Barbu, M.C.; Whalley, H.C.; Harris, S.E.; et al. DNA methylome-wide association study of genetic risk for depression implicates antigen processing and immune responses. Genome Med. 2021, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Feng, J.; Ji, C.; Mu, X.; Ma, Q.; Fan, Y.; Chen, C.; Gao, C.; Ma, X.C.; Zhu, F. Increased methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of patients with generalized anxiety disorder. J. Psychiatr. Res. 2017, 91, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Emeny, R.T.; Baumert, J.; Zannas, A.S.; Kunze, S.; Wahl, S.; Iurato, S.; Arloth, J.; Erhardt, A.; Balsevich, G.; Schmidt, M.V.; et al. Anxiety Associated Increased CpG Methylation in the Promoter of Asb1, A Translational Approach Evidenced by Epidemiological and Clinical Studies and a Murine Model. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, T.M.; O’Donovan, A.; Mullins, N.; O’Farrelly, C.; McCann, A.; Malone, K. Anxiety is associated with higher levels of global DNA methylation and altered expression of epigenetic and interleukin-6 genes. Psychiatr. Genet. 2015, 25, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Piletič, K.; Kunej, T. MicroRNA epigenetic signatures in human disease. Arch. Toxicol. 2016, 90, 2405–2419. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Q.; Song, R.; Kong, Y.; Zhang, Z. Non-coding RNAs in depression: Promising diagnostic and therapeutic biomarkers. EBioMedicine 2021, 71, 103569. [Google Scholar] [CrossRef]
- Zhang, H.P.; Liu, X.L.; Chen, J.J.; Cheng, K.; Bai, S.J.; Zheng, P.; Zhou, C.J.; Wang, W.; Wang, H.Y.; Zhong, L.M.; et al. Circulating microRNA 134 sheds light on the diagnosis of major depressive disorder. Transl. Psychiatry 2020, 10, 95. [Google Scholar] [CrossRef]
- Chen, S.D.; Sun, X.Y.; Niu, W.; Kong, L.M.; He, M.J.; Fan, H.M.; Li, W.S.; Zhong, A.F.; Zhang, L.Y.; Lu, J. Correlation between the level of microRNA expression in peripheral blood mononuclear cells and symptomatology in patients with generalized anxiety disorder. Compr. Psychiatry 2016, 69, 216–224. [Google Scholar] [CrossRef]
- Wang, X.; Sundquist, K.; Hedelius, A.; Palmér, K.; Memon, A.A.; Sundquist, J. Circulating microRNA-144-5p is associated with depressive disorders. Clin. Epigenetics 2015, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Kim, S.N.; Liu, X.; Zhang, H.; Zhang, C.; Seo, J.S.; Kim, Y.; Sun, T. miR-17-92 Cluster Regulates Adult Hippocampal Neurogenesis, Anxiety, and Depression. Cell Rep. 2016, 16, 1653–1663. [Google Scholar] [CrossRef] [Green Version]
- Hüls, A.; Robins, C.; Conneely, K.N.; De Jager, P.L.; Bennett, D.A.; Epstein, M.P.; Wingo, T.S.; Wingo, A.P. Association between DNA methylation levels in brain tissue and late-life depression in community-based participants. Transl. Psychiatry 2020, 10, 262. [Google Scholar] [CrossRef]
- Aberg, K.A.; Dean, B.; Shabalin, A.A.; Chan, R.F.; Han, L.K.M.; Zhao, M.; van Grootheest, G.; Xie, L.Y.; Milaneschi, Y.; Clark, S.L.; et al. Methylome-wide association findings for major depressive disorder overlap in blood and brain and replicate in independent brain samples. Mol. Psychiatry 2020, 25, 1344–1354. [Google Scholar] [CrossRef]
- Chan, R.F.; Turecki, G.; Shabalin, A.A.; Guintivano, J.; Zhao, M.; Xie, L.Y.; van Grootheest, G.; Kaminsky, Z.A.; Dean, B.; Penninx, B.; et al. Cell Type-Specific Methylome-wide Association Studies Implicate Neurotrophin and Innate Immune Signaling in Major Depressive Disorder. Biol. Psychiatry 2020, 87, 431–442. [Google Scholar] [CrossRef]
- Islam, S.A.; Lussier, A.A.; Kobor, M.S. Chapter 17—Epigenetic Analysis of Human Postmortem Brain Tissue. In Handbook of Clinical Neurology; Huitinga, I., Webster, M.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 150, pp. 237–261. [Google Scholar]
- Nishioka, M.; Bundo, M.; Kasai, K.; Iwamoto, K. DNA methylation in schizophrenia: Progress and challenges of epigenetic studies. Genome Med. 2012, 4, 96. [Google Scholar] [CrossRef] [Green Version]
- Rhein, M.; Hagemeier, L.; Klintschar, M.; Muschler, M.; Bleich, S.; Frieling, H. DNA methylation results depend on DNA integrity-role of post mortem interval. Front. Genet. 2015, 6, 182. [Google Scholar] [CrossRef] [Green Version]
- Rizzardi, L.F.; Hickey, P.F.; Idrizi, A.; Tryggvadóttir, R.; Callahan, C.M.; Stephens, K.E.; Taverna, S.D.; Zhang, H.; Ramazanoglu, S.; Hansen, K.D.; et al. Human brain region-specific variably methylated regions are enriched for heritability of distinct neuropsychiatric traits. Genome Biol. 2021, 22, 116. [Google Scholar] [CrossRef]
- Rizzardi, L.F.; Hickey, P.F.; Rodriguez DiBlasi, V.; Tryggvadóttir, R.; Callahan, C.M.; Idrizi, A.; Hansen, K.D.; Feinberg, A.P. Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat. Neurosci. 2019, 22, 307–316. [Google Scholar] [CrossRef]
- Rukova, B.; Staneva, R.; Hadjidekova, S.; Stamenov, G.; Milanova t Toncheva, D. Genome-wide methylation profiling of schizophrenia. Balk. J. Med. Genet. BJMG 2014, 17, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansell, G.; Gorrie-Stone, T.J.; Bao, Y.; Kumari, M.; Schalkwyk, L.S.; Mill, J.; Hannon, E. Guidance for DNA methylation studies: Statistical insights from the Illumina EPIC array. BMC Genom. 2019, 20, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.H. Beyond depression: The expanding role of inflammation in psychiatric disorders. World Psychiatry 2020, 19, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.; Chen, Y.; Xia, Y.; Dai, J.; Liu, C. Inflammation-related biomarkers in major psychiatric disorders: A cross-disorder assessment of reproducibility and specificity in 43 meta-analyses. Transl. Psychiatry 2019, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Pearlman, D.M.; Devinsky, O.; Najjar, A.; Zagzag, D. Neurovascular unit dysfunction with blood-brain barrier hyperpermeability contributes to major depressive disorder: A review of clinical and experimental evidence. J. Neuroinflammation 2013, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyf, M. Examining peripheral DNA methylation in behavioral epigenetic and epigenetic psychiatry: Opportunities and challenges. Epigenomics 2014, 6, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Walton, E.; Hass, J.; Liu, J.; Roffman, J.L.; Bernardoni, F.; Roessner, V.; Kirsch, M.; Schackert, G.; Calhoun, V.; Ehrlich, S. Correspondence of DNA Methylation Between Blood and Brain Tissue and Its Application to Schizophrenia Research. Schizophr. Bull 2016, 42, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Hannon, E.; Lunnon, K.; Schalkwyk, L.; Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: Implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 2015, 10, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.K.; Kilaru, V.; Klengel, T.; Mercer, K.B.; Bradley, B.; Conneely, K.N.; Ressler, K.J.; Binder, E.B. DNA extracted from saliva for methylation studies of psychiatric traits: Evidence tissue specificity and relatedness to brain. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2015, 168b, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Braun, P.R.; Han, S.; Hing, B.; Nagahama, Y.; Gaul, L.N.; Heinzman, J.T.; Grossbach, A.J.; Close, L.; Dlouhy, B.J.; Howard, M.A.; et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 2019, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [CrossRef] [Green Version]
- Ernst, J.; Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010, 28, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Abascal, F.; Acosta, R.; Addleman, N.J.; Adrian, J.; Afzal, V.; Aken, B.; Akiyama, J.A.; Jammal, O.A.; Amrhein, H.; Anderson, S.M.; et al. Perspectives on ENCODE. Nature 2020, 583, 693–698. [Google Scholar]
- Buitrago, D.; Labrador, M.; Arcon, J.P.; Lema, R.; Flores, O.; Esteve-Codina, A.; Blanc, J.; Villegas, N.; Bellido, D.; Gut, M.; et al. Impact of DNA methylation on 3D genome structure. Nat. Commun. 2021, 12, 3243. [Google Scholar] [CrossRef]
- Pérez, A.; Castellazzi, C.L.; Battistini, F.; Collinet, K.; Flores, O.; Deniz, O.; Ruiz, M.L.; Torrents, D.; Eritja, R.; Soler-López, M. Impact of methylation on the physical properties of DNA. Biophys. J. 2012, 102, 2140–2148. [Google Scholar] [CrossRef] [Green Version]
- Portella, G.; Battistini, F.; Orozco, M. Understanding the connection between epigenetic DNA methylation and nucleosome positioning from computer simulations. PLoS Comput. Biol. 2013, 9, e1003354. [Google Scholar] [CrossRef] [Green Version]
- Collings, C.K.; Anderson, J.N. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenetics Chromatin 2017, 10, 18. [Google Scholar] [CrossRef]
- Choy, J.S.; Wei, S.; Lee, J.Y.; Tan, S.; Chu, S.; Lee, T.H. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010, 132, 1782–1783. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Tsai, S.J. Epigenetics and Depression: An Update. Psychiatry Investig 2019, 16, 654–661. [Google Scholar] [CrossRef]
- Covington, H.E., 3rd; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J., 3rd; Wu, E.Y.; et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci 2009, 29, 11451–11460. [Google Scholar] [CrossRef]
- Park, H.-S.; Kim, J.; Ahn, S.H.; Ryu, H.-Y. Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression. Int. J. Mol. Sci. 2021, 22, 5398. [Google Scholar] [CrossRef] [PubMed]
- Martins de Carvalho, L.; Chen, W.Y.; Lasek, A.W. Epigenetic mechanisms underlying stress-induced depression. Int. Rev. Neurobiol. 2021, 156, 87–126. [Google Scholar] [PubMed]
- Hobara, T.; Uchida, S.; Otsuki, K.; Matsubara, T.; Funato, H.; Matsuo, K.; Suetsugi, M.; Watanabe, Y. Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatr. Res. 2010, 44, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Göndör, A.; Ohlsson, R. Chromosome crosstalk in three dimensions. Nature 2009, 461, 212–217. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Maricque, B.; Xie, M.; Li, D.; Sundaram, V.; Martin, E.A.; Koebbe, B.C.; Nielsen, C.; Hirst, M.; Farnham, P.; et al. The Human Epigenome Browser at Washington University. Nat. Methods 2011, 8, 989–990. [Google Scholar] [CrossRef] [Green Version]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Rauluseviciute, I.; Drabløs, F.; Rye, M.B. DNA methylation data by sequencing: Experimental approaches and recommendations for tools and pipelines for data analysis. Clin. Epigenetics 2019, 11, 193. [Google Scholar] [CrossRef] [Green Version]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Houtepen, L.C.; van Bergen, A.H.; Vinkers, C.H.; Boks, M.P. DNA methylation signatures of mood stabilizers and antipsychotics in bipolar disorder. Epigenomics 2016, 8, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.C.; Loh, M.; Lehne, B.; Drong, A.; Kriebel, J.; Motta, V.; Wahl, S.; Elliott, H.R.; Rota, F.; Scott, W.R.; et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: A nested case-control study. Lancet Diabetes Endocrinol 2015, 3, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Šestáková, Š.; Šálek, C.; Remešová, H. DNA Methylation Validation Methods: A Coherent Review with Practical Comparison. Biol. Proced. Online 2019, 21, 19. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Kraiczy, J.; Nayak, K.; Ross, A.; Raine, T.; Mak, T.N.; Gasparetto, M.; Cario, E.; Rakyan, V.; Heuschkel, R.; Zilbauer, M. Assessing DNA methylation in the developing human intestinal epithelium: Potential link to inflammatory bowel disease. Mucosal Immunol. 2016, 9, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.A.; Nagy-Szakal, D.; Mir, S.A.; Frank, E.; Szigeti, R.; Kaplan, J.L.; Bronsky, J.; Opekun, A.; Ferry, G.D.; Winter, H.; et al. DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics 2014, 9, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Gross, J.A.; Pacis, A.; Chen, G.G.; Drupals, M.; Lutz, P.E.; Barreiro, L.B.; Turecki, G. Gene-body 5-hydroxymethylation is associated with gene expression changes in the prefrontal cortex of depressed individuals. Transl. Psychiatry 2017, 7, e1119. [Google Scholar] [CrossRef] [Green Version]
- Yong, W.-S.; Hsu, F.-M.; Chen, P.-Y. Profiling genome-wide DNA methylation. Epigenetics Chromatin 2016, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Franks, J.M.; Whitfield, M.L.; Cheng, C. BioMethyl: An R package for biological interpretation of DNA methylation data. Bioinformatics 2019, 35, 3635–3641. [Google Scholar] [CrossRef]
- Halachev, K.; Bast, H.; Albrecht, F.; Lengauer, T.; Bock, C. EpiExplorer: Live exploration and global analysis of large epigenomic datasets. Genome Biol. 2012, 13, R96. [Google Scholar] [CrossRef] [Green Version]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; The Galaxy, T. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannon, E.; Mansell, G.; Walker, E.; Nabais, M.F.; Burrage, J.; Kepa, A.; Best-Lane, J.; Rose, A.; Heck, S.; Moffitt, T.E.; et al. Assessing the co-variability of DNA methylation across peripheral cells and tissues: Implications for the interpretation of findings in epigenetic epidemiology. PLoS Genet. 2021, 17, e1009443. [Google Scholar] [CrossRef] [PubMed]
- Perzel Mandell, K.A.; Eagles, N.J.; Wilton, R.; Price, A.J.; Semick, S.A.; Collado-Torres, L.; Ulrich, W.S.; Tao, R.; Han, S.; Szalay, A.S.; et al. Genome-wide sequencing-based identification of methylation quantitative trait loci and their role in schizophrenia risk. Nat. Commun. 2021, 12, 5251. [Google Scholar] [CrossRef]
- McCartney, D.L.; Hillary, R.F.; Stevenson, A.J.; Ritchie, S.J.; Walker, R.M.; Zhang, Q.; Morris, S.W.; Bermingham, M.L.; Campbell, A.; Murray, A.D.; et al. Epigenetic prediction of complex traits and death. Genome Biol. 2018, 19, 136. [Google Scholar] [CrossRef] [Green Version]
- Vickerstaff, V.; Omar, R.Z.; Ambler, G. Methods to adjust for multiple comparisons in the analysis and sample size calculation of randomised controlled trials with multiple primary outcomes. BMC Med. Res. Methodol. 2019, 19, 129. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D. A Direct Approach to False Discovery Rates. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2002, 64, 479–498. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Bai, L.; Cui, B.; Wu, L.; Wang, L.; An, Z.; Ruan, S.; Yu, Y.; Zhang, X.; Chen, J. Leveraging biological and statistical covariates improves the detection power in epigenome-wide association testing. Genome Biol. 2020, 21, 88. [Google Scholar] [CrossRef]
- Saffari, A.; Silver, M.J.; Zavattari, P.; Moi, L.; Columbano, A.; Meaburn, E.L.; Dudbridge, F. Estimation of a significance threshold for epigenome-wide association studies. Genet. Epidemiol 2018, 42, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Arslan, A.A.; Tuminello, S.; Yang, L.; Zhang, Y.; Durmus, N.; Snuderl, M.; Heguy, A.; Zeleniuch-Jacquotte, A.; Shao, Y.; Reibman, J. Genome-Wide DNA Methylation Profiles in Community Members Exposed to the World Trade Center Disaster. Int. J. Environ. Res. Public Health 2020, 17, 5493. [Google Scholar] [CrossRef]
- Kuan, P.F.; Waszczuk, M.A.; Kotov, R.; Marsit, C.J.; Guffanti, G.; Gonzalez, A.; Yang, X.; Koenen, K.; Bromet, E.; Luft, B.J. An epigenome-wide DNA methylation study of PTSD and depression in World Trade Center responders. Transl. Psychiatry 2017, 7, e1158. [Google Scholar] [CrossRef]
- Baharudin, R.; Ishak, M.; Muhamad Yusof, A.; Saidin, S.; Syafruddin, S.E.; Wan Mohamad Nazarie, W.F.; Lee, L.-H.; Ab Mutalib, N.-S. Epigenome-Wide DNA Methylation Profiling in Colorectal Cancer and Normal Adjacent Colon Using Infinium Human Methylation 450K. Diagnostics 2022, 12, 198. [Google Scholar] [CrossRef]
- Goodin, B.R.; Overstreet, D.S.; Penn, T.M.; Bakshi, R.; Quinn, T.L.; Sims, A.; Ptacek, T.; Jackson, P.; Long, D.L.; Aroke, E.N. Epigenome-wide DNA methylation profiling of conditioned pain modulation in individuals with non-specific chronic low back pain. Clin. Epigenetics 2022, 14, 45. [Google Scholar] [CrossRef]
- Castro de Moura, M.; Davalos, V.; Planas-Serra, L.; Alvarez-Errico, D.; Arribas, C.; Ruiz, M.; Aguilera-Albesa, S.; Troya, J.; Valencia-Ramos, J.; Vélez-Santamaria, V.; et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 2021, 66, 103339. [Google Scholar] [CrossRef]
- Witasp, A.; Luttropp, K.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Wennberg, L.; Ekström, T.J.; Shiels, P.G.; Stenvinkel, P.; Nordfors, L. Longitudinal genome-wide DNA methylation changes in response to kidney failure replacement therapy. Sci. Rep. 2022, 12, 470. [Google Scholar] [CrossRef]
- Bauer, M.; Fink, B.; Thürmann, L.; Eszlinger, M.; Herberth, G.; Lehmann, I. Tobacco smoking differently influences cell types of the innate and adaptive immune system—indications from CpG site methylation. Clin. Epigenetics 2016, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014, 15, R31. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Bringeland, N.; Nilsson, E.K.; Bandstein, M.; Olaya Búcaro, M.; Vogel, H.; Schürmann, A.; Hogenkamp, P.S.; Benedict, C.; Schiöth, H.B. Postprandial alterations in whole-blood DNA methylation are mediated by changes in white blood cell composition. Am. J. Clin. Nutr. 2016, 104, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.C.; Beck, S.; Jaffe, A.E.; Koestler, D.C.; Hansen, K.D.; Houseman, A.E.; Irizarry, R.A.; Teschendorff, A.E. Correcting for cell-type heterogeneity in epigenome-wide association studies: Revisiting previous analyses. Nat. Methods 2017, 14, 216–217. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.-L.; Zhang, K.; Zhang, K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Abu Hamdeh, S.; Ciuculete, D.M.; Sarkisyan, D.; Bakalkin, G.; Ingelsson, M.; Schiöth, H.B.; Marklund, N. Differential DNA Methylation of the Genes for Amyloid Precursor Protein, Tau, and Neurofilaments in Human Traumatic Brain Injury. J. Neurotrauma. 2021, 38, 1679–1688. [Google Scholar] [CrossRef]
- Ciuculete, D.M.; Voisin, S.; Kular, L.; Jonsson, J.; Rask-Andersen, M.; Mwinyi, J.; Schiöth, H.B. meQTL and ncRNA functional analyses of 102 GWAS-SNPs associated with depression implicate HACE1 and SHANK2 genes. Clin. Epigenetics 2020, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Voisin, S.; Almén, M.S.; Zheleznyakova, G.Y.; Lundberg, L.; Zarei, S.; Castillo, S.; Eriksson, F.E.; Nilsson, E.K.; Blüher, M.; Böttcher, Y.; et al. Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 2015, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Behnam, E.; Huang, J.; Moffatt, M.F.; Schaid, D.J.; Liang, L.; Lin, X. Fast and robust adjustment of cell mixtures in epigenome-wide association studies with SmartSVA. BMC Genom. 2017, 18, 413. [Google Scholar] [CrossRef] [PubMed]
- Lomvardas, S.; Barnea, G.; Pisapia, D.J.; Mendelsohn, M.; Kirkland, J.; Axel, R. Interchromosomal interactions and olfactory receptor choice. Cell 2006, 126, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, M.V.; Piovan, C.; Croce, C.M. Interplay between microRNAs and the epigenetic machinery: An intricate network. Biochim. Et Biophys. Acta 2010, 1799, 694–701. [Google Scholar] [CrossRef] [PubMed]
- McClay, J.L.; Shabalin, A.A.; Dozmorov, M.G.; Adkins, D.E.; Kumar, G.; Nerella, S.; Clark, S.L.; Bergen, S.E.; Hultman, C.M.; Magnusson, P.K.; et al. High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol. 2015, 16, 291. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Arcelus, M.; Ongen, H.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Yurovsky, A.; Bryois, J.; Padioleau, I.; Romano, L.; Planchon, A.; et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015, 11, e1004958. [Google Scholar] [CrossRef] [Green Version]
- Benitez, J.A.; Cheng, S.; Deng, Q. Revealing allele-specific gene expression by single-cell transcriptomics. Int. J. Biochem. Cell Biol. 2017, 90, 155–160. [Google Scholar] [CrossRef]
- Zaina, S.; Pérez-Luque, E.L.; Lund, G. Genetics talks to epigenetics? The interplay between sequence variants and chromatin structure. Curr. Genom. 2010, 11, 359–367. [Google Scholar] [CrossRef]
- Schiele, M.A.; Domschke, K. Epigenetics at the crossroads between genes, environment and resilience in anxiety disorders. Genes Brain Behav. 2018, 17, e12423. [Google Scholar] [CrossRef] [Green Version]
- Maddox, S.A.; Schafe, G.E.; Ressler, K.J. Exploring epigenetic regulation of fear memory and biomarkers associated with post-traumatic stress disorder. Front. Psychiatry 2013, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.D.; Jones, M.J.; Meaney, M.J.; Turecki, G.; Kobor, M.S. BECon: A tool for interpreting DNA methylation findings from blood in the context of brain. Transl. Psychiatry 2017, 7, e1187. [Google Scholar] [CrossRef] [Green Version]
- Brazma, A.; Parkinson, H.; Sarkans, U.; Shojatalab, M.; Vilo, J.; Abeygunawardena, N.; Holloway, E.; Kapushesky, M.; Kemmeren, P.; Lara, G.G.; et al. ArrayExpress—A public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003, 31, 68–71. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Website (accessed on 11 May 2022) | Developer | Query | Reference |
|---|---|---|---|
| http://mqtldb.godmc.org.uk/ | Josine L Min et al. | DNAm-meQTL | [52] |
| http://epigenetics.essex.ac.uk/bloodbrain/ | Hannon E. et al. | Blood–brain DNAm correlation | [93] |
| https://redgar598.shinyapps.io/BECon/ | Edgar et al. | Blood–brain DNAm correlation | [159] |
| http://epigenomegateway.wustl.edu/browser/ | Xin Zhou et al. | Exploration of genomic data | [111] |
| https://www.ebi.ac.uk/arrayexpress/ | Alvis Brazma et al. | Open-access data | [160] |
| https://epigenetics.essex.ac.uk/shiny/EPICDNAmPowerCalcs/ | Mansell et al. | Power calculation | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manu, D.M.; Mwinyi, J.; Schiöth, H.B. Challenges in Analyzing Functional Epigenetic Data in Perspective of Adolescent Psychiatric Health. Int. J. Mol. Sci. 2022, 23, 5856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105856
Manu DM, Mwinyi J, Schiöth HB. Challenges in Analyzing Functional Epigenetic Data in Perspective of Adolescent Psychiatric Health. International Journal of Molecular Sciences. 2022; 23(10):5856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105856
Chicago/Turabian StyleManu, Diana M., Jessica Mwinyi, and Helgi B. Schiöth. 2022. "Challenges in Analyzing Functional Epigenetic Data in Perspective of Adolescent Psychiatric Health" International Journal of Molecular Sciences 23, no. 10: 5856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105856