
NKG2C+ NK Cells for Immunotherapy of Glioblastoma Multiforme

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
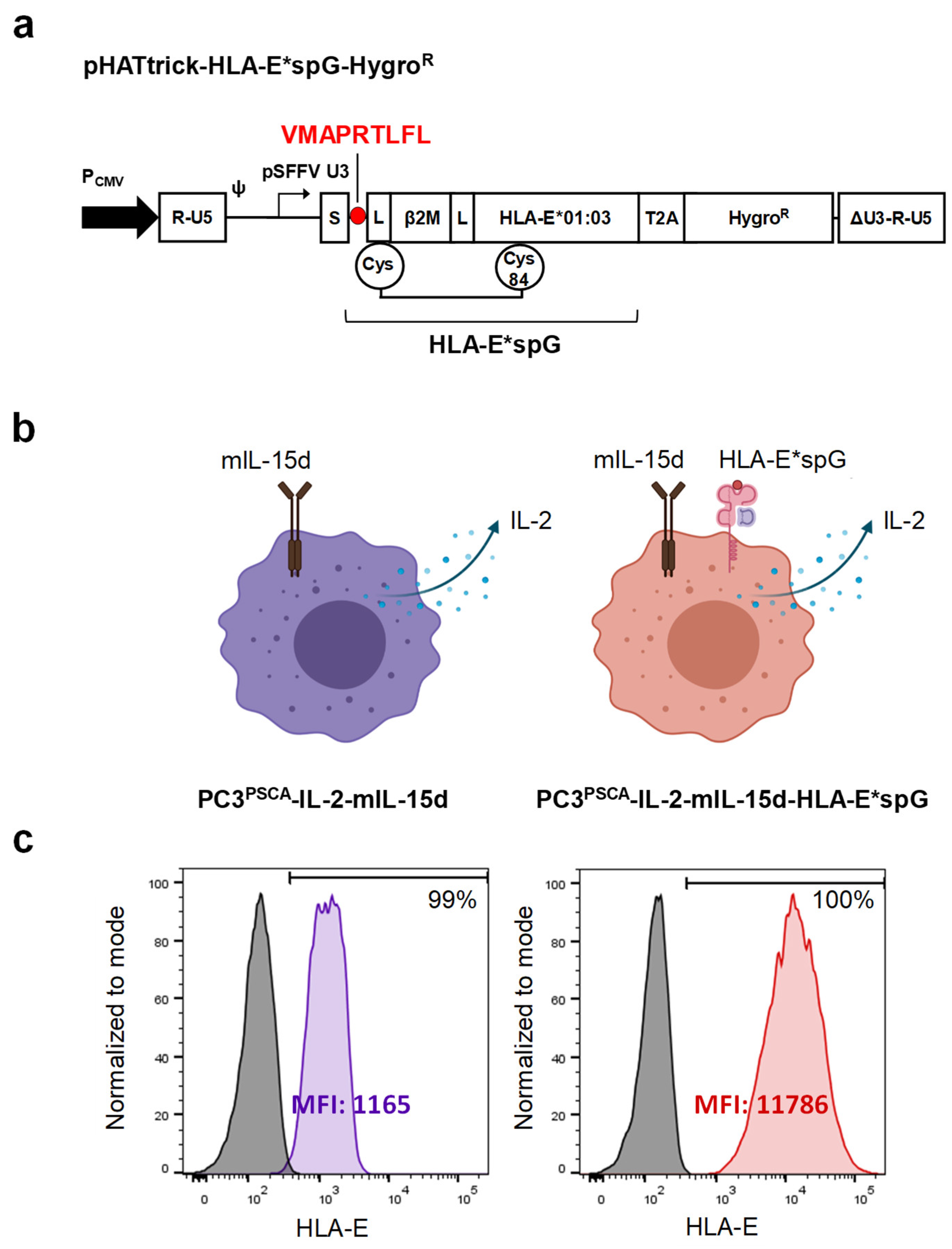
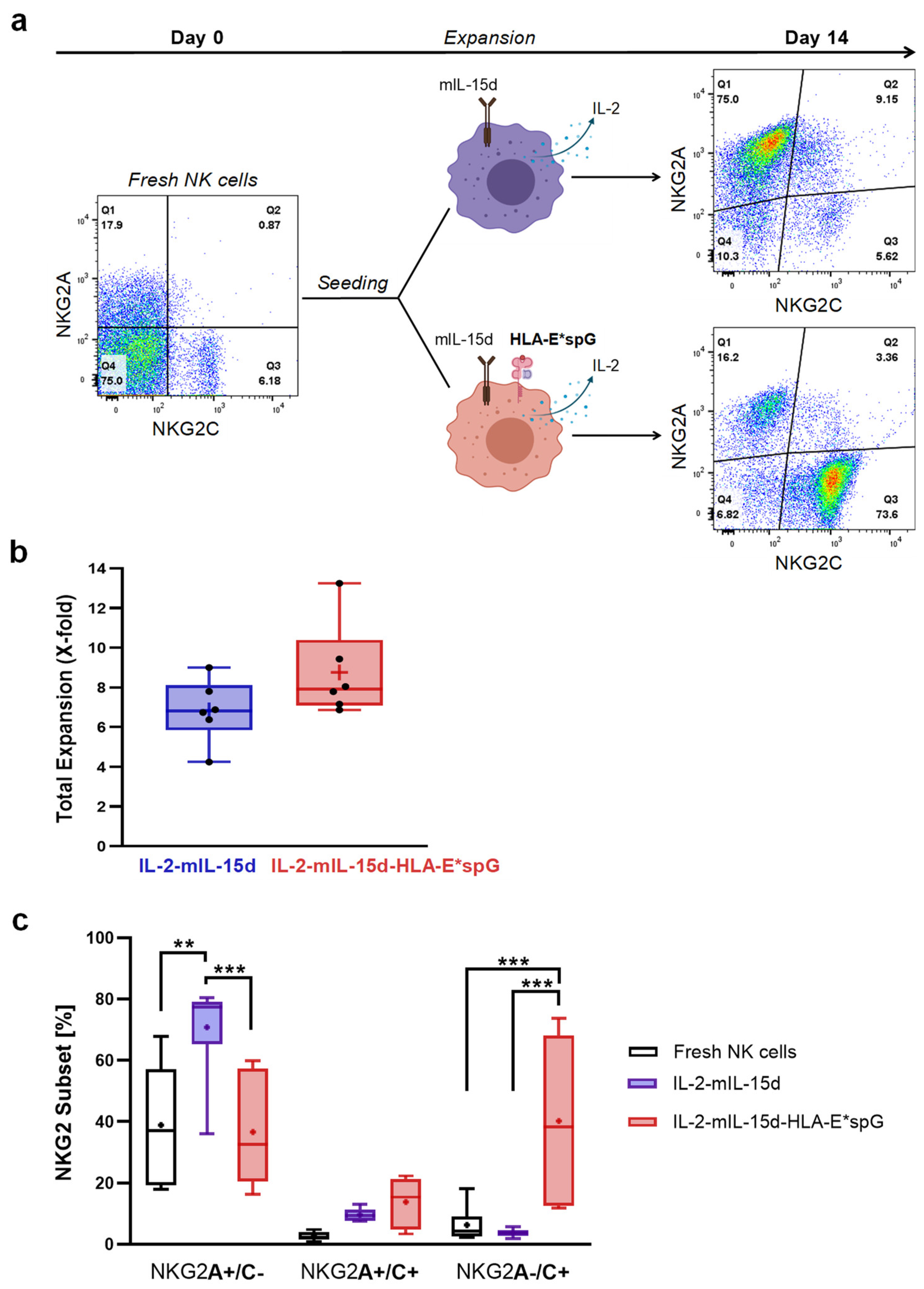
2.1. Expansion of NKG2C+ NK Cells Using Feeder Cells
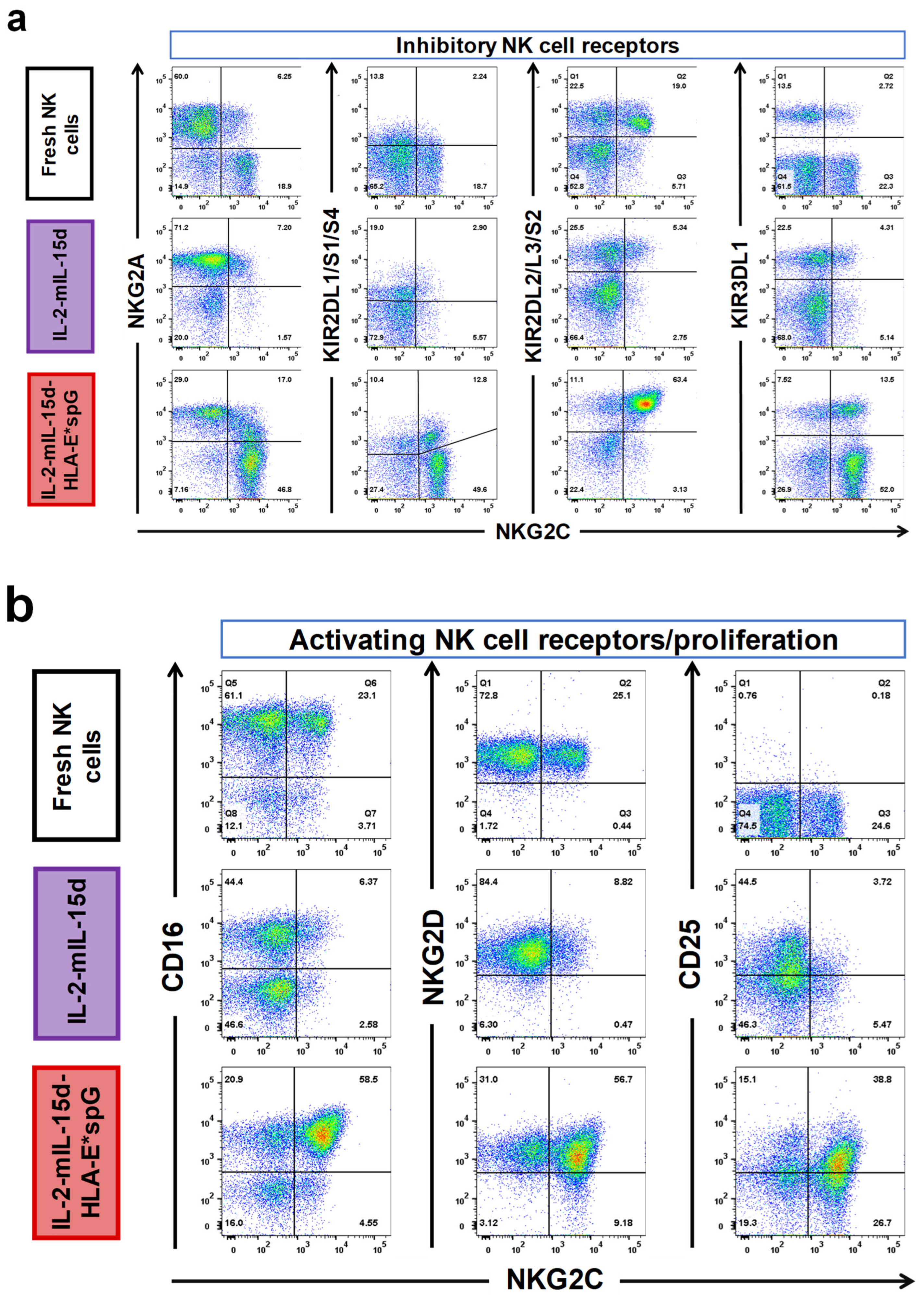
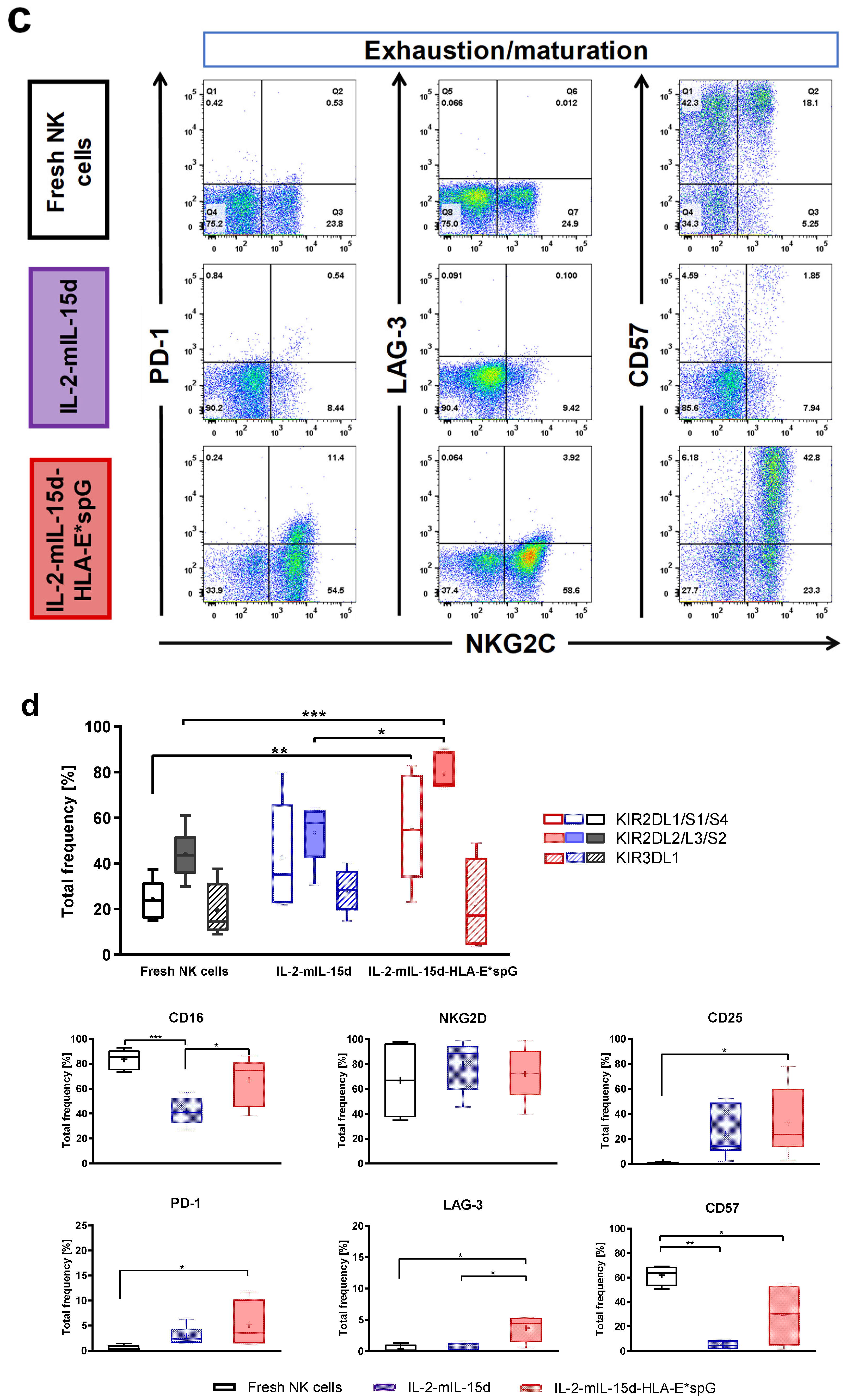
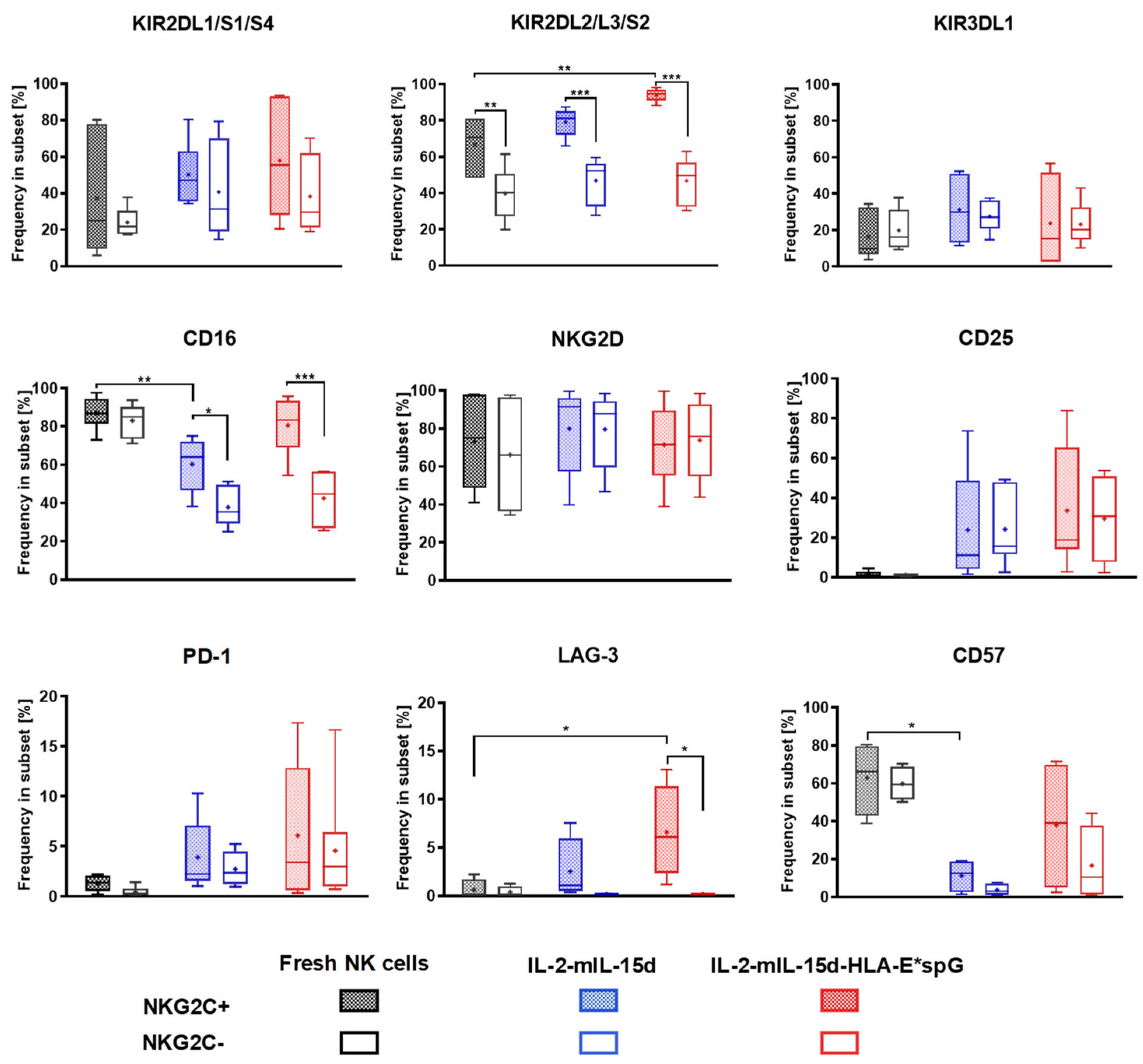
2.2. Expansion by HLA-E*spG-Modified Feeder Cells Results in Increased Frquencies of NK Cells Expressing CD16, KIR2DL2/L3/S2 and CD25
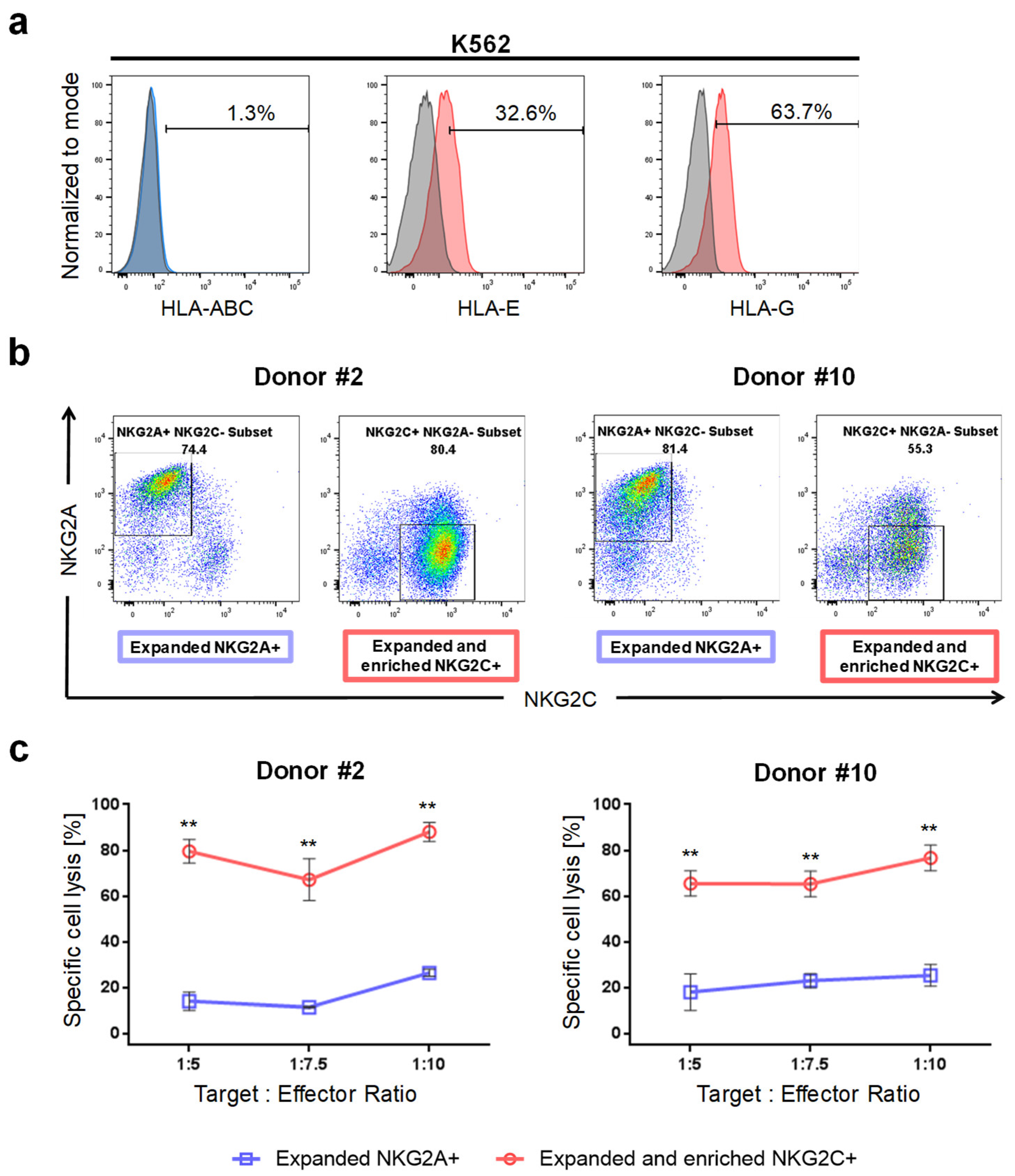
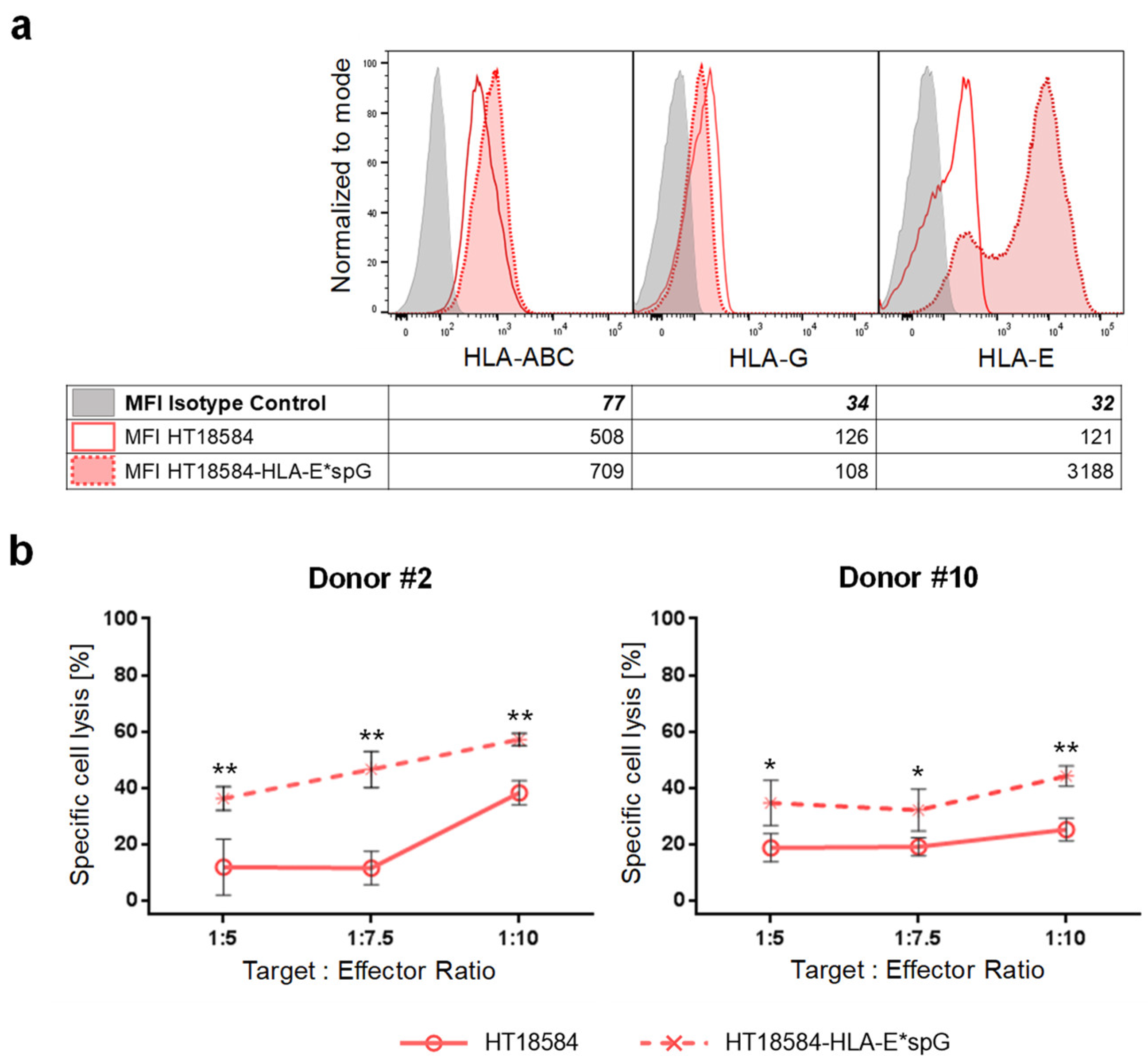
2.3. Expanded NKG2C+ NK Cells React against K562 Target Cells and Kill Primary Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Lentiviral Vectors Encoding HLA-E*spG and Transduction of Cells
4.3. Isolation and Expansion NK Cells
4.4. HLA and KIR Genotyping
4.5. Flow Cytometry Analyses of Cell Surface Proteins
4.6. Chromium (51Cr) Release Cytotoxicity Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Sulman, E.P.; Ismaila, N.; Armstrong, T.S.; Tsien, C.; Batchelor, T.T.; Cloughesy, T.; Galanis, E.; Gilbert, M.; Gondi, V.; Lovely, M.; et al. Radiation Therapy for Glioblastoma: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the American Society for Radiation Oncology Guideline. J. Clin. Oncol. 2017, 35, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma the CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Tietze, S.; Michen, S.; Schackert, G.; Temme, A. Prospects of immune checkpoint blockade and vaccine-based immunotherapy for glioblastoma. Innov. Surg. Sci. 2021, 6, 35–48. [Google Scholar] [CrossRef]
- Topfer, K.; Kempe, S.; Muller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed. Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2015, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Michen, S.; Temme, A. Genetically Engineered Natural Killer Cells as a Means for Adoptive Tumor Immunotherapy. Crit. Rev. Immunol. 2016, 36, 329–347. [Google Scholar] [CrossRef] [PubMed]
- Shifrin, N.; Raulet, D.H.; Ardolino, M. NK cell self tolerance, responsiveness and missing self recognition. Semin. Immunol. 2014, 26, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Petrie, E.J.; Clements, C.S.; Lin, J.; Sullivan, L.C.; Johnson, D.; Huyton, T.; Heroux, A.; Hoare, H.L.; Beddoe, T.; Reid, H.H.; et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J. Exp. Med. 2008, 205, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L.; Corliss, B.; Wu, J.; Phillips, J.H. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity 1998, 8, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Abi-Rached, L.; Parham, P. Natural selection drives recurrent formation of activating killer cell immunoglobulin-like receptor and Ly49 from inhibitory homologues. J. Exp. Med. 2005, 201, 1319–1332. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.A.; Laugier-Anfossi, F.; Vely, F.; Saulquin, X.; Riedmuller, J.; Tisserant, A.; Gauthier, L.; Romagne, F.; Ferracci, G.; Arosa, F.A.; et al. Recognition of peptide-MHC class I complexes by activating killer immunoglobulin-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 13224–13229. [Google Scholar] [CrossRef] [Green Version]
- Wischhusen, J.; Friese, M.A.; Mittelbronn, M.; Meyermann, R.; Weller, M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: Implications for immune escape in vivo. J. Neuropathol. Exp. Neurol. 2005, 64, 523–528. [Google Scholar] [CrossRef]
- Wolpert, F.; Roth, P.; Lamszus, K.; Tabatabai, G.; Weller, M.; Eisele, G. HLA-E contributes to an immune-inhibitory phenotype of glioblastoma stem-like cells. J. Neuroimmunol. 2012, 250, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Wastowski, I.J.; Simoes, R.T.; Yaghi, L.; Donadi, E.A.; Pancoto, J.T.; Poras, I.; Lechapt-Zalcman, E.; Bernaudin, M.; Valable, S.; Carlotti, C.G., Jr.; et al. Human leukocyte antigen-G is frequently expressed in glioblastoma and may be induced in vitro by combined 5-aza-2’-deoxycytidine and interferon-gamma treatments: Results from a multicentric study. Am. J. Pathol. 2013, 182, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Rolle, A.; Meyer, M.; Calderazzo, S.; Jager, D.; Momburg, F. Distinct HLA-E Peptide Complexes Modify Antibody-Driven Effector Functions of Adaptive NK Cells. Cell Rep. 2018, 24, 1967–1976.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vales-Gomez, M.; Reyburn, H.T.; Erskine, R.A.; Lopez-Botet, M.; Strominger, J.L. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J. 1999, 18, 4250–4260. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L.; Corliss, B.C.; Wu, J.; Leong, C.; Phillips, J.H. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 1998, 391, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Topfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Muller, N.; Lindemann, D.; Bachmann, M.; Fussel, M.; Schackert, G.; Temme, A. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Muller, N.; Michen, S.; Tietze, S.; Topfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified with an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1alpha-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Guma, M.; Angulo, A.; Vilches, C.; Gomez-Lozano, N.; Malats, N.; Lopez-Botet, M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Q.; Ruckert, T.; Borst, E.M.; Dunst, J.; Haubner, A.; Durek, P.; Heinrich, F.; Gasparoni, G.; Babic, M.; Tomic, A.; et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 2018, 19, 453–463. [Google Scholar] [CrossRef]
- Guma, M.; Budt, M.; Saez, A.; Brckalo, T.; Hengel, H.; Angulo, A.; Lopez-Botet, M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006, 107, 3624–3631. [Google Scholar] [CrossRef] [Green Version]
- Sivori, S.; Pende, D.; Quatrini, L.; Pietra, G.; Della Chiesa, M.; Vacca, P.; Tumino, N.; Moretta, F.; Mingari, M.C.; Locatelli, F.; et al. NK cells and ILCs in tumor immunotherapy. Mol. Asp. Med. 2021, 80, 100870. [Google Scholar] [CrossRef]
- Michen, S.; Frosch, J.; Fussel, M.; Schackert, G.; Momburg, F.; Temme, A. Artificial feeder cells expressing ligands for killer cell immunoglobulin-like receptors and CD94/NKG2A for expansion of functional primary natural killer cells with tolerance to self. Cytotherapy 2020, 22, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, N.; Wieten, L.; Popeijus, H.E.; Voorter, C.E.M.; Tilanus, M.G.J. HLA-E regulates NKG2C+natural killer cell function through presentation of a restricted peptide repertoire. Hum. Immunol. 2015, 76, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Graef, T.; Moesta, A.K.; Norman, P.J.; Abi-Rached, L.; Vago, L.; Older Aguilar, A.M.; Gleimer, M.; Hammond, J.A.; Guethlein, L.A.; Bushnell, D.A.; et al. KIR2DS4 is a product of gene conversion with KIR3DL2 that introduced specificity for HLA-A*11 while diminishing avidity for HLA-C. J. Exp. Med. 2009, 206, 2557–2572. [Google Scholar] [CrossRef] [Green Version]
- Beziat, V.; Liu, L.L.; Malmberg, J.A.; Ivarsson, M.A.; Sohlberg, E.; Bjorklund, A.T.; Retiere, C.; Sverremark-Ekstrom, E.; Traherne, J.; Ljungman, P.; et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013, 121, 2678–2688. [Google Scholar] [CrossRef]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.Y.; Chiang, S.C.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus Infection Drives Adaptive Epigenetic Diversification of NK Cells with Altered Signaling and Effector Function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.F.; Hultin, L.E.; Hultin, P.; Hausner, M.A.; Hirji, K.; Jewett, A.; Bonavida, B.; Detels, R.; Giorgi, J.V. Natural-Killer-Cell Immunodeficiency in Hiv Disease Is Manifest by Profoundly Decreased Numbers of Cd16(+)Cd56(+) Cells and Expansion of a Population of Cd16(Dim)Cd56(-) Cells with Low Lytic Activity. J. Acq. Immun. Def. Synd. 1995, 10, 331–340. [Google Scholar] [CrossRef]
- Gonzalez, V.D.; Falconer, K.; Bjorkstrom, N.K.; Blom, K.G.; Weiland, O.; Ljunggren, H.G.; Alaeus, A.; Sandberg, J.K. Expansion of Functionally Skewed CD56-Negative NK Cells in Chronic Hepatitis C Virus Infection: Correlation with Outcome of Pegylated IFN-alpha and Ribavirin Treatment. J. Immunol. 2009, 183, 6612–6618. [Google Scholar] [CrossRef] [Green Version]
- Muller-Durovic, B.; Grahlert, J.; Devine, O.P.; Akbar, A.N.; Hess, C. CD56-negative NK cells with impaired effector function expand in CMV and EBV co-infected healthy donors with age. Aging 2019, 11, 724–740. [Google Scholar] [CrossRef]
- Merino, A.; Zhang, B.; Dougherty, P.; Luo, X.; Wang, J.; Blazar, B.R.; Miller, J.S.; Cichocki, F. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Investig. 2019, 129, 3770–3785. [Google Scholar] [CrossRef] [Green Version]
- Oei, V.Y.S.; Siernicka, M.; Graczyk-Jarzynka, A.; Hoel, H.J.; Yang, W.W.; Palacios, D.; Almasbak, H.; Bajor, M.; Clement, D.; Brandt, L.; et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer Immunol. Res. 2018, 6, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.; Goodlett, D.R.; Ishitani, A.; Marquardt, H.; Geraghty, D.E. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J. Immunol. 1998, 160, 4951–4960. [Google Scholar] [PubMed]
- Friese, M.A.; Platten, M.; Lutz, S.Z.; Naumann, U.; Aulwurm, S.; Bischof, F.; Buhring, H.J.; Dichgans, J.; Rammensee, H.G.; Steinle, A.; et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003, 63, 8996–9006. [Google Scholar] [PubMed]
- Dominguez-Valentin, M.; Gras, N.A.; Rahman, A.M.; Kumar, S.; Retiere, C.; Ulvestad, E.; Kristensen, V.; Lund-Johansen, M.; Lie, B.A.; Enger, P.O.; et al. Identification of a Natural Killer Cell Receptor Allele That Prolongs Survival of Cytomegalovirus-Positive Glioblastoma Patients. Cancer Res. 2016, 76, 5326–5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendruschk, S.; Wiedemuth, R.; Aigner, A.; Topfer, K.; Cartellieri, M.; Martin, D.; Kirsch, M.; Ikonomidou, C.; Schackert, G.; Temme, A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro Oncol. 2011, 13, 1074–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murad, S.; Michen, S.; Becker, A.; Füssel, M.; Schackert, G.; Tonn, T.; Momburg, F.; Temme, A. NKG2C+ NK Cells for Immunotherapy of Glioblastoma Multiforme. Int. J. Mol. Sci. 2022, 23, 5857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105857
Murad S, Michen S, Becker A, Füssel M, Schackert G, Tonn T, Momburg F, Temme A. NKG2C+ NK Cells for Immunotherapy of Glioblastoma Multiforme. International Journal of Molecular Sciences. 2022; 23(10):5857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105857
Chicago/Turabian StyleMurad, Shafiq, Susanne Michen, Alexander Becker, Monika Füssel, Gabriele Schackert, Torsten Tonn, Frank Momburg, and Achim Temme. 2022. "NKG2C+ NK Cells for Immunotherapy of Glioblastoma Multiforme" International Journal of Molecular Sciences 23, no. 10: 5857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105857