
Reference-Guided De Novo Genome Assembly of the Flour Beetle Tribolium freemani

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Genome Size Evaluation
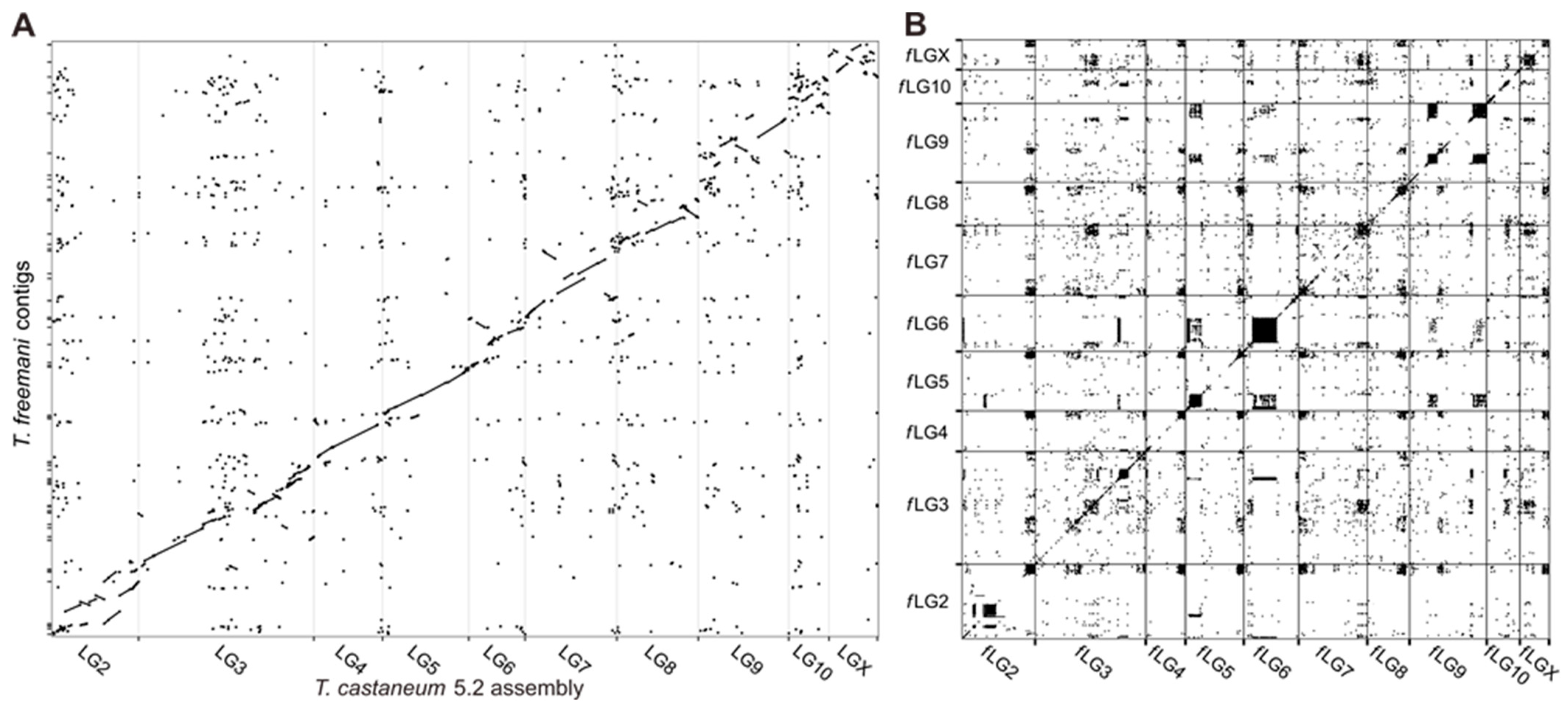
2.2. Genome Assembly
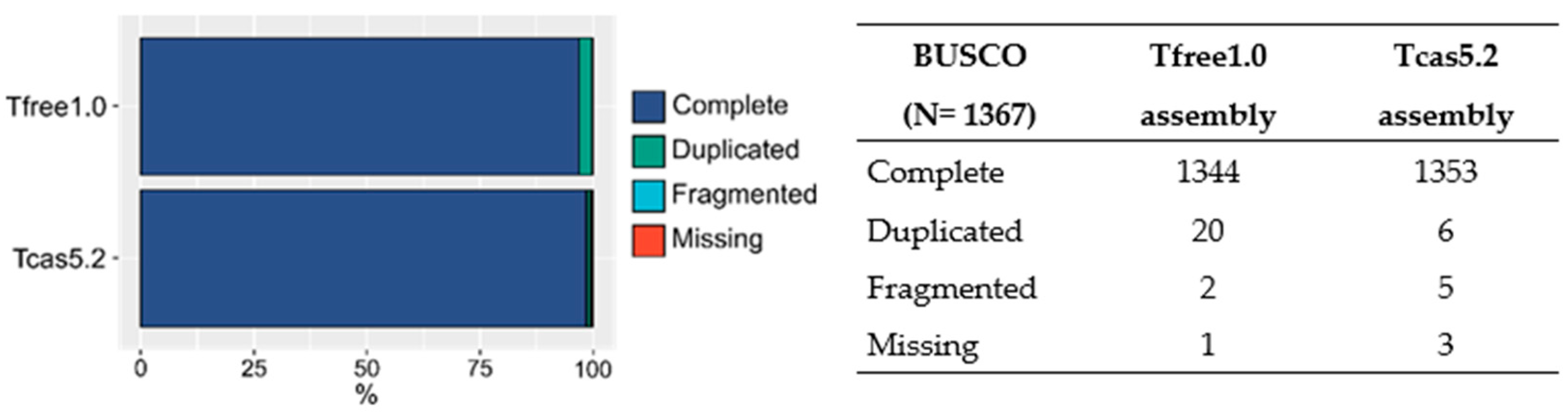
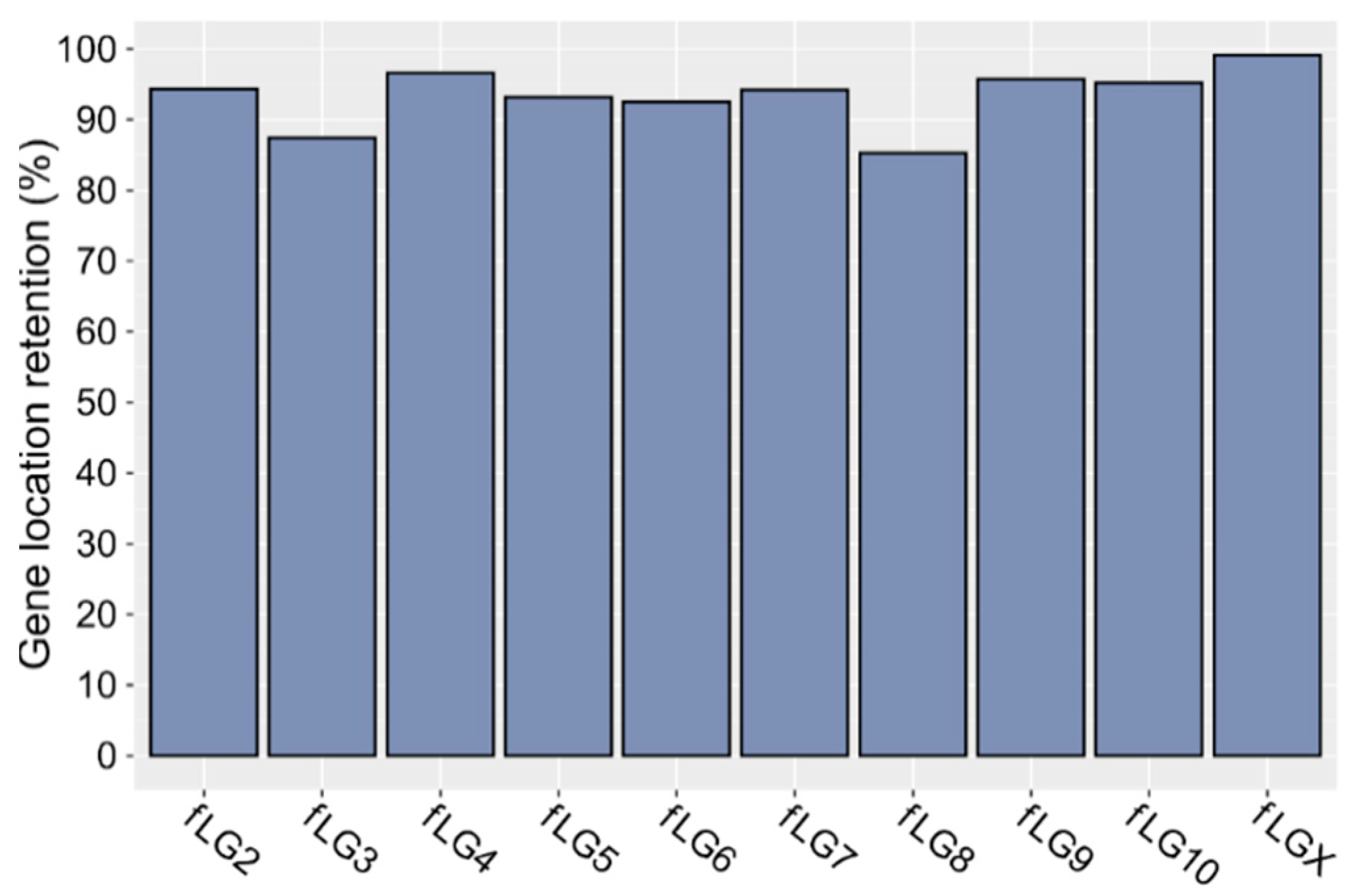
2.3. Gene Annotation
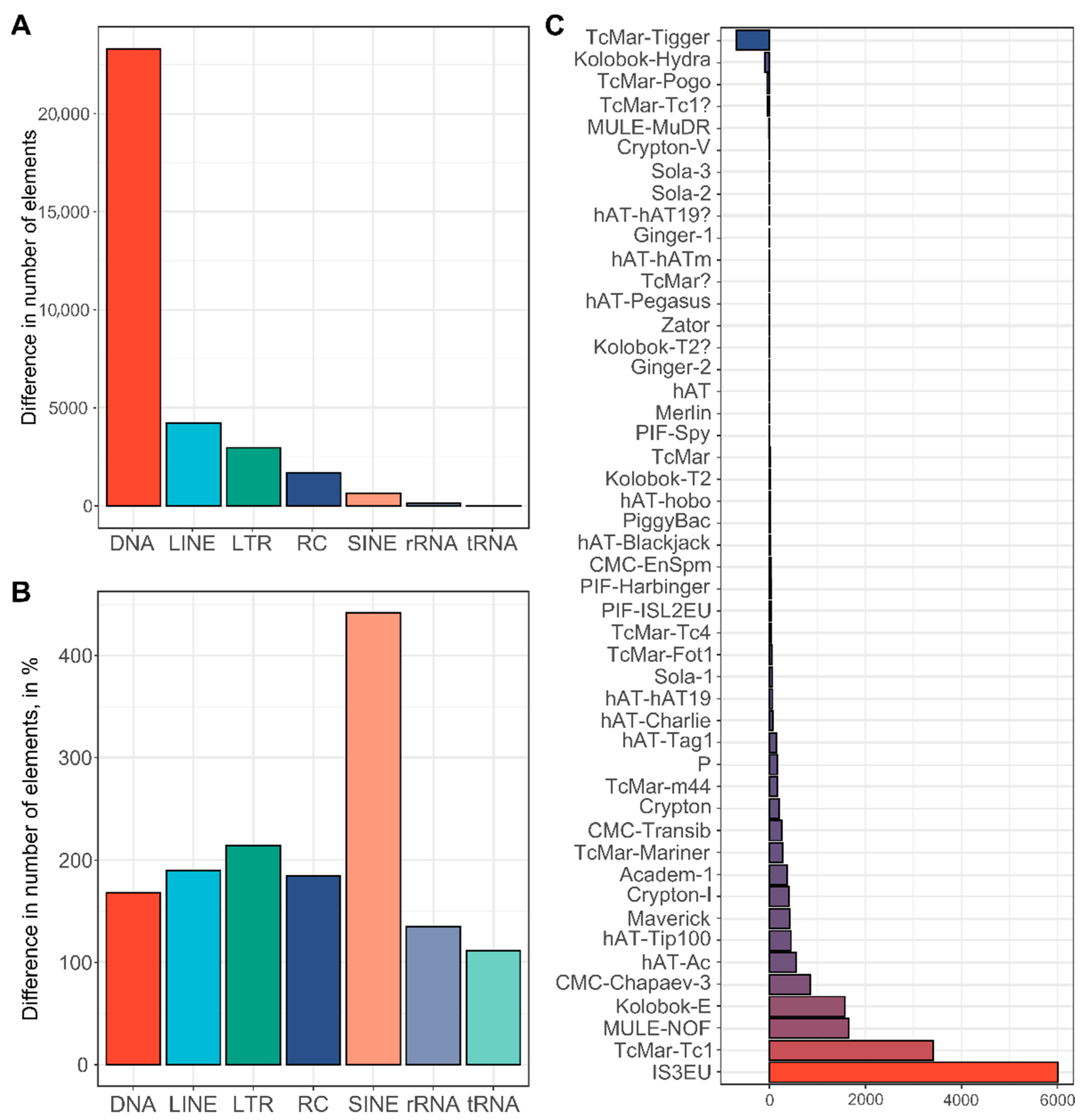
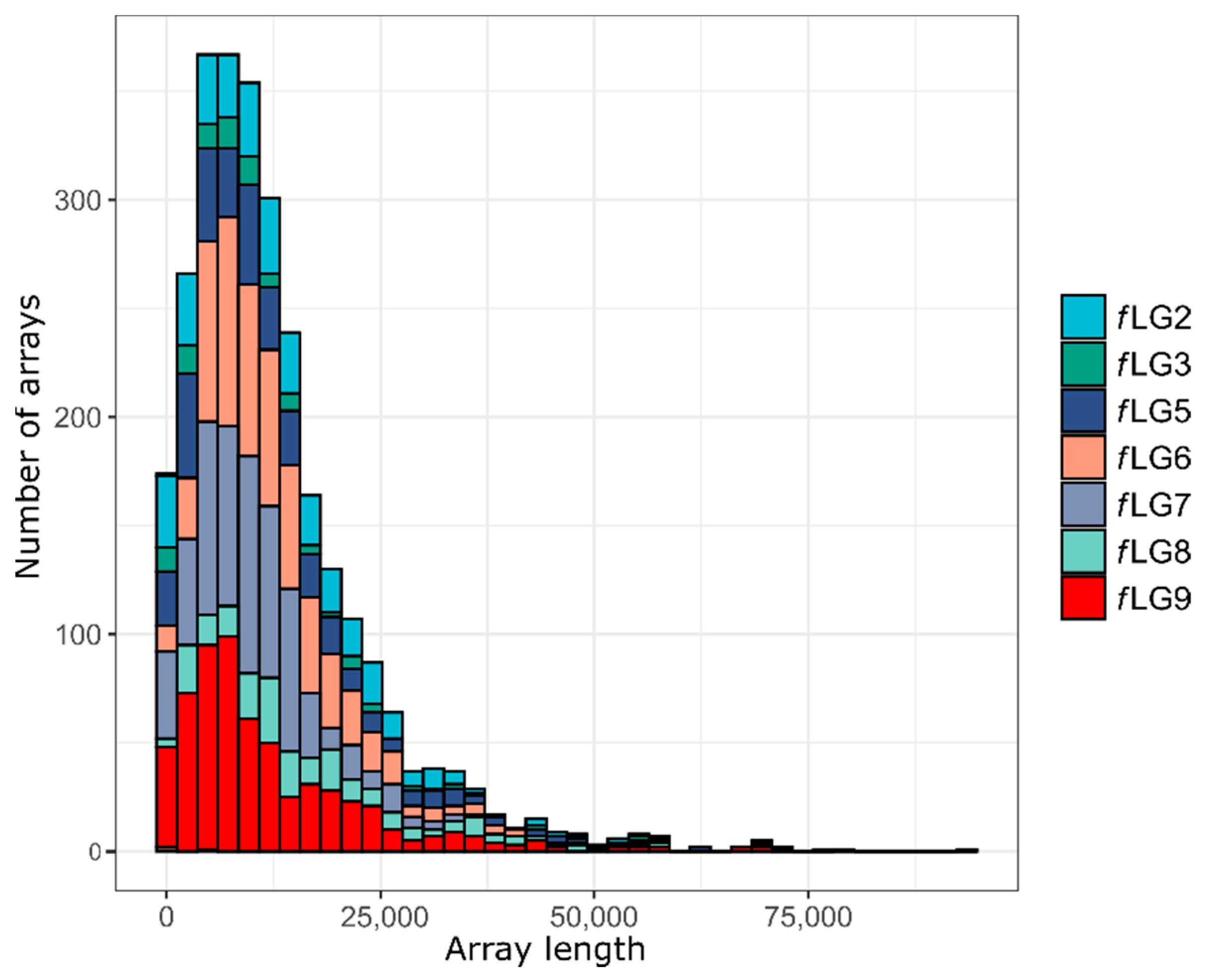
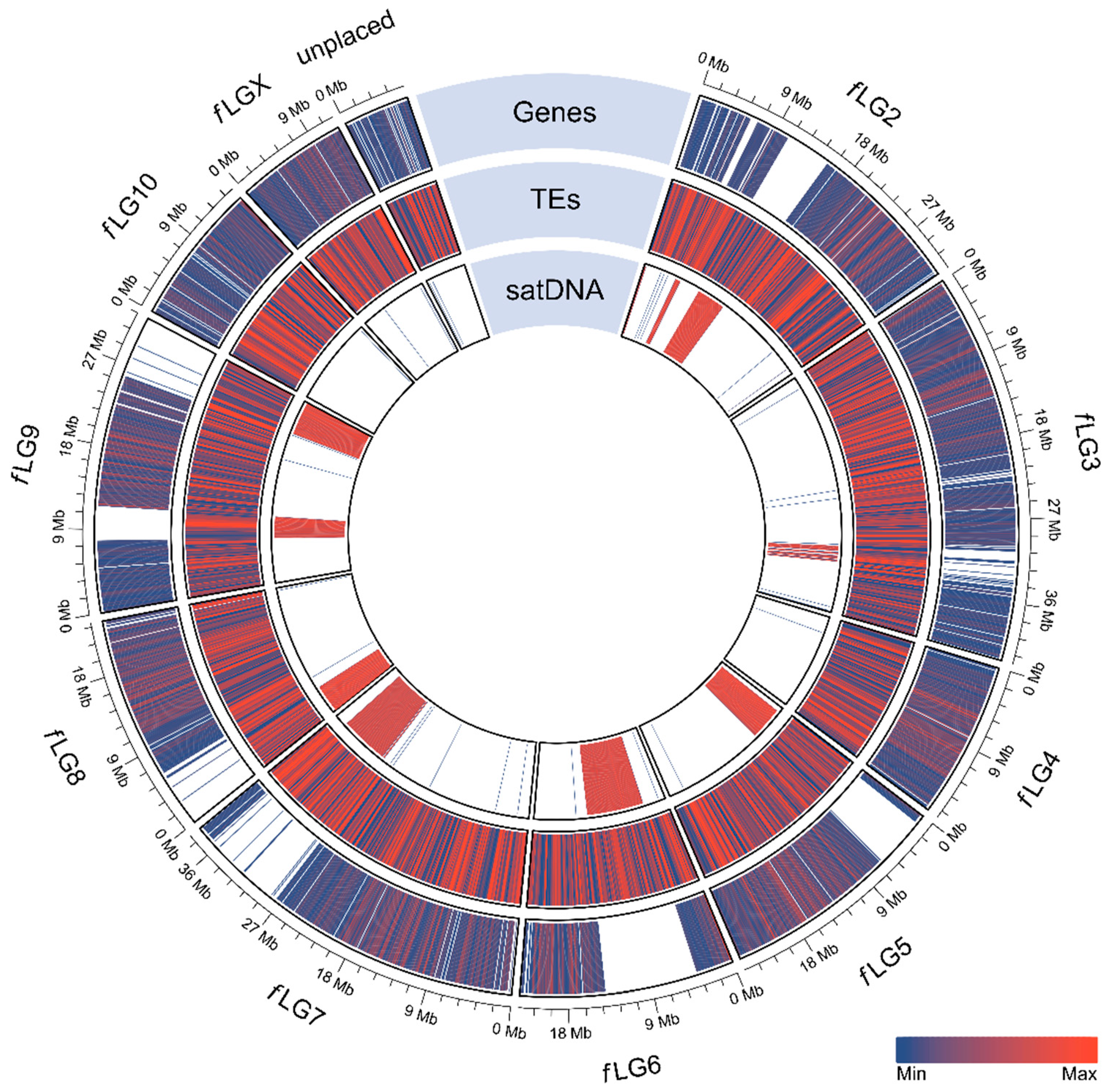
2.4. Repeat Annotation
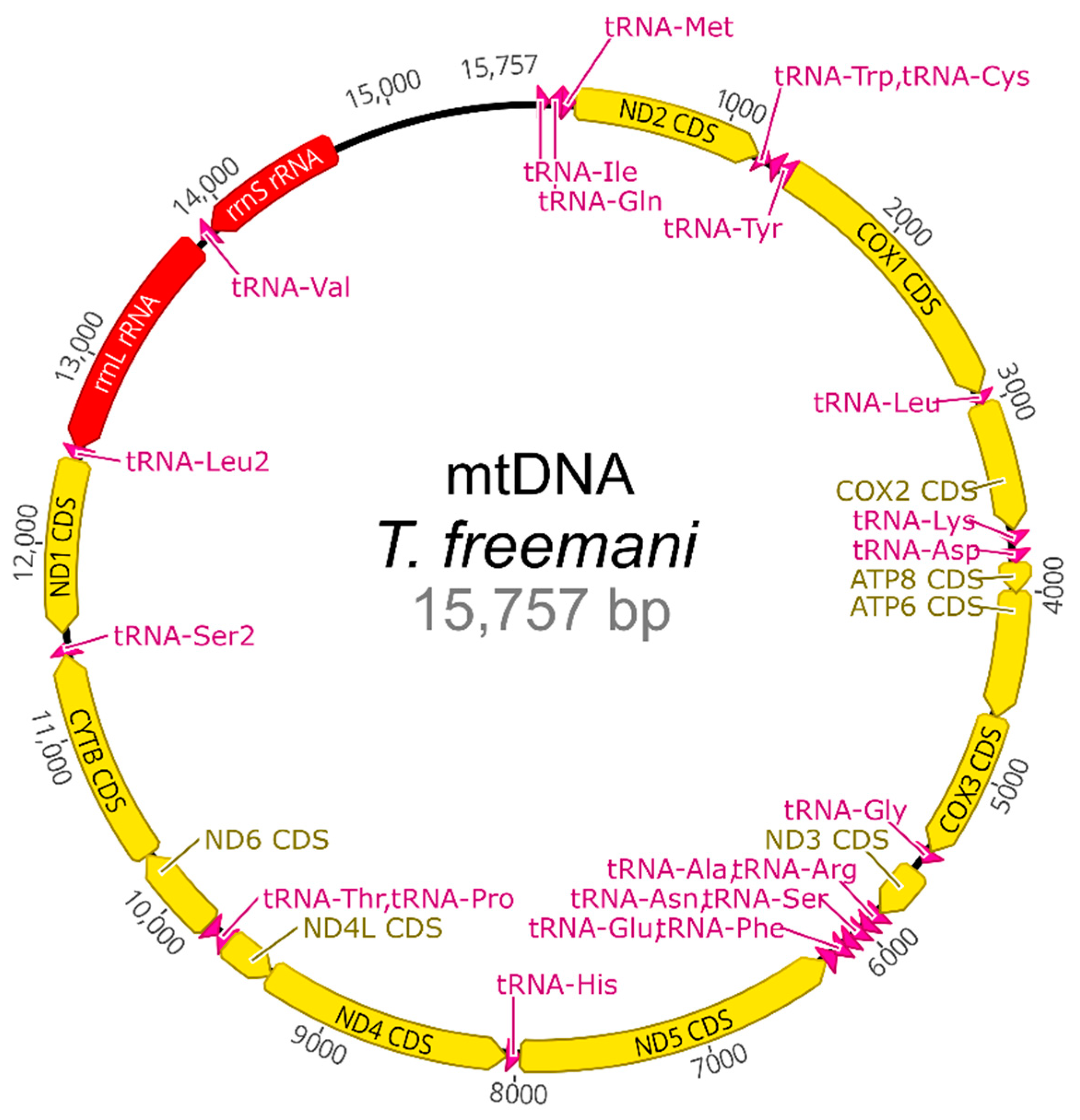
2.5. Mitochondrial DNA
3. Discussion
4. Materials and Methods
4.1. Insect Material
4.2. DNA Extraction and PacBio HiFi Sequencing
4.3. Genome Size Estimation
4.4. Reference Sequence
4.5. T. castaneum Assembly Gap Filling
4.6. GenomeAassembly
4.7. GeneAanalysis and Liftoff
4.8. Repeat Analyses
4.9. Mitochondrial DNA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Lawniczak, M.K.N.; Durbin, R.; Flicek, P.; Lindblad-toh, K.; Wei, X. Standards Recommendations for the Earth BioGenome Project. Proc. Natl. Acad. Sci. USA 2022, 119, e2115639118. [Google Scholar] [CrossRef]
- Childers, A.K.; Geib, S.M.; Sim, S.B.; Poelchau, M.F.; Coates, B.S.; Simmonds, T.J.; Scully, E.D.; Smith, T.P.L.; Childers, C.P.; Corpuz, R.L.; et al. The Usda-Ars Ag100pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research. Insects 2021, 12, 626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q. Animal Biodiversity: An Introduction to Higher-Level Classification and Taxonomic Richness. Zootaxa 2011, 12, 7–12. [Google Scholar] [CrossRef]
- Adamski, Z.; Bufo, S.A.; Chowański, S.; Falabella, P.; Lubawy, J.; Marciniak, P.; Pacholska-Bogalska, J.; Salvia, R.; Scrano, L.; Słocińska, M.; et al. Beetles as Model Organisms in Physiological, Biomedical and Environmental Studies—A Review. Front. Physiol. 2019, 10, 319. [Google Scholar] [CrossRef] [Green Version]
- Hotaling, S.; Sproul, J.S.; Heckenhauer, J.; Powell, A.; Larracuente, A.M.; Pauls, S.U.; Kelley, J.L.; Frandsen, P.B. Long Reads Are Revolutionizing 20 Years of Insect Genome Sequencing. Genome Biol. Evol. 2021, 13, evab138. [Google Scholar] [CrossRef]
- McKenna, D.D. Beetle Genomes in the 21st Century: Prospects, Progress and Priorities. Curr. Opin. Insect Sci. 2018, 25, 76–82. [Google Scholar] [CrossRef]
- Sokoloff, A. The Biology of Tribolium, with Special Emphasis on Genetic Aspects; Clarendon Press: Oxford, UK, 1972; Volume 1, ISBN 0198573537. [Google Scholar]
- Richards, S.; Gibbs, R.A.; Weinstock, G.M.; Brown, S.; Denell, R.; Beeman, R.W.; Gibbs, R.; Bucher, G.; Friedrich, M.; Grimmelikhuijzen, C.J.P.; et al. The Genome of the Model Beetle and Pest Tribolium Castaneum. Nature 2008, 452, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Herndon, N.; Shelton, J.; Gerischer, L.; Ioannidis, P.; Ninova, M.; Dönitz, J.; Waterhouse, R.M.; Liang, C.; Damm, C.; Siemanowski, J.; et al. Enhanced Genome Assembly and a New Official Gene Set for Tribolium Castaneum. BMC Genom. 2020, 21, 47. [Google Scholar] [CrossRef] [Green Version]
- Hinton, H.E. A Synopsis of the Genus Tribolium Macleay, with Some Remarks on the Evolution of Its Species-Groups (Coleoptera, Tenebrionidae). Bull. Entomol. Res. 1948, 39, 13–55. [Google Scholar] [CrossRef] [PubMed]
- Nakakita, H.; Imura, O.; Winks, R.G. Hybridization between Tribolium Freemani Hinton and Tribolium Castaneum (Herbst), and Some Preliminary Studies on the Biology of Tribolium Freemani (Coleoptera: Tenebrionidae). Appl. Entomol. Zool. 1981, 16, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, A.; Sokoloff, A. Transmission of Tribolium castaneum (Herbst) Mutants to T. castaneum- T. freemani Hinton Hybrids (Coleoptera: Tenebrionidae). J. Stored Prod. Res. 1988, 24, 145–150. [Google Scholar] [CrossRef]
- Wade, M.J.; Johnson, N.A. Reproductive Isolation between Two Species of Flour Beetles, Tribolium castaneum and T. freemani: Variation within and among Geographical Populations of T. Castaneum. Heredity 1994, 72, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Fuster, A.; Juan, C.; Petitpierre, E. Genome Size in Tribolium Flour-Beetles: Inter-and Intraspecific Variation. Genet. Res. 1991, 58, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Shimeld, L. A Cytogenetic Examination of Eight Species of Tribolium. Tribolium Inf. Bull. 1989, 29, 102–107. [Google Scholar]
- Ugarković, D.; Podnar, M.; Plohl, M. Satellite DNA of the Red Flour Beetle Tribolium Castaneum—Comparative Study of Satellites from the Genus Tribolium. Mol. Biol. Evol. 1996, 13, 1059–1066. [Google Scholar] [CrossRef] [Green Version]
- Juan, C.; Vazquez, P.; Rubio, J.M.; Petitpierre, E.; Hewitt, G.M. Presence of Highly Repetitive DNA Sequences in Tribolium Flour-Beetles. Heredity 1993, 70, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast Reference-Free Genome Profiling from Short Reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; DIng, J.; Piednoël, M.; Schneeberger, K. FindGSE: Estimating Genome Size Variation within Human and Arabidopsis Using k -Mer Frequencies. Bioinformatics 2018, 34, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Hozza, M.; Vinař, T.; Brejová, B. How Big Is That Genome? Estimating Genome Size and Coverage from k-Mer Abundance Spectra. In String Processing and Information Retrieval; Springer: Cham, Switzerland, 2015; Volume 9309, pp. 199–209. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Aganezov, S.; Wang, X.; Lippman, Z.B.; Schatz, M.C.; Soyk, S. Automated Assembly Scaffolding Elevates a New Tomato System for High-Throughput Genome Editing. bioRxiv 2021. 2021.11.18.469135. [Google Scholar] [CrossRef]
- Shumate, A.; Salzberg, S.L. Liftoff: Accurate Mapping of Gene Annotations. Bioinformatics 2021, 37, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2015. Available online: https://www.repeatmasker.org (accessed on 21 January 2022).
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaxter, M.; Mieszkowska, N.; Di Palma, F.; Holland, P.; Durbin, R.; Richards, T.; Berriman, M.; Kersey, P.; Hollingsworth, P.; Wilson, W.; et al. Sequence Locally, Think Globally: The Darwin Tree of Life Project. Proc. Natl. Acad. Sci. USA 2022, 119, e2115642118. [Google Scholar] [CrossRef]
- Miga, K.H. Centromere Studies in the Era of ‘Telomere-to-Telomere’ Genomics. Exp. Cell Res. 2020, 394, 112127. [Google Scholar] [CrossRef]
- Lischer, H.E.L.; Shimizu, K.K. Reference-Guided de Novo Assembly Approach Improves Genome Reconstruction for Related Species. BMC Bioinform. 2017, 18, 474. [Google Scholar] [CrossRef] [Green Version]
- Seixas, F.A.; Edelman, N.B.; Mallet, J. Synteny-Based Genome Assembly for 16 Species of Heliconius Butterflies, and an Assessment of Structural Variation across the Genus. Genome Biol. Evol. 2021, 13, evab069. [Google Scholar] [CrossRef]
- Wang, S.; Lorenzen, M.D.; Beeman, R.W.; Brown, S.J. Analysis of Repetitive DNA Distribution Patterns in the Tribolium Castaneum Genome. Genome Biol. 2008, 9, R61. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, N.; Tsai, I.J.; Tanaka, R.; Hunt, V.L.; Liu, D.; Tsuyama, K.; Maeda, Y.; Namai, S.; Kumagai, R.; Tracey, A.; et al. Biology and Genome of a Newly Discovered Sibling Species of Caenorhabditis Elegans. Nat. Commun. 2018, 9, 3216. [Google Scholar] [CrossRef]
- Plohl, M.; Lucijanic-Justic, V.; Ugarkovic, D.; Petitpierre, E.; Juan, C. Satellite DNA and Heterochromatin of the Flour Beetle Tribolium Confusum. Genome 1993, 36, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Ugarković, D.; Durajlija, S.; Plohl, M. Evolution of Tribolium Madens (Insecta, Coleoptera) Satellite DNA through DNA Inversion and Insertion. J. Mol. Evol. 1996, 42, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Mravinac, B.; Plohl, M.; Ugarković, D. Conserved Patterns in the Evolution of Tribolium Satellite DNAs. Gene 2004, 332, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Mravinac, B.; Ugarković, D.; Franjević, D.; Plohl, M. Long Inversely Oriented Subunits Form a Complex Monomer of Tribolium Brevicornis Satellite DNA. J. Mol. Evol. 2005, 60, 513–525. [Google Scholar] [CrossRef]
- Mravinac, B.; Plohl, M. Parallelism in Evolution of Highly Repetitive DNAs in Sibling Species. Mol. Biol. Evol. 2010, 27, 1857–1867. [Google Scholar] [CrossRef] [Green Version]
- Pavlek, M.; Gelfand, Y.; Plohl, M.; Meštrović, N. Genome-Wide Analysis of Tandem Repeats in Tribolium Castaneum Genome Reveals Abundant and Highly Dynamic Tandem Repeat Families with Satellite DNA Features in Euchromatic Chromosomal Arms. DNA Res. 2015, 22, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, B.; Firneno, T.J.; Demuth, J.P. Divergence Time Estimation of Genus Tribolium by Extensive Sampling of Highly Conserved Orthologs. Mol. Phylogenet. Evol. 2021, 159, 107084. [Google Scholar] [CrossRef]
- Ferree, P.M.; Barbash, D.A. Species-Specific Heterochromatin Prevents Mitotic Chromosome Segregation to Cause Hybrid Lethality in Drosophila. PLoS Biol. 2009, 7, e1000234. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Kinney, N.A.; Timoshevskiy, V.A.; Sharakhova, M.V.; Sharakhov, I.V. Structural Variation of the x Chromosome Heterochromatin in the Anopheles Gambiae Complex. Genes 2020, 11, 327. [Google Scholar] [CrossRef] [Green Version]
- Turner, L.M.; White, M.A.; Tautz, D.; Payseur, B.A. Genomic Networks of Hybrid Sterility. PLoS Genet. 2014, 10, e1004162. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Ramírez, S.; Weiss, J.G.; Thomas, C.G.; Cutter, A.D. Widespread Misregulation of Inter-Species Hybrid Transcriptomes Due to Sex-Specific and Sex-Chromosome Regulatory Evolution. PLoS Genet. 2021, 17, e1009409. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.R.; Yan, Z.T.; Si, F.L.; Li, X.D.; Mao, Q.M.; Asghar, S.; Chen, B. Mitochondrial Genes Associated with Pyrethroid Resistance Revealed by Mitochondrial Genome and Transcriptome Analyses in the Malaria Vector Anopheles Sinensis (Diptera: Culicidae). Pest Manag. Sci. 2020, 76, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Steele, L.D.; Coates, B.S.; Seong, K.M.; Valero, M.C.; Mittapalli, O.; Sun, W.; Clark, J.; Pittendrigh, B.R. Variation in Mitochondria-Derived Transcript Levels Associated with DDT Resistance in the 91-R Strain of Drosophila Melanogaster (Diptera: Drosophilidae). J. Insect Sci. 2018, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Oppert, B.; Guedes, R.N.C.; Aikins, M.J.; Perkin, L.; Chen, Z.; Phillips, T.W.; Zhu, K.Y.; Opit, G.P.; Hoon, K.; Sun, Y.; et al. Genes Related to Mitochondrial Functions Are Differentially Expressed in Phosphine-Resistant and -Susceptible Tribolium Castaneum. BMC Genom. 2015, 16, 968. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Koren, S.; Arang, R.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Hon, T.; Mars, K.; Young, G.; Tsai, Y.C.; Karalius, J.W.; Landolin, J.M.; Maurer, N.; Kudrna, D.; Hardigan, M.A.; Steiner, C.C.; et al. Highly Accurate Long-Read HiFi Sequencing Data for Five Complex Genomes. Sci. Data 2020, 7, 399. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L.; et al. TGS-GapCloser: A Fast and Accurate Gap Closer for Large Genomes with Low Coverage of Error-Prone Long Reads. Gigascience 2020, 9, giaa094. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoit, M.; Drost, H.G. A Predictive Approach to Infer the Activity and Natural Variation of Retrotransposon Families in Plants. In Plant Transposable Elements; Springer: Berlin/Heidelberg, Germany, 2021; Volume 2250, pp. 1–14. [Google Scholar] [CrossRef]
- Hammel, M.; Simon, A.; Arbiol, C.; Villalba, A.; Burioli, E.A.V.; Pépin, J.F.; Lamy, J.B.; Benabdelmouna, A.; Bernard, I.; Houssin, M.; et al. Prevalence and Polymorphism of a Mussel Transmissible Cancer in Europe. Mol. Ecol. 2022, 31, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Allio, R.; Schomaker-Bastos, A.; Romiguier, J.; Prosdocimi, F.; Nabholz, B.; Delsuc, F. MitoFinder: Efficient Automated Large-Scale Extraction of Mitogenomic Data in Target Enrichment Phylogenomics. Mol. Ecol. Resour. 2020, 20, 892–905. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.; Muqim, N. Sequence and Phylogenetic Analysis of the Complete Mitochondrial Genome of the Flour Beetle Tribolium Castanaeum. Mol. Phylogenet. Evol. 2003, 26, 502–512. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithm | Genome Size Prediction (Mb) | Estimated Repeat Ratio (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| k-mer Size | Average | k-mer Size | Average | |||||
| 21 | 31 | 41 | 21 | 31 | 41 | |||
| GenomeScope | 189.6 | 202.2 | 212.8 | 201.5 | 30.4 | 28.4 | 28 | 28.9 |
| findGSE | 225.6 | 240.4 | 255.7 | 240.6 | 33.9 | 32.4 | 32.4 | 32.9 |
| CovEST RE | 238.7 | 305.5 | 412.3 | 318.8 | ||||
| Raw PacBio Data | Hifiasm Output | Tfree1.0 | |||
|---|---|---|---|---|---|
| Number of reads | 1,617,087 | Number of contigs | 679 | Number of contigs | 110 |
| Total length (bp) | 23,796,436,578 | Total length (bp) | 465,826,150 | Total length (bp) | 269,018,543 |
| Number of reads <5000 bp | 301 | Min contig length (bp) | 13,227 | Min contig length (bp) | 22,590 |
| Largest read (bp) | 42,982 | Max contig length (bp) | 23,376,958 | Max contig length (bp) | 23,376,958 |
| GC (%) | 32 | GC (%) | 31.52 | GC (%) | 32.53 |
| N50 | 14,965 | N50 | 5,522,289 | N50 | 8,487,211 |
| N90 | 11,545 | N90 | 345,137 | N90 | 1,794,767 |
| L50 | 675,343 | L50 | 23 | L50 | 10 |
| L90 | 1,394,918 | L90 | 160 | L90 | 30 |
| Genomic Feature | Tcas5.2 | Tfree1.0 | Retained Data (%) |
|---|---|---|---|
| Gene | 14,467 | 13,845 | 95.70 |
| mRNA | 22,598 | 21,936 | 97.07 |
| Exon | 171,320 | 149,045 | 87.00 |
| CDS | 22,611 | 21,827 | 96.53 |
| Transcript | 317 | 289 | 91.17 |
| lncRNA | 1364 | 1165 | 85.41 |
| Primary transcript | 220 | 144 | 65.45 |
| tRNA | 247 | 237 | 95.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volarić, M.; Despot-Slade, E.; Veseljak, D.; Meštrović, N.; Mravinac, B. Reference-Guided De Novo Genome Assembly of the Flour Beetle Tribolium freemani. Int. J. Mol. Sci. 2022, 23, 5869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115869
Volarić M, Despot-Slade E, Veseljak D, Meštrović N, Mravinac B. Reference-Guided De Novo Genome Assembly of the Flour Beetle Tribolium freemani. International Journal of Molecular Sciences. 2022; 23(11):5869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115869
Chicago/Turabian StyleVolarić, Marin, Evelin Despot-Slade, Damira Veseljak, Nevenka Meštrović, and Brankica Mravinac. 2022. "Reference-Guided De Novo Genome Assembly of the Flour Beetle Tribolium freemani" International Journal of Molecular Sciences 23, no. 11: 5869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115869