
Sphk1 and Sphk2 Differentially Regulate Erythropoietin Synthesis in Mouse Renal Interstitial Fibroblast-like Cells

 ,
, 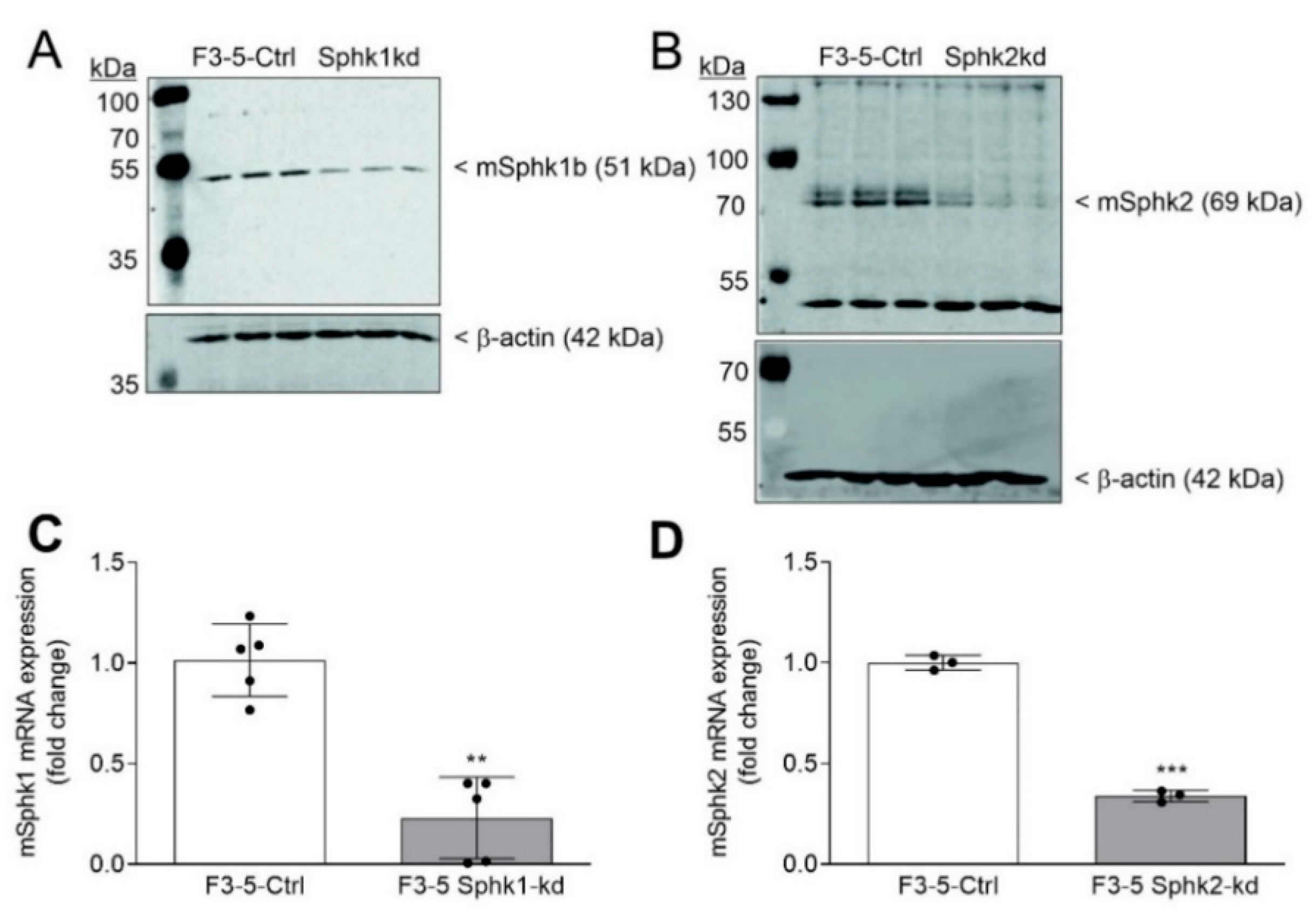{kind=link}
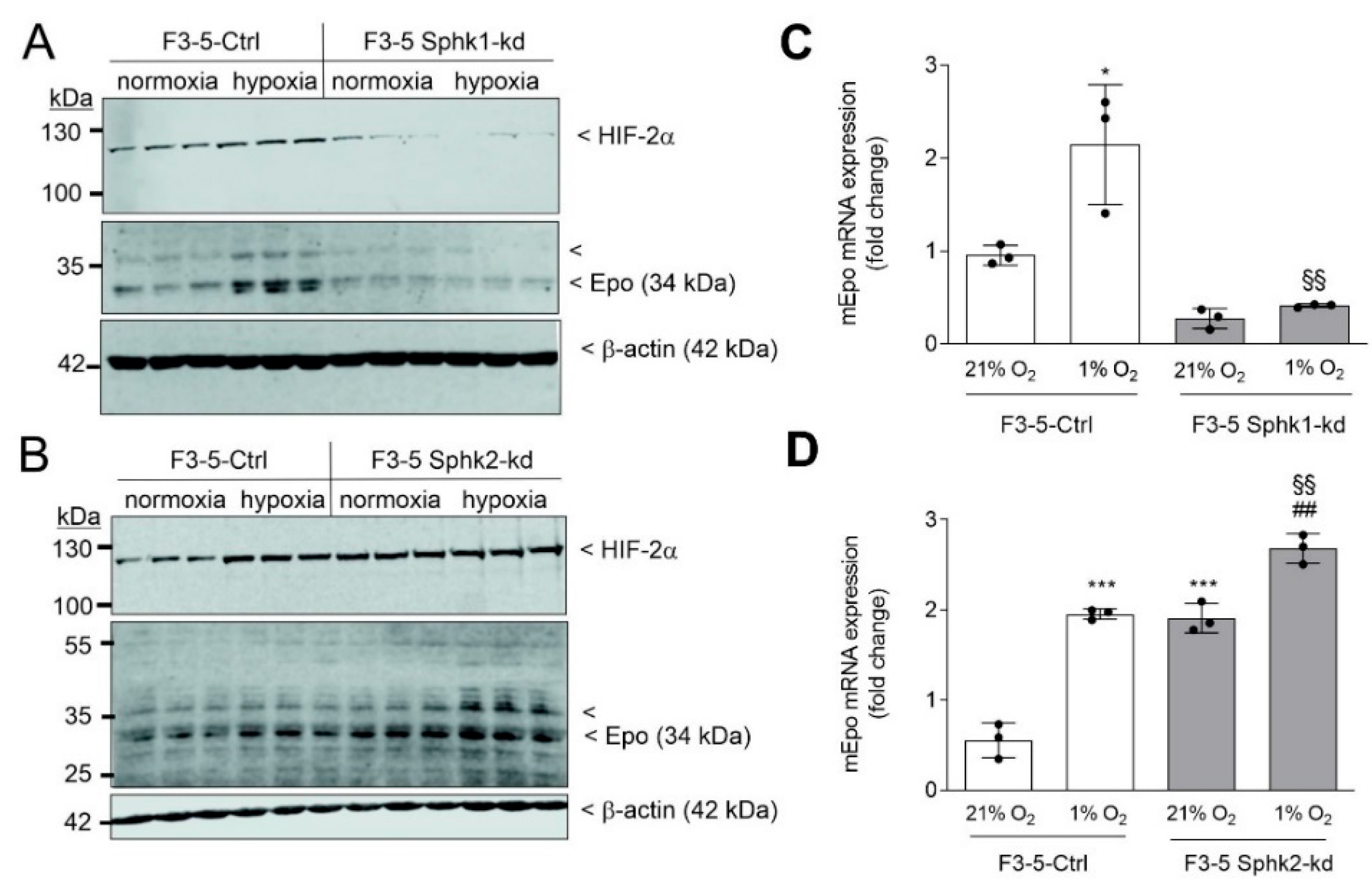{kind=link}
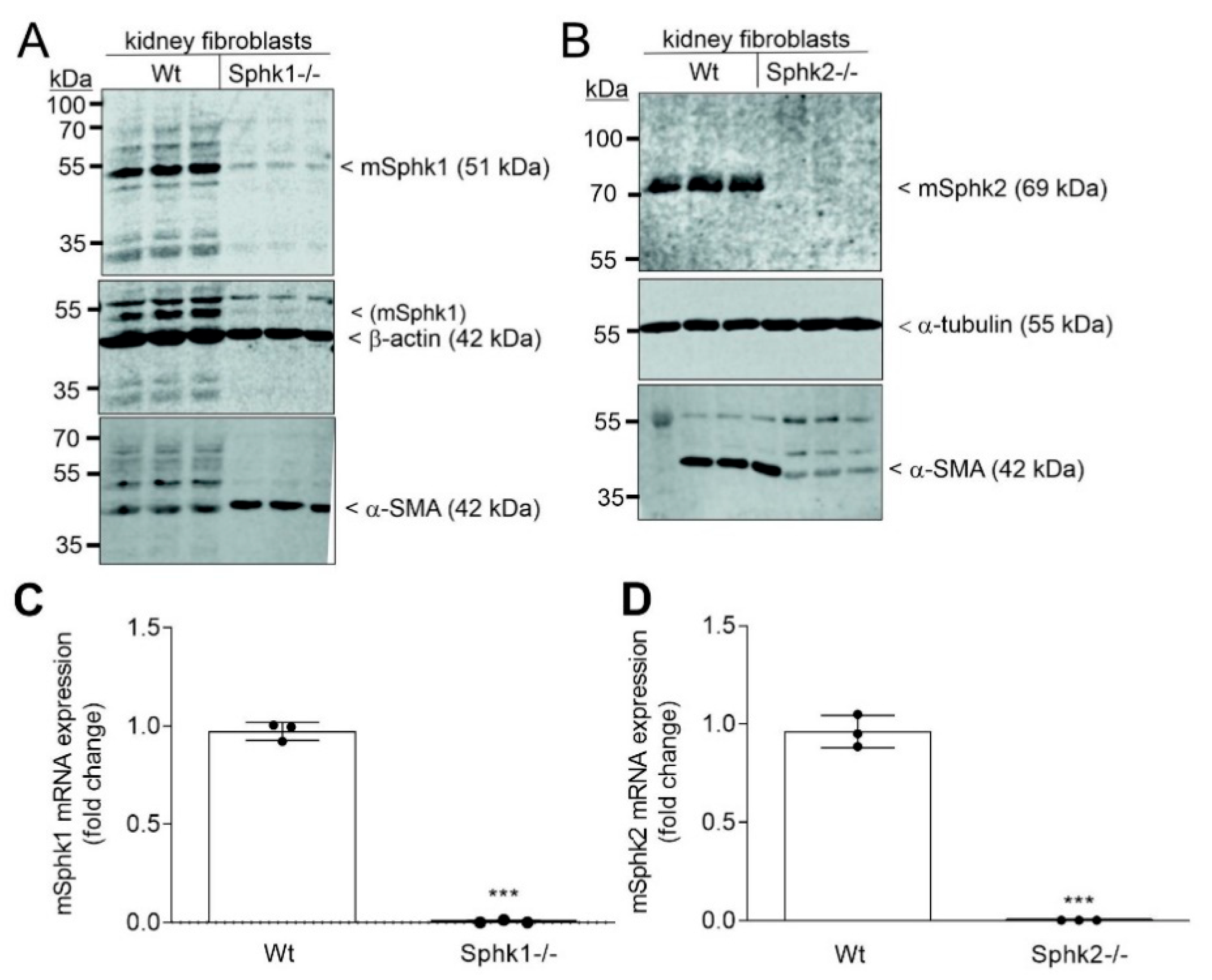{kind=link}
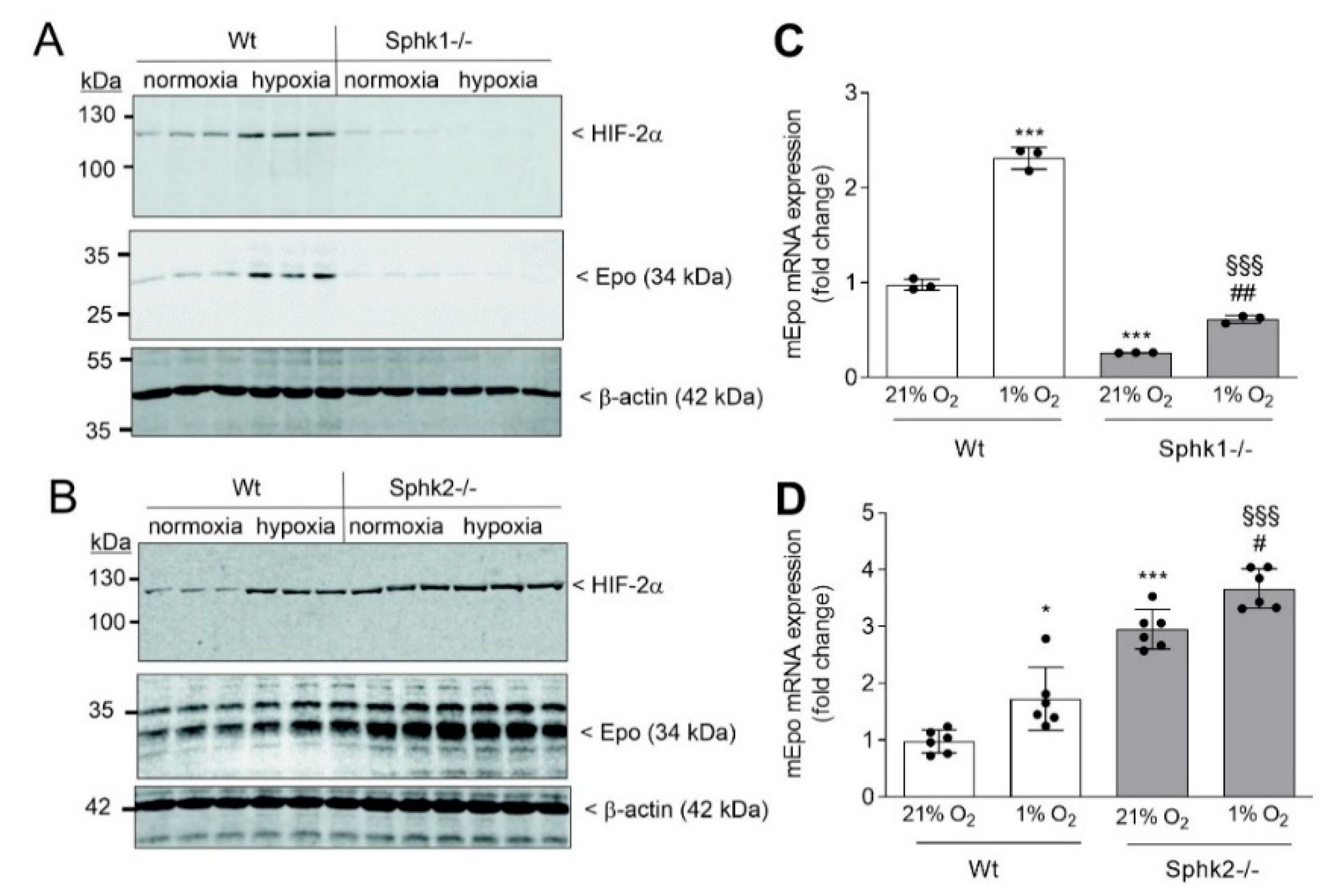{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
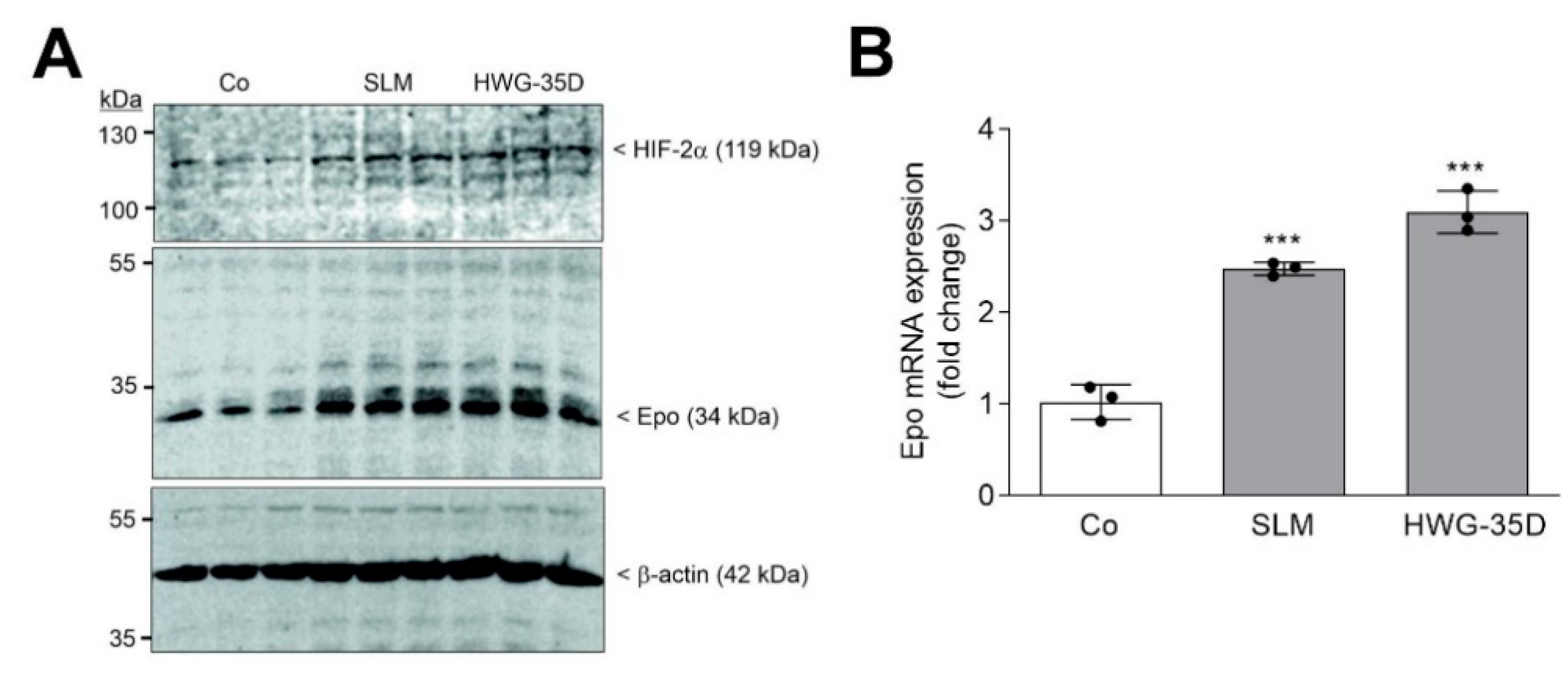
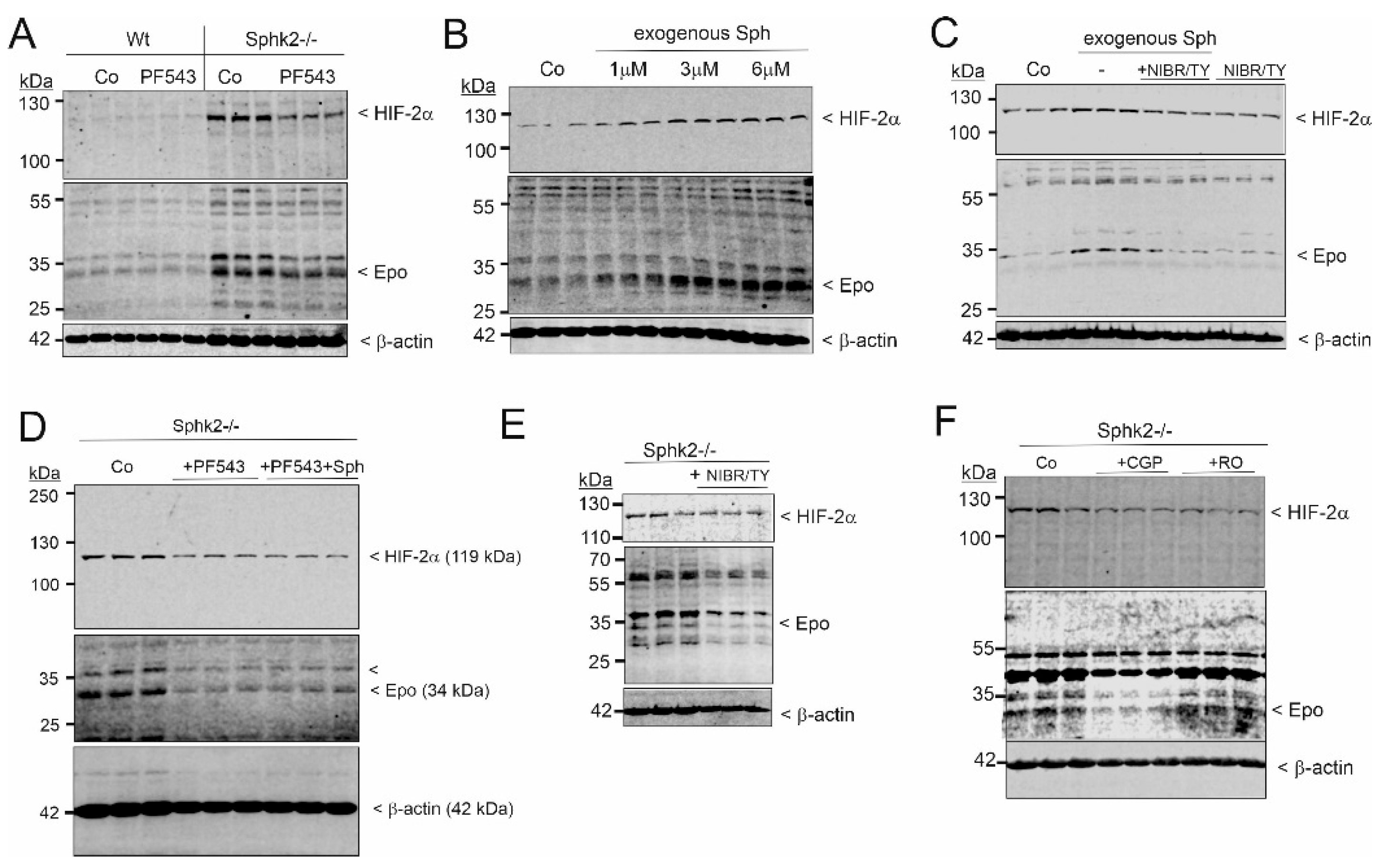
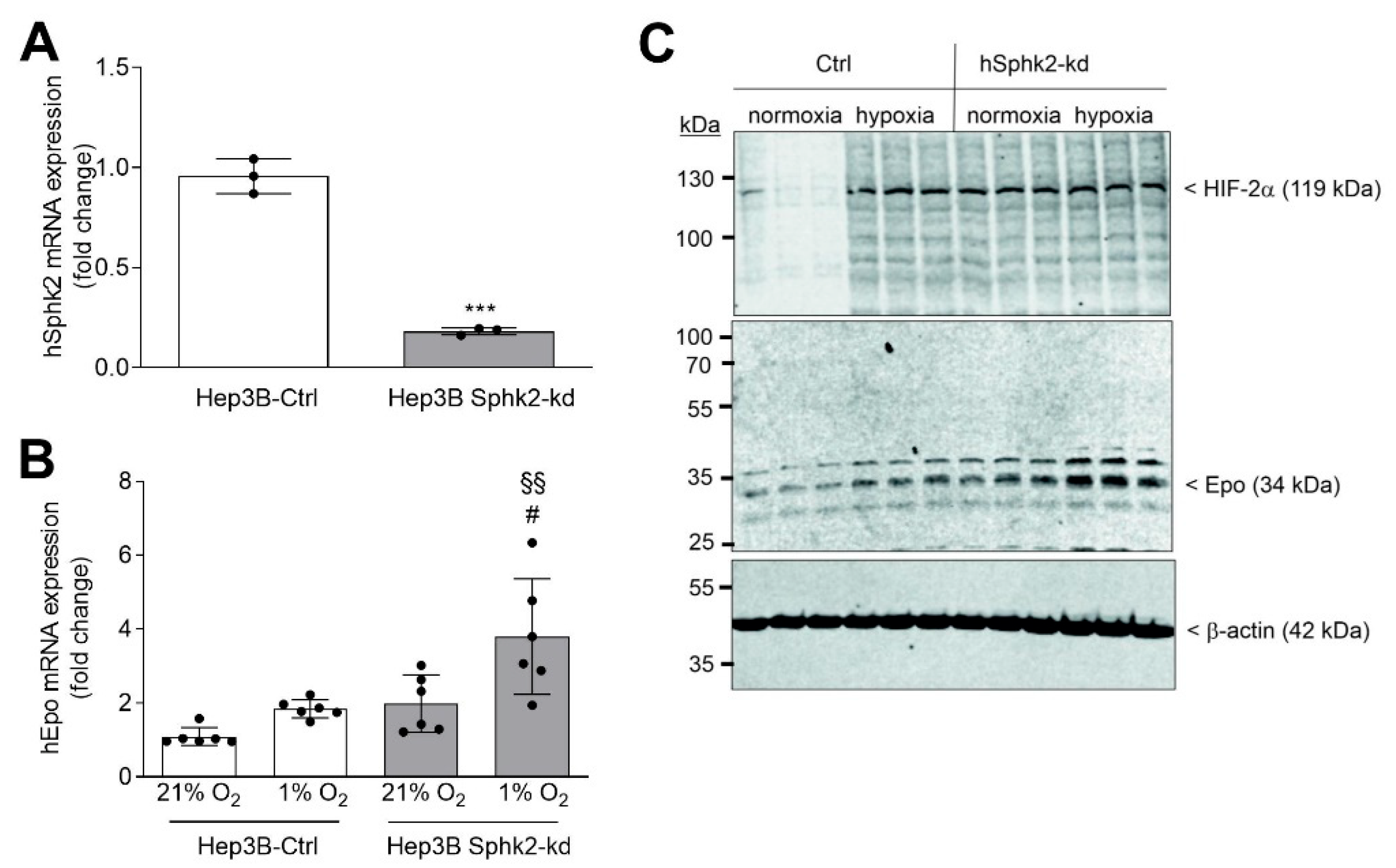
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Cell Culture Conditions
4.3. Stable mRNA Knockdown in F3-5 and Hep3B Cells
4.4. Cell Stimulation, Homogenization, and Immunoblotting
4.5. RNA Extraction and Quantitative PCR Analysis
4.6. Sphingolipid Quantification by Mass Spectrometry
4.7. Red Blood Cell Counting
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| CKD | chronic kidney disease |
| CTGF | connective tissue growth factor |
| DMEM | Dulbecco’s modified Eagle medium |
| Epo | erythropoietin |
| FBS | fetal bovine serum |
| GPCR | G protein-coupled receptor |
| HIF | hypoxia-inducible factor |
| kd | knockdown |
| MAPK | mitogen-activated protein kinase |
| PBS | phosphate-buffered saline |
| PKC | protein kinase C |
| qPCR | quantitative polymerase chain reaction |
| REPC | renal Epo-producing cells |
| S1P | sphingosine 1-phosphate |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| shRNA | small hairpin RNA |
| Sph | sphingosine |
| SphK | sphingosine kinase |
| UUO | unilateral ureteral obstruction |
References
- Proia, L.R.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huwiler, A.; Pfeilschifter, J. New players on the center stage: Sphingosine 1-phosphate and its receptors as drug targets. Biochem. Pharm. 2008, 75, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Pfeilschifter, J. Sphingolipid signaling in renal fibrosis. Matrix Biol. 2018, 68, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell. Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyne, S.; Lee, S.C.; Long, J.; Pyne, N.J. Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell. Signal. 2009, 21, 14–21. [Google Scholar] [CrossRef]
- Saba, J.D. Fifty years of lyase and a moment of truth: Sphingosine phosphate lyase from discovery to disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef] [Green Version]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharm. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharm. 2014, 171, 3575–3594. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-phosphate in renal diseases. Cell. Physiol. Biochem. 2013, 31, 745–760. [Google Scholar] [CrossRef]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine-1-phosphate: A Janus-faced mediator of fibrotic diseases. Biochim. Biophys. Acta 2013, 1831, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Beyer, S.; Frey, H.; Haceni, R.; Grammatikos, G.; Thomas, D.; Geisslinger, G.; Schaefer, L.; Huwiler, A.; Pfeilschifter, J. Sphingosine Kinase-2 Deficiency Ameliorates Kidney Fibrosis by Up-Regulating Smad7 in a Mouse Model of Unilateral Ureteral Obstruction. Am. J. Pathol. 2017, 187, 2413–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, A.; Huang, L.; Kurmaeva, E.; Ye, H.; Dondeti, K.R.; Chroscicki, P.; Foley, L.S.; Balogun, Z.A.; Alexander, K.J.; Park, H.; et al. Sphingosine Kinase 2 Deficiency Attenuates Kidney Fibrosis via IFN-gamma. J. Am. Soc. Nephrol. 2017, 28, 1145–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Thangada, S.; Dasgupta, O.; Khanna, K.M.; Yamase, H.T.; Kashgarian, M.; Hla, T.; Shapiro, L.H.; Ferrer, F.A. Cell-intrinsic sphingosine kinase 2 promotes macrophage polarization and renal inflammation in response to unilateral ureteral obstruction. PLoS ONE 2018, 13, e0194053. [Google Scholar] [CrossRef]
- Imeri, F.; Stepanovska Tanturovska, B.; Schwalm, S.; Saha, S.; Zeng-Brouwers, J.; Pavenstadt, H.; Pfeilschifter, J.; Schaefer, L.; Huwiler, A. Loss of sphingosine kinase 2 enhances Wilm’s tumor suppressor gene 1 and nephrin expression in podocytes and protects from streptozotocin-induced podocytopathy and albuminuria in mice. Matrix Biol. 2021, 98, 32–48. [Google Scholar] [CrossRef]
- Du, C.; Ren, Y.; Yao, F.; Duan, J.; Zhao, H.; Du, Y.; Xiao, X.; Duan, H.; Shi, Y. Sphingosine kinase 1 protects renal tubular epithelial cells from renal fibrosis via induction of autophagy. Int. J. Biochem. Cell. Biol. 2017, 90, 17–28. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Ji, X.Y.; Ritter, J.K.; Li, N. Knockout of Sphingosine Kinase 1 Attenuates Renal Fibrosis in Unilateral Ureteral Obstruction Model. Am. J. Nephrol. 2019, 50, 196–203. [Google Scholar] [CrossRef]
- Allende, M.L.; Sasaki, T.; Kawai, H.; Olivera, A.; Mi, Y.; van Echten-Deckert, G.; Hajdu, R.; Rosenbach, M.; Keohane, C.A.; Mandala, S.; et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 2004, 279, 52487–52492. [Google Scholar] [CrossRef] [Green Version]
- Zemann, B.; Urtz, N.; Reuschel, R.; Mechtcheriakova, D.; Bornancin, F.; Badegruber, R.; Baumruker, T.; Billich, A. Normal neutrophil functions in sphingosine kinase type 1 and 2 knockout mice. Immunol. Lett. 2007, 109, 56–63. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.Y.; Smith, T.D.; Meli, V.S.; Tran, T.N.; Botvinick, E.L.; Liu, W.F. Differential regulation of macrophage inflammatory activation by fibrin and fibrinogen. Acta Biomater. 2017, 47, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalm, S.; Timcheva, T.M.; Filipenko, I.; Ebadi, M.; Hofmann, L.P.; Zangemeister-Wittke, U.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase 2 deficiency increases proliferation and migration of renal mouse mesangial cells and fibroblasts. Biol. Chem. 2015, 396, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuma, S.; Ruike, Y.; Yano, T.; Kimura, M.; Hirasawa, A.; Tsujimoto, G. Transcriptional regulation of connective tissue growth factor by sphingosine 1-phosphate in rat cultured mesangial cells. FEBS Lett. 2005, 579, 2576–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, C.; Ren, S.; Kleuser, B.; Shabahang, S.; Eberhardt, W.; Radeke, H.; Schafer-Korting, M.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J. Biol. Chem. 2004, 279, 35255–35262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, C.D.; Rivera Gil, P.; Tolle, M.; van der Giet, M.; Chun, J.; Radeke, H.H.; Schafer-Korting, M.; Kleuser, B. Immunomodulator FTY720 induces myofibroblast differentiation via the lysophospholipid receptor S1P3 and Smad3 signaling. Am. J. Pathol. 2007, 170, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Gellings Lowe, N.; Swaney, J.S.; Moreno, K.M.; Sabbadini, R.A. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc. Res. 2009, 82, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Priyadarsini, S.; Nicholas, S.E.; Sarker-Nag, A.; Allegood, J.; Chalfant, C.E.; Mandal, N.A.; Karamichos, D. Analysis of sphingolipids in human corneal fibroblasts from normal and keratoconus patients. J. Lipid Res. 2017, 58, 636–648. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Babelova, A.; Moreth, K.; Xin, C.; Eberhardt, W.; Doller, A.; Pavenstadt, H.; Schaefer, L.; Pfeilschifter, J.; Huwiler, A. Transforming growth factor-beta2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009, 76, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008, 173, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, O.; Stepanovska, B.; Starck, M.; Erhardt, M.; Romer, I.; Meyer Zu Heringdorf, D.; Pfeilschifter, J.; Zangemeister-Wittke, U.; Huwiler, A. Downregulation of the S1P Transporter Spinster Homology Protein 2 (Spns2) Exerts an Anti-Fibrotic and Anti-Inflammatory Effect in Human Renal Proximal Tubular Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imeri, F.; Nolan, K.A.; Bapst, A.M.; Santambrogio, S.; Abreu-Rodriguez, I.; Spielmann, P.; Pfundstein, S.; Libertini, S.; Crowther, L.; Orlando, I.M.C.; et al. Generation of renal Epo-producing cell lines by conditional gene tagging reveals rapid HIF-2 driven Epo kinetics, cell autonomous feedback regulation, and a telocyte phenotype. Kidney Int. 2019, 95, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Castrop, H.; Kurtz, A. Functional evidence confirmed by histological localization: Overlapping expression of erythropoietin and HIF-2alpha in interstitial fibroblasts of the renal cortex. Kidney Int. 2010, 77, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broeker, K.A.E.; Fuchs, M.A.A.; Schrankl, J.; Kurt, B.; Nolan, K.A.; Wenger, R.H.; Kramann, R.; Wagner, C.; Kurtz, A. Different subpopulations of kidney interstitial cells produce erythropoietin and factors supporting tissue oxygenation in response to hypoxia in vivo. Kidney Int. 2020, 98, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Wenger, H.R.; Kurtz, A. Erythropoietin. Compr. Physiol. 2011, 1, 1759–1794. [Google Scholar]
- Oba, S.; Suzuki, E.; Nishimatsu, H.; Kumano, S.; Hosoda, C.; Homma, Y.; Hirata, Y. Renoprotective effect of erythropoietin in ischemia/reperfusion injury: Possible roles of the Akt/endothelial nitric oxide synthase-dependent pathway. Int. J. Urol. 2012, 19, 248–255. [Google Scholar] [CrossRef]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, A.; Johnson, R.S. Nonrenal regulation of EPO synthesis. Kidney Int. 2009, 75, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Souma, T.; Yamazaki, S.; Moriguchi, T.; Suzuki, N.; Hirano, I.; Pan, X.; Minegishi, N.; Abe, M.; Kiyomoto, H.; Ito, S.; et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1599–1616. [Google Scholar] [CrossRef] [Green Version]
- Babitt, L.J.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [Green Version]
- Eschbach, J.W.; Abdulhadi, M.H.; Browne, J.K.; Delano, B.G.; Downing, M.R.; Egrie, J.C.; Evans, R.W.; Friedman, E.A.; Graber, S.E.; Haley, N.R.; et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann. Intern. Med. 1989, 111, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Eckardt, K.U. Pathogenesis of renal anemia. Semin. Nephrol. 2006, 26, 261–268. [Google Scholar] [CrossRef] [PubMed]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Hafizi, R.; Imeri, F.; Wenger, R.H.; Huwiler, A. S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P1 and S1P3 Receptor Activation and HIF-2alpha Stabilization. Int. J. Mol. Sci. 2021, 22, 9468. [Google Scholar] [CrossRef]
- Yasuoka, Y.; Fukuyama, T.; Izumi, Y.; Nakayama, Y.; Inoue, H.; Yanagita, K.; Oshima, T.; Yamazaki, T.; Uematsu, T.; Kobayashi, N.; et al. Erythropoietin production by the kidney and the liver in response to severe hypoxia evaluated by Western blotting with deglycosylation. Physiol. Rep. 2020, 8, e14485. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Tawati, S.; Berretta, G.; Rivas, P.L.; Baiget, J.; Jiang, Z.; Alsfouk, A.; Mackay, S.P.; Pyne, N.J.; Pyne, S. Topographical Mapping of Isoform-Selectivity Determinants for J-Channel-Binding Inhibitors of Sphingosine Kinases 1 and 2. J. Med. Chem. 2019, 62, 3658–3676. [Google Scholar] [CrossRef] [Green Version]
- Kharel, Y.; Morris, E.A.; Congdon, M.D.; Thorpe, S.B.; Tomsig, J.L.; Santos, W.L.; Lynch, K.R. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J. Pharmacol. Exp. Ther. 2015, 355, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Schwalm, S.; Beyer, S.; Hafizi, R.; Trautmann, S.; Geisslinger, G.; Adams, D.R.; Pyne, S.; Pyne, N.; Schaefer, L.; Huwiler, A.; et al. Validation of highly selective sphingosine kinase 2 inhibitors SLM6031434 and HWG-35D as effective anti-fibrotic treatment options in a mouse model of tubulointerstitial fibrosis. Cell. Signal. 2021, 79, 109881. [Google Scholar] [CrossRef]
- Quancard, J.; Bollbuck, B.; Janser, P.; Angst, D.; Berst, F.; Buehlmayer, P.; Streiff, M.; Beerli, C.; Brinkmann, V.; Guerini, D.; et al. A potent and selective S1P(1) antagonist with efficacy in experimental autoimmune encephalomyelitis. Chem. Biol. 2012, 19, 1142–1151. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: Investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 2010, 77, 704–713. [Google Scholar] [CrossRef]
- Wang, L.G.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imeri, F.; Stepanovska Tanturovska, B.; Zivkovic, A.; Enzmann, G.; Schwalm, S.; Pfeilschifter, J.; Homann, T.; Kleuser, B.; Engelhardt, B.; Stark, H.; et al. Novel compounds with dual S1P receptor agonist and histamine H3 receptor antagonist activities act protective in a mouse model of multiple sclerosis. Neuropharmacology 2021, 186, 108464. [Google Scholar] [CrossRef] [PubMed]
- Stepanovska Tanturovska, B.; Zivkovic, A.; Imeri, F.; Homann, T.; Kleuser, B.; Stark, H.; Huwiler, A. ST-2191, an Anellated Bismorpholino Derivative of Oxy-Fingolimod, Shows Selective S1P1 Agonist and Functional Antagonist Potency In Vitro and In Vivo. Molecules 2021, 26, 5134. [Google Scholar] [CrossRef] [PubMed]
- Stepanovska, B.; Zivkovic, A.; Enzmann, G.; Tietz, S.; Homann, T.; Kleuser, B.; Engelhardt, B.; Stark, H.; Huwiler, A. Morpholino Analogues of Fingolimod as Novel and Selective S1P1 Ligands with In Vivo Efficacy in a Mouse Model of Experimental Antigen-Induced Encephalomyelitis. Int. J. Mol. Sci. 2020, 21, 6463. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.A.; Glass, G.A.; Cunningham, J.M.; Bunn, H.F. The regulated expression of erythropoietin by two human hepatoma cell lines. Proc. Natl. Acad. Sci. USA 1987, 84, 7972–7976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ader, I.; Malavaud, B.; Cuvillier, O. When the sphingosine kinase 1/sphingosine 1-phosphate pathway meets hypoxia signaling: New targets for cancer therapy. Cancer Res. 2009, 69, 3723–3726. [Google Scholar] [CrossRef] [Green Version]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [Green Version]
- Bouquerel, P.; Gstalder, C.; Muller, D.; Laurent, J.; Brizuela, L.; Sabbadini, R.A.; Malavaud, B.; Pyronnet, S.; Martineau, Y.; Ader, I.; et al. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2alpha expression and activity in cancer. Oncogenesis 2016, 5, e209. [Google Scholar] [CrossRef] [Green Version]
- Gstalder, C.; Ader, I.; Cuvillier, O. FTY720 (Fingolimod) Inhibits HIF1 and HIF2 Signaling, Promotes Vascular Remodeling, and Chemosensitizes in Renal Cell Carcinoma Animal Model. Mol. Cancer Ther. 2016, 15, 2465–2474. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Maiti, A.; Xu, P.; Qi, Q.; Kawaguchi, T.; Okano, M.; Takabe, K.; Yan, L.; Luo, C. Regulation of hypoxia-inducible factor functions in the nucleus by sphingosine-1-phosphate. FASEB J. 2020, 34, 4293–4310. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr.; Nimkar, S.; Menaldino, D.; Hannun, Y.A.; Loomis, C.; Bell, R.M.; Tyagi, S.R.; Lambeth, J.D.; Stevens, V.L.; Hunter, R.; et al. Structural requirements for long-chain (sphingoid) base inhibition of protein kinase C in vitro and for the cellular effects of these compounds. Biochemistry 1989, 28, 3138–3145. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W.; Huwiler, A.; Fandrey, J.; Pfeilschifter, J. Inhibition of erythropoietin production by phorbol ester is associated with down-regulation of protein kinase C-alpha isoenzyme in hepatoma cells. Biochem. Biophys. Res. Commun. 1991, 179, 1441–1448. [Google Scholar] [CrossRef]
- Kim, H.; Na, Y.R.; Kim, S.Y.; Yang, E.G. Protein Kinase C Isoforms Differentially Regulate Hypoxia-Inducible Factor-1alpha Accumulation in Cancer Cells. J. Cell. Biochem. 2016, 117, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Ring, A.; Maier, M.; Gess, B.; Fabbro, D.; Kurtz, A. Hypoxia-induced accumulation of erythropoietin mRNA in isolated hepatocytes is inhibited by protein kinase C. Pflugers Arch. 1994, 426, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Gaspersic, J.; Kristan, A.; Kunej, T.; Zupan, I.P.; Debeljak, N. Erythrocytosis: Genes and pathways involved in disease development. Blood Transfus. 2021, 19, 518–532. [Google Scholar]
- Nagatoya, K.; Moriyama, T.; Kawada, N.; Takeji, M.; Oseto, S.; Murozono, T.; Ando, A.; Imai, E.; Hori, M. Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney Int. 2002, 61, 1684–1695. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafizi, R.; Imeri, F.; Stepanovska Tanturovska, B.; Manaila, R.; Schwalm, S.; Trautmann, S.; Wenger, R.H.; Pfeilschifter, J.; Huwiler, A. Sphk1 and Sphk2 Differentially Regulate Erythropoietin Synthesis in Mouse Renal Interstitial Fibroblast-like Cells. Int. J. Mol. Sci. 2022, 23, 5882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115882
Hafizi R, Imeri F, Stepanovska Tanturovska B, Manaila R, Schwalm S, Trautmann S, Wenger RH, Pfeilschifter J, Huwiler A. Sphk1 and Sphk2 Differentially Regulate Erythropoietin Synthesis in Mouse Renal Interstitial Fibroblast-like Cells. International Journal of Molecular Sciences. 2022; 23(11):5882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115882
Chicago/Turabian StyleHafizi, Redona, Faik Imeri, Bisera Stepanovska Tanturovska, Roxana Manaila, Stephanie Schwalm, Sandra Trautmann, Roland H. Wenger, Josef Pfeilschifter, and Andrea Huwiler. 2022. "Sphk1 and Sphk2 Differentially Regulate Erythropoietin Synthesis in Mouse Renal Interstitial Fibroblast-like Cells" International Journal of Molecular Sciences 23, no. 11: 5882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115882