
Molecular and Genetic Interactions between CCN2 and CCN3 behind Their Yin–Yang Collaboration

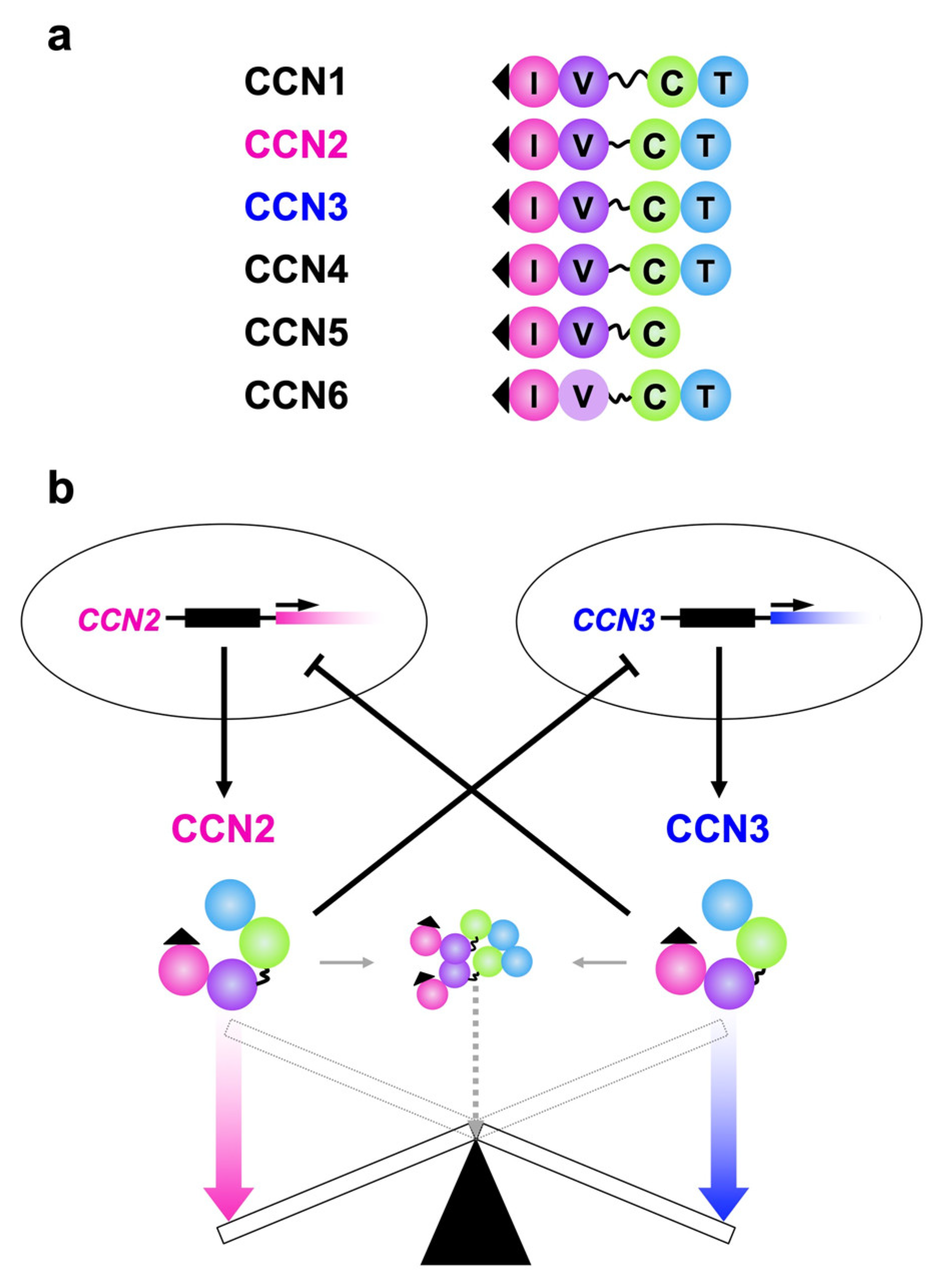{kind=link}
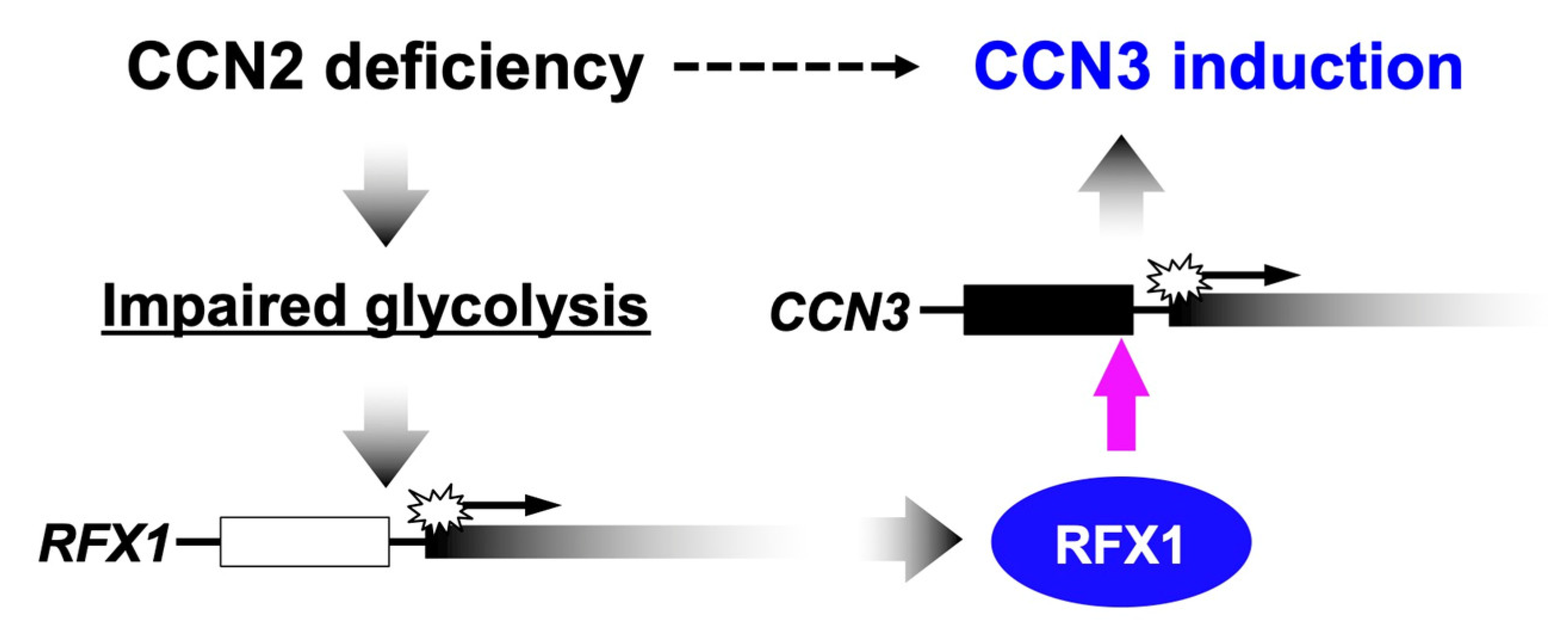{kind=link}
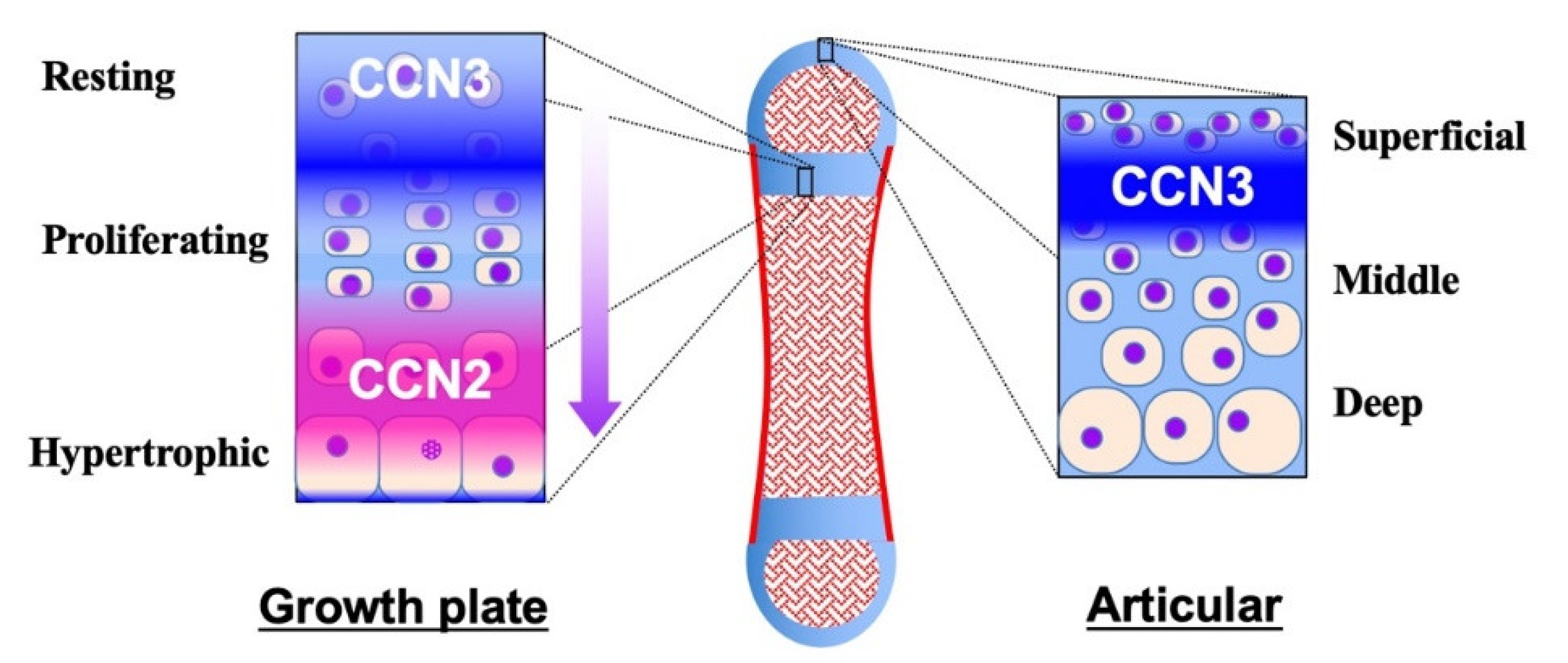{kind=link}
{kind=link}
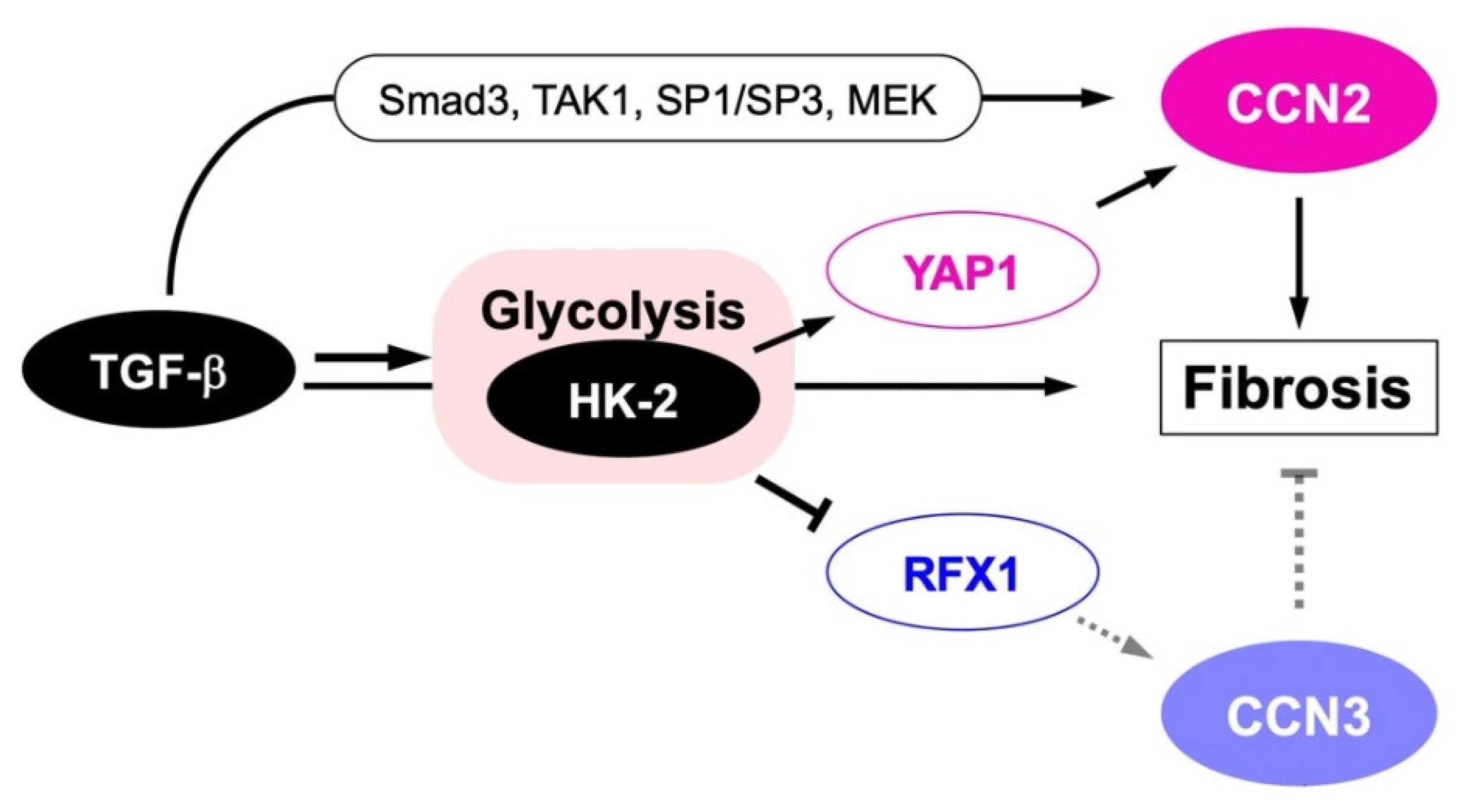{kind=link}
Abstract
:1. CCN2 and CCN3 in CCN Family
2. Yin–Yang Actions of CCN2 and CCN3
3. Molecular Interaction between CCN2 and CCN3
4. CCN2-CCN3 Genetic Interaction and Its Mechanism
5. CCN2-CCN3 Collaboration in Cartilage
6. CCN2-CCN3 Interaction in Fibrosis and Inflammation
7. CCN2-CCN3 Interplay in Malignancies
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bradham, D.M.; Igarashi, A.; Potter, R.L.; Grotendorst, G.R. Connective tissue growth factor: A cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J. Cell Biol. 1991, 114, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joliot, V.; Martinerie, C.; Dambrine, G.; Plassiart, G.; Brisac, M.; Crochet, J.; Perbal, B. Proviral rearrangements and overexpression of a new cellular gene (nov) in myeloblastosis-associated virus type 1-induced nephroblastomas. Mol. Cell Biol. 1992, 12, 10–21. [Google Scholar] [PubMed] [Green Version]
- Simmons, D.L.; Levy, D.B.; Yannoni, Y.; Erikson, R.L. Identification of a phorbol ester-repressible v-src-inducible gene. Proc. Natl. Acad. Sci. USA 1989, 86, 1178–1182. [Google Scholar] [CrossRef] [Green Version]
- Bork, P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993, 327, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef]
- Bleau, A.M.; Planque, N.; Lazar, N.; Zambelli, D.; Ori, A.; Quan, T.; Fisher, G.; Scotlandi, K.; Perbal, B. Antiproliferative activity of CCN3: Involvement of the C-terminal module and post-translational regulation. J. Cell Biochem. 2007, 101, 1475–1491. [Google Scholar] [CrossRef] [Green Version]
- Kawaki, H.; Kubota, S.; Suzuki, A.; Lazar, N.; Yamada, T.; Matsumura, T.; Ohgawara, T.; Maeda, T.; Perbal, B.; Lyons, K.M.; et al. Cooperative regulation of chondrocyte differentiation by CCN2 and CCN3 shown by a comprehensive analysis of the CCN family proteins in cartilage. J. Bone Miner. Res. 2008, 23, 1751–1764. [Google Scholar] [CrossRef]
- Kuwahara, M.; Kadoya, K.; Kondo, S.; Fu, S.; Miyake, Y.; Ogo, A.; Ono, M.; Furumatsu, T.; Nakata, E.; Sasaki, T.; et al. CCN3 (NOV) Drives Degradative Changes in Aging Articular Cartilage. Int. J. Mol. Sci. 2020, 21, 7556. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Shindo-Okada, N.; Tani, M.; Nagamachi, Y.; Takeuchi, K.; Shiroishi, T.; Toma, H.; Yokota, J. Expression of the Elm1 gene, a novel gene of the CCN (connective tissue growth factor, Cyr61/Cef10, and neuroblastoma overexpressed gene) family, suppresses In vivo tumor growth and metastasis of K-1735 murine melanoma cells. J. Exp. Med. 1998, 187, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Averboukh, L.; Zhu, W.; Zhang, H.; Jo, H.; Dempsey, P.J.; Coffey, R.J.; Pardee, A.B.; Liang, P. Identification of rCop-1, a new member of the CCN protein family, as a negative regulator for cell transformation. Mol. Cell Biol. 1998, 18, 6131–6141. [Google Scholar] [CrossRef] [Green Version]
- Pennica, D.; Swanson, T.A.; Welsh, J.W.; Roy, M.A.; Lawrence, D.A.; Lee, J.; Brush, J.; Taneyhill, L.A.; Deuel, B.; Lew, M.; et al. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc. Natl. Acad. Sci. USA 1998, 95, 14717–14722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigstock, D.R. The CCN family: A new stimulus package. J. Endocrinol. 2003, 178, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [Green Version]
- Kubota, S.; Takigawa, M. The CCN family acting throughout the body: Recent research developments. Biomol. Concepts 2013, 4, 477–494. [Google Scholar] [CrossRef]
- Perbal, B. CCN proteins: A centralized communication network. J. Cell Commun. Signal. 2013, 7, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Kubota, S.; Takigawa, M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin. Sci. 2015, 128, 181–196. [Google Scholar] [CrossRef]
- Perbal, B. The concept of the CCN protein family revisited: A centralized coordination network. J. Cell Commun. Signal. 2018, 12, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Perbal, B.; Tweedie, S.; Bruford, E. The official unified nomenclature adopted by the HGNC calls for the use of the acronyms, CCN1-6, and discontinuation in the use of CYR61, CTGF, NOV and WISP 1-3 respectively. J. Cell Commun. Signal. 2018, 12, 625–629. [Google Scholar] [CrossRef] [Green Version]
- Inoki, I.; Shiomi, T.; Hashimoto, G.; Enomoto, H.; Nakamura, H.; Makino, K.; Ikeda, E.; Takata, S.; Kobayashi, K.; Okada, Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002, 16, 219–221. [Google Scholar] [CrossRef] [Green Version]
- Abd El Kader, T.; Kubota, S.; Anno, K.; Tanaka, S.; Nishida, T.; Furumatsu, T.; Aoyama, E.; Kuboki, T.; Takigawa, M. Direct interaction between CCN family protein 2 and fibroblast growth factor 1. J. Cell Commun. Signal. 2014, 8, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, T.; Kubota, S.; Aoyama, E.; Janune, D.; Maeda, A.; Takigawa, M. Effect of CCN2 on FGF2-induced proliferation and MMP9 and MMP13 productions by chondrocytes. Endocrinology 2011, 152, 4232–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, A.; Nishida, T.; Aoyama, E.; Kubota, S.; Lyons, K.M.; Kuboki, T.; Takigawa, M. CCN family 2/connective tissue growth factor modulates BMP signalling as a signal conductor, which action regulates the proliferation and differentiation of chondrocytes. J. Biochem. 2009, 145, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Nagalla, S.R.; Oh, Y.; Wilson, E.; Roberts, C.T., Jr.; Rosenfeld, R.G. Identification of a family of low-affinity insulin-like growth factor binding proteins (IGFBPs): Characterization of connective tissue growth factor as a member of the IGFBP superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 12981–12986. [Google Scholar] [CrossRef] [Green Version]
- Hoshijima, M.; Hattori, T.; Inoue, M.; Araki, D.; Hanagata, H.; Miyauchi, A.; Takigawa, M. CT domain of CCN2/CTGF directly interacts with fibronectin and enhances cell adhesion of chondrocytes through integrin alpha5beta1. FEBS Lett. 2006, 580, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, E.; Hattori, T.; Hoshijima, M.; Araki, D.; Nishida, T.; Kubota, S.; Takigawa, M. N-terminal domains of CCN family 2/connective tissue growth factor bind to aggrecan. Biochem. J. 2009, 420, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Kubota, S.; Fukunaga, T.; Kondo, S.; Yosimichi, G.; Nakanishi, T.; Takano-Yamamoto, T.; Takigawa, M. CTGF/Hcs24, hypertrophic chondrocyte-specific gene product, interacts with perlecan in regulating the proliferation and differentiation of chondrocytes. J. Cell Physiol. 2003, 196, 265–275. [Google Scholar] [CrossRef]
- Babic, A.M.; Chen, C.C.; Lau, L.F. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol. Cell Biol. 1999, 19, 2958–2966. [Google Scholar] [CrossRef] [Green Version]
- Kiwanuka, E.; Andersson, L.; Caterson, E.J.; Junker, J.P.; Gerdin, B.; Eriksson, E. CCN2 promotes keratinocyte adhesion and migration via integrin alpha5beta1. Exp. Cell Res. 2013, 319, 2938–2946. [Google Scholar] [CrossRef]
- Kubota, S.; Takigawa, M. CCN family proteins and angiogenesis: From embryo to adulthood. Angiogenesis 2007, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Son, S.; Ko, Y.; Shin, I. CTGF regulates cell proliferation, migration, and glucose metabolism through activation of FAK signaling in triple-negative breast cancer. Oncogene 2021, 40, 2667–2681. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, E.; Kubota, S.; Takigawa, M. CCN2/CTGF binds to fibroblast growth factor receptor 2 and modulates its signaling. FEBS Lett. 2012, 586, 4270–4275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayego-Mateos, S.; Rodrigues-Díez, R.; Morgado-Pascual, J.L.; Rodrigues Díez, R.R.; Mas, S.; Lavoz, C.; Alique, M.; Pato, J.; Keri, G.; Ortiz, A.; et al. Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J. Mol. Cell Biol. 2013, 5, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Yang, M.H.; Lin, B.R.; Chen, S.T.; Pan, S.H.; Hsiao, M.; Lai, T.C.; Lin, S.K.; Jeng, Y.M.; Chu, C.Y.; et al. CCN2 inhibits lung cancer metastasis through promoting DAPK-dependent anoikis and inducing EGFR degradation. Cell Death Differ. 2013, 20, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Wahab, N.A.; Weston, B.S.; Mason, R.M. Connective tissue growth factor CCN2 interacts with and activates the tyrosine kinase receptor TrkA. J. Am. Soc. Nephrol. 2005, 16, 340–351. [Google Scholar] [CrossRef]
- Kawata, K.; Kubota, S.; Eguchi, T.; Aoyama, E.; Moritani, N.H.; Kondo, S.; Nishida, T.; Takigawa, M. Role of LRP1 in transport of CCN2 protein in chondrocytes. J. Cell Sci. 2012, 125, 2965–2972. [Google Scholar]
- Mercurio, S.; Latinkic, B.; Itasaki, N.; Krumlauf, R.; Smith, J.C. Connective-tissue growth factor modulates WNT signalling and interacts with the WNT receptor complex. Development 2004, 131, 2137–2147. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, E.; Kubota, S.; Khattab, H.M.; Nishida, T.; Takigawa, M. CCN2 enhances RANKL-induced osteoclast differentiation via direct binding to RANK and OPG. Bone 2015, 73, 242–248. [Google Scholar] [CrossRef]
- Surmann-Schmitt, C.; Sasaki, T.; Hattori, T.; Eitzinger, N.; Schett, G.; von der Mark, K.; Stock, M. The Wnt antagonist Wif-1 interacts with CTGF and inhibits CTGF activity. J. Cell Physiol. 2012, 227, 2207–2216. [Google Scholar] [CrossRef]
- Pi, L.; Shenoy, A.K.; Liu, J.; Kim, S.; Nelson, N.; Xia, H.; Hauswirth, W.W.; Petersen, B.E.; Schultz, G.S.; Scott, E.W. CCN2/CTGF regulates neovessel formation via targeting structurally conserved cystine knot motifs in multiple angiogenic regulators. FASEB J. 2012, 26, 3365–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshijima, M.; Hattori, T.; Aoyama, E.; Nishida, T.; Yamashiro, T.; Takigawa, M. Roles of heterotypic CCN2/CTGF-CCN3/NOV and homotypic CCN2-CCN2 interactions in expression of the differentiated phenotype of chondrocytes. FEBS J. 2012, 279, 3584–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perbal, B.; Martinerie, C.; Sainson, R.; Werner, M.; He, B.; Roizman, B. The C-terminal domain of the regulatory protein NOVH is sufficient to promote interaction with fibulin 1C: A clue for a role of NOVH in cell-adhesion signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Yamaguchi, S.; Ando, R.; Miyawaki, A.; Kabasawa, Y.; Takagi, M.; Li, C.L.; Perbal, B.; Katsube, K. The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J. Biol. Chem. 2002, 277, 29399–29405. [Google Scholar] [CrossRef] [Green Version]
- Takayama, I.; Tanabe, H.; Nishiyama, T.; Ito, H.; Amizuka, N.; Li, M.; Katsube, K.I.; Kii, I.; Kudo, A. Periostin is required for matricellular localization of CCN3 in periodontal ligament of mice. J. Cell Commun. Signal. 2017, 11, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riser, B.L.; Najmabadi, F.; Perbal, B.; Rambow, J.A.; Riser, M.L.; Sukowski, E.; Yeger, H.; Riser, S.C.; Peterson, D.R. CCN3/CCN2 regulation and the fibrosis of diabetic renal disease. J. Cell Commun. Signal. 2010, 4, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leask, A. Yin and Yang: CCN3 inhibits the pro-fibrotic effects of CCN2. J. Cell Commun. Signal. 2009, 3, 161–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peidl, A.; Perbal, B.; Leask, A. Yin/Yang expression of CCN family members: Transforming growth factor beta 1, via ALK5/FAK/MEK, induces CCN1 and CCN2, yet suppresses CCN3, expression in human dermal fibroblasts. PLoS ONE 2019, 14, e0218178. [Google Scholar] [CrossRef]
- Shimo, T.; Nakanishi, T.; Nishida, T.; Asano, M.; Kanyama, M.; Kuboki, T.; Tamatani, T.; Tezuka, K.; Takemura, M.; Matsumura, T.; et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J. Biochem. 1999, 126, 137–145. [Google Scholar] [CrossRef]
- Nakanishi, T.; Nishida, T.; Shimo, T.; Kobayashi, K.; Kubo, T.; Tamatani, T.; Tezuka, K.; Takigawa, M. Effects of CTGF/Hcs24, a product of a hypertrophic chondrocyte-specific gene, on the proliferation and differentiation of chondrocytes in culture. Endocrinology 2000, 141, 264–273. [Google Scholar] [CrossRef]
- Nishida, T.; Nakanishi, T.; Asano, M.; Shimo, T.; Takigawa, M. Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J. Cell Physiol. 2000, 184, 197–206. [Google Scholar] [CrossRef]
- Asano, M.; Kubota, S.; Nakanishi, T.; Nishida, T.; Yamaai, T.; Yosimichi, G.; Ohyama, K.; Sugimoto, T.; Murayama, Y.; Takigawa, M. Effect of connective tissue growth factor (CCN2/CTGF) on proliferation and differentiation of mouse periodontal ligament-derived cells. Cell Commun. Signal. 2005, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.J.; Xu, L.Y.; Wu, J.Y.; Shen, Z.Y.; Zhao, Q.; Du, Z.P.; Lv, Z.; Gu, W.; Pan, F.; Xu, X.E.; et al. Involvement of CYR61 and CTGF in the fascin-mediated proliferation and invasiveness of esophageal squamous cell carcinomas cells. Am. J. Pathol. 2010, 176, 939–951. [Google Scholar] [CrossRef] [Green Version]
- Kothapalli, D.; Grotendorst, G.R. CTGF modulates cell cycle progression in cAMP-arrested NRK fibroblasts. J. Cell Physiol. 2000, 182, 119–126. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Duncan, M.R. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 2005, 19, 729–738. [Google Scholar] [CrossRef]
- Kawaki, H.; Kubota, S.; Suzuki, A.; Suzuki, M.; Kohsaka, K.; Hoshi, K.; Fujii, T.; Lazar, N.; Ohgawara, T.; Maeda, T.; et al. Differential roles of CCN family proteins during osteoblast differentiation: Involvement of Smad and MAPK signaling pathways. Bone 2011, 49, 975–989. [Google Scholar] [CrossRef]
- Shimoyama, T.; Hiraoka, S.; Takemoto, M.; Koshizaka, M.; Tokuyama, H.; Tokuyama, T.; Watanabe, A.; Fujimoto, M.; Kawamura, H.; Sato, S.; et al. CCN3 inhibits neointimal hyperplasia through modulation of smooth muscle cell growth and migration. Arter. Thromb. Vasc. Biol. 2010, 30, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Wang, H.; McLeod, T.L.; Naus, C.C.; Kyurkchiev, S.; Advani, S.; Yu, J.; Perbal, B.; Weichselbaum, R.R. Inhibition of glioma cell growth and tumorigenic potential by CCN3 (NOV). Mol. Pathol. 2001, 54, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Gellhaus, A.; Dong, X.; Propson, S.; Maass, K.; Klein-Hitpass, L.; Kibschull, M.; Traub, O.; Willecke, K.; Perbal, B.; Lye, S.J.; et al. Connexin43 interacts with NOV: A possible mechanism for negative regulation of cell growth in choriocarcinoma cells. J. Biol. Chem. 2004, 279, 36931–36942. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.T.; Bechberger, J.F.; Ozog, M.A.; Perbal, B.; Naus, C.C. CCN3 (NOV) interacts with connexin43 in C6 glioma cells: Possible mechanism of connexin-mediated growth suppression. J. Biol. Chem. 2004, 279, 36943–36950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crean, J.K.; Furlong, F.; Finlay, D.; Mitchell, D.; Murphy, M.; Conway, B.; Brady, H.R.; Godson, C.; Martin, F. Connective tissue growth factor [CTGF]/CCN2 stimulates mesangial cell migration through integrated dissolution of focal adhesion complexes and activation of cell polarization. FASEB J. 2004, 18, 1541–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benini, S.; Perbal, B.; Zambelli, D.; Colombo, M.P.; Manara, M.C.; Serra, M.; Parenza, M.; Martinez, V.; Picci, P.; Scotlandi, K. In Ewing’s sarcoma CCN3(NOV) inhibits proliferation while promoting migration and invasion of the same cell type. Oncogene 2005, 24, 4349–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, Z.; Bi, D.; Yuan, X.; Liu, X.; Ding, S.; Lu, J.; Niu, Z. CCN3 (NOV) regulates proliferation, adhesion, migration and invasion in clear cell renal cell carcinoma. Oncol. Lett. 2012, 3, 1099–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, H.E.; Chen, J.C.; Tsai, C.H.; Kuo, C.C.; Hsu, H.C.; Hwang, W.L.; Fong, Y.C.; Tang, C.H. CCN3 increases cell motility and MMP-13 expression in human chondrosarcoma through integrin-dependent pathway. J. Cell Physiol. 2011, 226, 3181–3189. [Google Scholar] [CrossRef]
- Lin, C.G.; Chen, C.C.; Leu, S.J.; Grzeszkiewicz, T.M.; Lau, L.F. Integrin-dependent functions of the angiogenic inducer NOV (CCN3): Implication in wound healing. J. Biol. Chem. 2005, 280, 8229–8237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leu, S.J.; Lam, S.C.; Lau, L.F. Pro-angiogenic activities of CYR61 (CCN1) mediated through integrins alphavbeta3 and alpha6beta1 in human umbilical vein endothelial cells. J. Biol. Chem. 2002, 277, 46248–46255. [Google Scholar] [CrossRef] [Green Version]
- Leu, S.J.; Liu, Y.; Chen, N.; Chen, C.C.; Lam, S.C.; Lau, L.F. Identification of a novel integrin alpha 6 beta 1 binding site in the angiogenic inducer CCN1 (CYR61). J. Biol. Chem. 2003, 278, 33801–33808. [Google Scholar] [CrossRef] [Green Version]
- Khodosevich, K.; Lazarini, F.; von Engelhardt, J.; Kaneko, H.; Lledo, P.M.; Monyer, H. Connective tissue growth factor regulates interneuron survival and information processing in the olfactory bulb. Neuron 2013, 79, 1136–1151. [Google Scholar] [CrossRef] [Green Version]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 2003, 130, 2779–2791. [Google Scholar] [CrossRef] [Green Version]
- Takigawa, M.; Nakanishi, T.; Kubota, S.; Nishida, T. Role of CTGF/HCS24/ecogenin in skeletal growth control. J. Cell Physiol. 2003, 194, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Ohkawara, B.; Kobayakawa, A.; Kanbara, S.; Hattori, T.; Kubota, S.; Ito, M.; Masuda, A.; Takigawa, M.; Lyons, K.M.; Ishiguro, N.; et al. CTGF/CCN2 facilitates LRP4-mediated formation of the embryonic neuromuscular junction. EMBO Rep. 2020, 21, e48462. [Google Scholar] [CrossRef] [PubMed]
- Charrier, A.; Brigstock, D.R. Regulation of pancreatic function by connective tissue growth factor (CTGF, CCN2). Cytokine Growth Factor Rev. 2013, 24, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Leask, A. CCN2 modulates hair follicle cycling in mice. Mol. Biol. Cell 2013, 24, 3939–3944. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, M.S.; Reis, A.H.; Aguiar, D.P.; Lyons, K.M.; Abreu, J.G. Dynamic analysis of the expression of the TGFbeta/SMAD2 pathway and CCN2/CTGF during early steps of tooth development. Cells Tissues Organs. 2008, 187, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Hong, D.; Iborra, F.; Sarno, S.; Enver, T. NOV (CCN3) functions as a regulator of human hematopoietic stem or progenitor cells. Science 2007, 316, 590–593. [Google Scholar] [CrossRef]
- Ishihara, J.; Umemoto, T.; Yamato, M.; Shiratsuchi, Y.; Takaki, S.; Petrich, B.G.; Nakauchi, H.; Eto, K.; Kitamura, T.; Okano, T. Nov/CCN3 regulates long-term repopulating activity of murine hematopoietic stem cells via integrin alphavbeta3. Int. J. Hematol. 2014, 99, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Turati, V.; Brian, D.; Thrussel, C.; Wilbourn, B.; May, G.; Enver, T. Nov/CCN3 Enhances Cord Blood Engraftment by Rapidly Recruiting Latent Human Stem Cell Activity. Cell Stem. Cell 2020, 26, 527–541. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Roestenberg, P.; van Nieuwenhoven, F.A.; Bovenschen, N.; Li, Z.; Xu, L.; Oliver, N.; Aten, J.; Joles, J.A.; Vial, C.; et al. CTGF inhibits BMP-7 signaling in diabetic nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2098–2107. [Google Scholar] [CrossRef] [Green Version]
- Minamizato, T.; Sakamoto, K.; Liu, T.; Kokubo, H.; Katsube, K.; Perbal, B.; Nakamura, S.; Yamaguchi, A. CCN3/NOV inhibits BMP-2-induced osteoblast differentiation by interacting with BMP and Notch signaling pathways. Biochem. Biophys. Res Commun. 2007, 354, 567–573. [Google Scholar] [CrossRef]
- Rydziel, S.; Stadmeyer, L.; Zanotti, S.; Durant, D.; Smerdel-Ramoya, A.; Canalis, E. Nephroblastoma overexpressed (Nov) inhibits osteoblastogenesis and causes osteopenia. J. Biol. Chem. 2007, 282, 19762–19772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, Y.; Sakamoto, K.; Tamamura, Y.; Shibata, Y.; Minamizato, T.; Kihara, T.; Ito, M.; Katsube, K.; Hiraoka, S.; Koseki, H.; et al. CCN3 protein participates in bone regeneration as an inhibitory factor. J. Biol. Chem. 2013, 288, 19973–19985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Wei, Y.; Cao, J.; Wu, X.; Mou, D.; Luo, J.; Li, A.; Zuo, G.W.; Tang, M. CCN3 and DLL1 co-regulate osteogenic differentiation of mouse embryonic fibroblasts in a Hey1-dependent manner. Cell Death Dis. 2018, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Liu, J.F.; Fong, Y.C.; Huang, Y.L.; Chao, C.C.; Tang, C.H. CCN3 Facilitates Runx2 and Osterix Expression by Inhibiting miR-608 through PI3K/Akt Signaling in Osteoblasts. Int. J. Mol. Sci. 2019, 20, 3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda-Uematsu, A.; Kubota, S.; Kawaki, H.; Kawata, K.; Miyake, Y.; Hattori, T.; Nishida, T.; Moritani, N.; Lyons, K.M.; Iida, S.; et al. CCN2 as a novel molecule supporting energy metabolism of chondrocytes. J. Cell Biochem. 2014, 115, 854–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, S.; Maeda-Uematsu, A.; Nishida, T.; Takigawa, M. New functional aspects of CCN2 revealed by trans-omic approaches. J. Oral. Biosci. 2015, 57, 37–43. [Google Scholar] [CrossRef]
- Akashi, S.; Nishida, T.; El-Seoudi, A.; Takigawa, M.; Iida, S.; Kubota, S. Metabolic regulation of the CCN family genes by glycolysis in chondrocytes. J. Cell Commun. Signal. 2018, 12, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Mizukawa, T.; Nishida, T.; Akashi, S.; Kawata, K.; Kikuchi, S.; Kawaki, H.; Takigawa, M.; Kamioka, H.; Kubota, S. RFX1-mediated CCN3 induction that may support chondrocyte survival under starved conditions. J. Cell Physiol. 2021, 236, 6884–6896. [Google Scholar] [CrossRef]
- Kular, L.; Pakradouni, J.; Kitabgi, P.; Laurent, M.; Martinerie, C. The CCN family: A new class of inflammation modulators? Biochimie 2011, 93, 377–388. [Google Scholar] [CrossRef]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.H.; Zhao, Y.P.; Xue, M.; Ji, C.B.; Gao, C.L.; Zhu, J.G.; Qin, D.N.; Kou, C.Z.; Qin, X.H.; Tong, M.L.; et al. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2010, 328, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.M. Regulation and bioactivity of the CCN family of genes and proteins in obesity and diabetes. J. Cell Commun. Signal. 2018, 12, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Y.; Wang, Y.D.; Qi, X.Y.; Ran, L.; Hong, T.; Yang, J.; Yan, B.; Liao, Z.Z.; Liu, J.H.; Xiao, X.H. Serum CCN3 levels are increased in type 2 diabetes mellitus and associated with obesity, insulin resistance and inflammation. Clin. Chim. Acta 2019, 494, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Nagao, Y.; Hashitani, S.; Yamanaka, N.; Takigawa, M.; Kubota, S. Suppression of adipocyte differentiation by low-intensity pulsed ultrasound via inhibition of insulin signaling and promotion of CCN family protein 2. J. Cell Biochem. 2020, 121, 4724–4740. [Google Scholar] [CrossRef]
- Córdova, G.; Rochard, A.; Riquelme-Guzmán, C.; Cofré, C.; Scherman, D.; Bigey, P.; Brandan, E. SMAD3 and SP1/SP3 transcription factors collaborate to regulate connective tissue growth factor gene expression in myoblasts in response to transforming growth factor β. J. Cell Biochem. 2015, 116, 1880–1887. [Google Scholar] [CrossRef]
- Tran, C.M.; Smith, H.E.; Symes, A.; Rittié, L.; Perbal, B.; Shapiro, I.M.; Risbud, M.V. Transforming growth factor β controls CCN3 expression in nucleus pulposus cells of the intervertebral disc. Arthritis Rheum. 2011, 63, 3022–3031. [Google Scholar] [CrossRef] [Green Version]
- Kubota, S.; Kawaki, H.; Perbal, B.; Kawata, K.; Hattori, T.; Nishida, T. Cellular communication network factor 3 in cartilage development and maintenance. J. Cell Commun. Signal. 2021, 15, 533–543. [Google Scholar] [CrossRef]
- Yu, C.; Le, A.T.; Yeger, H.; Perbal, B.; Alman, B.A. NOV (CCN3) regulation in the growth plate and CCN family member expression in cartilage neoplasia. J. Pathol. 2003, 201, 609–615. [Google Scholar] [CrossRef]
- Itoh, S.; Hattori, T.; Tomita, N.; Aoyama, E.; Yutani, Y.; Yamashiro, T.; Takigawa, M. CCN family member 2/connective tissue growth factor (CCN2/CTGF) has anti-aging effects that protect articular cartilage from age-related degenerative changes. PLoS ONE 2013, 8, e71156. [Google Scholar] [CrossRef]
- Janune, D.; Abd El Kader, T.; Aoyama, E.; Nishida, T.; Tabata, Y.; Kubota, S.; Takigawa, M. Novel role of CCN3 that maintains the differentiated phenotype of articular cartilage. J. Bone Miner. Metab. 2017, 35, 582–597. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Kojima, S.; Kuboki, T.; Nakao, K.; Kushibiki, T.; Tabata, Y.; Takigawa, M. Regeneration of defects in articular cartilage in rat knee joints by CCN2 (connective tissue growth factor). J. Bone Miner. Res. 2004, 19, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Abd El Kader, T.; Kubota, S.; Nishida, T.; Hattori, T.; Aoyama, E.; Janune, D.; Hara, E.S.; Ono, M.; Tabata, Y.; Kuboki, T.; et al. The regenerative effects of CCN2 independent modules on chondrocytes in vitro and osteoarthritis models in vivo. Bone 2014, 59, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, A.; Nashiro, K.; Kikuchi, K.; Sato, S.; Ihn, H.; Grotendorst, G.R.; Takehara, K. Significant correlation between connective tissue growth factor gene expression and skin sclerosis in tissue sections from patients with systemic sclerosis. J. Invest. Dermatol. 1995, 105, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, A.; Nashiro, K.; Kikuchi, K.; Sato, S.; Ihn, H.; Fujimoto, M.; Grotendorst, G.R.; Takehara, K. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J. Invest. Dermatol. 1996, 106, 729–733. [Google Scholar] [CrossRef] [Green Version]
- Dziadzio, M.; Usinger, W.; Leask, A.; Abraham, D.; Black, C.M.; Denton, C.; Stratton, R. N-terminal connective tissue growth factor is a marker of the fibrotic phenotype in scleroderma. QJM 2005, 98, 485–492. [Google Scholar] [CrossRef]
- Leask, A.; Parapuram, S.K.; Shi-Wen, X.; Abraham, D.J. Connective tissue growth factor (CTGF, CCN2) gene regulation: A potent clinical bio-marker of fibroproliferative disease? J. Cell Commun. Signal. 2009, 3, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Ikawa, Y.; Ng, P.S.; Endo, K.; Kondo, M.; Chujo, S.; Ishida, W.; Shirasaki, F.; Fujimoto, M.; Takehara, K. Neutralizing monoclonal antibody to human connective tissue growth factor ameliorates transforming growth factor-beta-induced mouse fibrosis. J. Cell Physiol. 2008, 216, 680–687. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kubota, S.; Murakashi, E.; Zhou, Y.; Endo, K.; Ng, P.S.; Takigawa, M.; Numabe, Y. Nicotine-induced CCN2: From smoking to periodontal fibrosis. J. Dent. Res. 2010, 89, 34–39. [Google Scholar] [CrossRef]
- Uzel, M.I.; Kantarci, A.; Hong, H.H.; Uygur, C.; Sheff, M.C.; Firatli, E.; Trackman, P.C. Connective tissue growth factor in drug-induced gingival overgrowth. J. Periodontol. 2001, 72, 921–931. [Google Scholar] [CrossRef]
- Trackman, P.C.; Kantarci, A. Molecular and clinical aspects of drug-induced gingival overgrowth. J. Dent Res. 2015, 94, 540–546. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, K.; Igarashi-Takeuchi, H.; Numabe, Y. Hepatocyte growth factor exhibits anti-fibrotic effects in an in vitro model of nifedipine-induced gingival overgrowth. J. Oral. Sci. 2022, 64, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.A.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. 1998, 275, L365–L371. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Flore, M.; Siciliano, M.; Richeldi, L. Antibody-based therapies for idiopathic pulmonary fibrosis. Expert Opin. Biol. Ther. 2020, 20, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Koitabashi, N.; Arai, M.; Niwano, K.; Watanabe, A.; Endoh, M.; Suguta, M.; Yokoyama, T.; Tada, H.; Toyama, T.; Adachi, H.; et al. Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur. J. Heart Fail. 2008, 10, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Charrier, A.L.; Leask, A.; French, S.W.; Brigstock, D.R. Ethanol-stimulated differentiated functions of human or mouse hepatic stellate cells are mediated by connective tissue growth factor. J. Hepatol. 2011, 55, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Mola, F.F.; Friess, H.; Martignoni, M.E.; Di Sebastiano, P.; Zimmermann, A.; Innocenti, P.; Graber, H.; Gold, L.I.; Korc, M.; Büchler, M.W. Connective tissue growth factor is a regulator for fibrosis in human chronic pancreatitis. Ann. Surg. 1999, 230, 63–71. [Google Scholar] [CrossRef]
- Rebolledo, D.L.; Lipson, K.E.; Brandan, E. Driving fibrosis in neuromuscular diseases: Role and regulation of Connective tissue growth factor (CCN2/CTGF). Matrix Biol. Plus. 2021, 11, 100059. [Google Scholar] [CrossRef]
- Cicha, I.; Yilmaz, A.; Klein, M.; Raithel, D.; Brigstock, D.R.; Daniel, W.G.; Goppelt-Struebe, M.; Garlichs, C.D. Connective tissue growth factor is overexpressed in complicated atherosclerotic plaques and induces mononuclear cell chemotaxis in vitro. Arter. Thromb. Vasc. Biol 2005, 25, 1008–1013. [Google Scholar] [CrossRef]
- Chintala, H.; Liu, H.; Parmar, R.; Kamalska, M.; Kim, Y.J.; Lovett, D.; Grant, M.B.; Chaqour, B. Connective tissue growth factor regulates retinal neovascularization through p53 protein-dependent transactivation of the matrix metalloproteinase (MMP)-2 gene. J. Biol. Chem. 2012, 287, 40570–40585. [Google Scholar] [CrossRef] [Green Version]
- Barbe, M.F.; Hilliard, B.A.; Amin, M.; Harris, M.Y.; Hobson, L.J.; Cruz, G.E.; Popoff, S.N. Blocking CTGF/CCN2 reduces established skeletal muscle fibrosis in a rat model of overuse injury. FASEB J. 2020, 34, 6554–6569. [Google Scholar] [CrossRef] [Green Version]
- NCT02606136. Trial of Pamrevlumab (FG-3019), in Non-Ambulatory Participants with Duchenne muscular Dystrophy (DMD). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02606136 (accessed on 1 May 2022).
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leask, A. CCN2: A novel, specific and valid target for anti-fibrotic drug intervention. Expert Opin. Targets 2013, 17, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Mukoyama, M.; Sugawara, A.; Mori, K.; Nagae, T.; Makino, H.; Suganami, T.; Yahata, K.; Fujinaga, Y.; Tanaka, I.; et al. Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. Am. J. Physiol. Renal. Physiol. 2002, 282, F933–F942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, H.; Kikuta, T.; Kobayashi, T.; Inoue, T.; Kanno, Y.; Takigawa, M.; Sugaya, T.; Kopp, J.B.; Suzuki, H. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J. Am. Soc. Nephrol. 2005, 16, 133–143. [Google Scholar] [CrossRef]
- Zhang, C.; van der Voort, D.; Shi, H.; Zhang, R.; Qing, Y.; Hiraoka, S.; Takemoto, M.; Yokote, K.; Moxon, J.V.; Norman, P.; et al. Matricellular protein CCN3 mitigates abdominal aortic aneurysm. J. Clin. Invest 2016, 126, 1282–1299. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Díez, R.R.; Tejera-Muñoz, A.; Esteban, V.; Steffensen, L.B.; Rodrigues-Díez, R.; Orejudo, M.; Rayego-Mateos, S.; Falke, L.L.; Cannata-Ortiz, P.; Ortiz, A.; et al. CCN2 (cellular communication network factor 2) deletion alters vascular integrity and function predisposing to aneurysm formation. Hypertension 2022, 79, e42–e55. [Google Scholar] [CrossRef]
- Ren, J.; Wang, X.; Parry, S.N.; Yee, C.; Gorrell, M.D.; McLennan, S.V.; Twigg, S.M. Targeting CCN2 protects against progressive non-alcoholic steatohepatitis in a preclinical model induced by high-fat feeding and type 2 diabetes. J. Cell Commun. Signal. 2022; in press. [Google Scholar] [CrossRef]
- Abd El Kader, T.; Kubota, S.; Janune, D.; Nishida, T.; Hattori, T.; Aoyama, E.; Perbal, B.; Kuboki, T.; Takigawa, M. Anti-fibrotic effect of CCN3 accompanied by altered gene expression profile of the CCN family. J. Cell Commun. Signal. 2013, 7, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Borkham-Kamphorst, E.; van Roeyen, C.R.; Van de Leur, E.; Floege, J.; Weiskirchen, R. CCN3/NOV small interfering RNA enhances fibrogenic gene expression in primary hepatic stellate cells and cirrhotic fat storing cell line CFSC. J. Cell Commun. Signal. 2012, 6, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Choudhury, M.; Kang, J.H.; Schaefbauer, K.J.; Jung, M.Y.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci. Signal. 2019, 12, eaax4067. [Google Scholar] [CrossRef]
- Yoon, P.O.; Lee, M.A.; Cha, H.; Jeong, M.H.; Kim, J.; Jang, S.P.; Choi, B.Y.; Jeong, D.; Yang, D.K.; Hajjar, R.J.; et al. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J. Mol. Cell Cardiol. 2010, 49, 294–303. [Google Scholar] [CrossRef]
- Jeong, D.; Lee, M.A.; Li, Y.; Yang, D.K.; Kho, C.; Oh, J.G.; Hong, G.; Lee, A.; Song, M.H.; LaRocca, T.J.; et al. Matricellular protein CCN5 reverses established cardiac fibrosis. J. Am. Coll. Cardiol. 2016, 67, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Batmunkh, R.; Nishioka, Y.; Aono, Y.; Azuma, M.; Kinoshita, K.; Kishi, J.; Makino, H.; Kishi, M.; Takezaki, A.; Sone, S. CCN6 as a profibrotic mediator that stimulates the proliferation of lung fibroblasts via the integrin beta1/focal adhesion kinase pathway. J. Med. Investig. 2011, 58, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.P.; Huang, H.Y.; Wu, D.M.; Dong, N.; Dong, L.; Chen, C.S.; Chen, C.L.; Chen, Y.G. Regulatory mechanism of NOV/CCN3 in the inflammation and apoptosis of lung epithelial alveolar cells upon lipopolysaccharide stimulation. Mol. Med. Rep. 2020, 21, 1872–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowinski, D.; Koskela, A.; Kiwanuka, E.; Boström, M.; Gerdin, B.; Ivarsson, M. Inhibition of connective tissue growth factor/CCN2 expression in human dermal fibroblasts by interleukin-1alpha and beta. J. Cell Biochem. 2010, 110, 1226–1233. [Google Scholar] [CrossRef]
- Fukunaga-Kalabis, M.; Martinez, G.; Liu, Z.J.; Kalabis, J.; Mrass, P.; Weninger, W.; Firth, S.M.; Planque, N.; Perbal, B.; Herlyn, M. CCN3 controls 3D spatial localization of melanocytes in the human skin through DDR1. J. Cell Biol. 2006, 175, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Yeger, H.; Perbal, B. The CCN axis in cancer development and progression. J. Cell Commun. Signal. 2021, 15, 491–517. [Google Scholar] [CrossRef]
- Li, J.; Ye, L.; Sun, P.-H.; Zheng, F.; Ruge, F.; Satherley, L.K.; Feng, Y.; Zhao, H.; Du, G.; Wang, T.; et al. Reduced NOV expression correlates with disease progression in colorectal cancer and is associated with survival, invasion and chemoresistance of cancer cells. Oncotarget 2017, 8, 26231–26244. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, W.; Huang, P.; Lin, L.; Ye, H.; Lin, D.; Koeffler, H.P.; Wang, J.; Yin, D. Expression of CCN family members correlates with the clinical features of hepatocellular carcinoma. Oncol. Rep. 2015, 33, 1481–1492. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.G.; Watkins, G.; Fodstad, O.; Douglas-Jones, A.; Mokbel, K.; Mansel, R.E. Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr. Relat. Cancer 2004, 11, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 30, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akashi, S.; Nishida, T.; Mizukawa, T.; Kawata, K.; Takigawa, M.; Iida, S.; Kubota, S. Regulation of cellular communication factor 2 (CCN2) in breast cancer cells via the cell-type dependent interplay between CCN2 and glycolysis. J. Oral. Biosci. 2020, 62, 280–288. [Google Scholar] [CrossRef] [PubMed]
- NCT03955146. Evaluation of Efficacy and Safety of Pamrevlumab in Patients with Idiopathic Pulmonary Fibrosis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03955146?term=FG-3019&draw=3&rank=17 (accessed on 11 May 2022).
- NCT00102297. Study of the Safety of FG-3019 in Incipient Nephropathy Due to Type 1 or Type 2 Diabetes Mellitus. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00102297?term=FG-3019&draw=4&rank=10 (accessed on 11 May 2022).
- NCT03941093. Evaluation of Efficacy and Safety of Neoadjuvant Treatment with Pamrevlumab in Combination with CHEMOTHERAPY (either Gemcitabine Plus Nab-Paclitaxel or FOLFIRINOX) in Participants with Locally Advanced Pancreatic Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03941093?term=FG-3019&draw=5&rank=18 (accessed on 11 May 2022).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubota, S.; Kawata, K.; Hattori, T.; Nishida, T. Molecular and Genetic Interactions between CCN2 and CCN3 behind Their Yin–Yang Collaboration. Int. J. Mol. Sci. 2022, 23, 5887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115887
Kubota S, Kawata K, Hattori T, Nishida T. Molecular and Genetic Interactions between CCN2 and CCN3 behind Their Yin–Yang Collaboration. International Journal of Molecular Sciences. 2022; 23(11):5887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115887
Chicago/Turabian StyleKubota, Satoshi, Kazumi Kawata, Takako Hattori, and Takashi Nishida. 2022. "Molecular and Genetic Interactions between CCN2 and CCN3 behind Their Yin–Yang Collaboration" International Journal of Molecular Sciences 23, no. 11: 5887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115887