
Artemisinin Attenuates Amyloid-Induced Brain Inflammation and Memory Impairments by Modulating TLR4/NF-κB Signaling

 ,
,
Abstract
:1. Introduction
2. Results
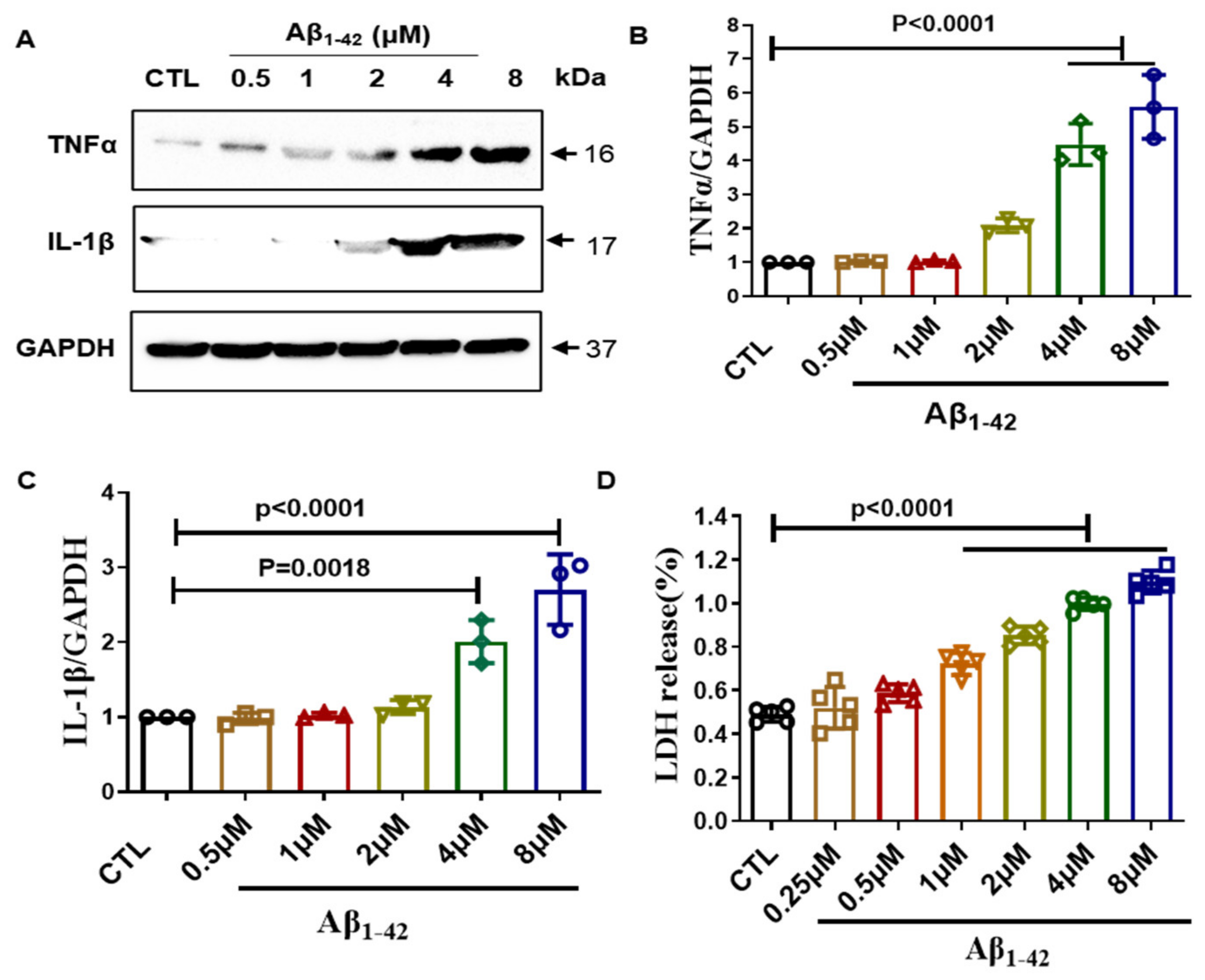
2.1. Establishment of Inflammation Model in BV2 Cells
2.2. Artemisinin Decreased the Production of ROS and iNOS in BV2 Cells
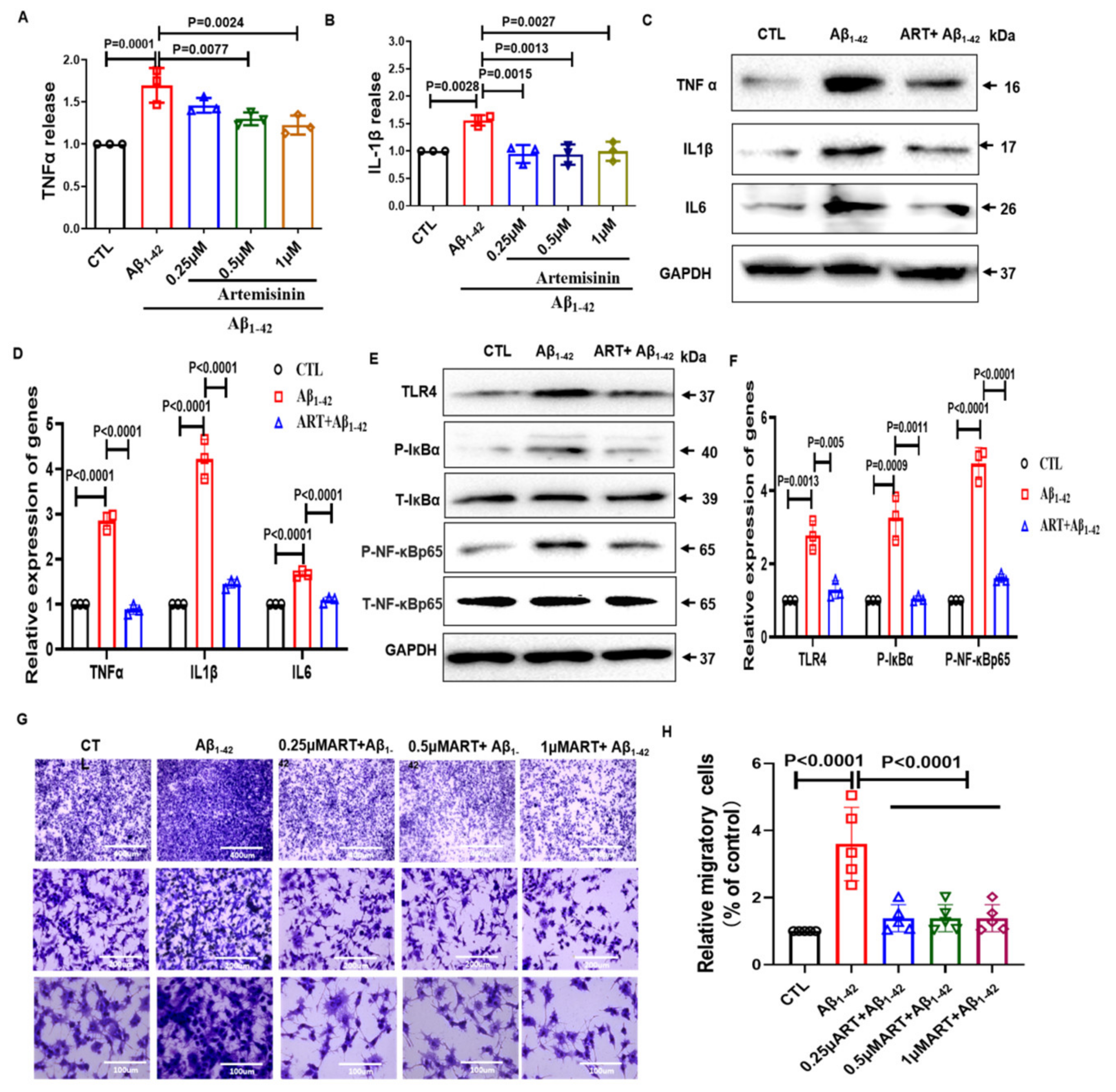
2.3. Artemisinin Reduced the Release of Inflammatory Factors and Inhibited the Migratory Ability of BV2 Cells
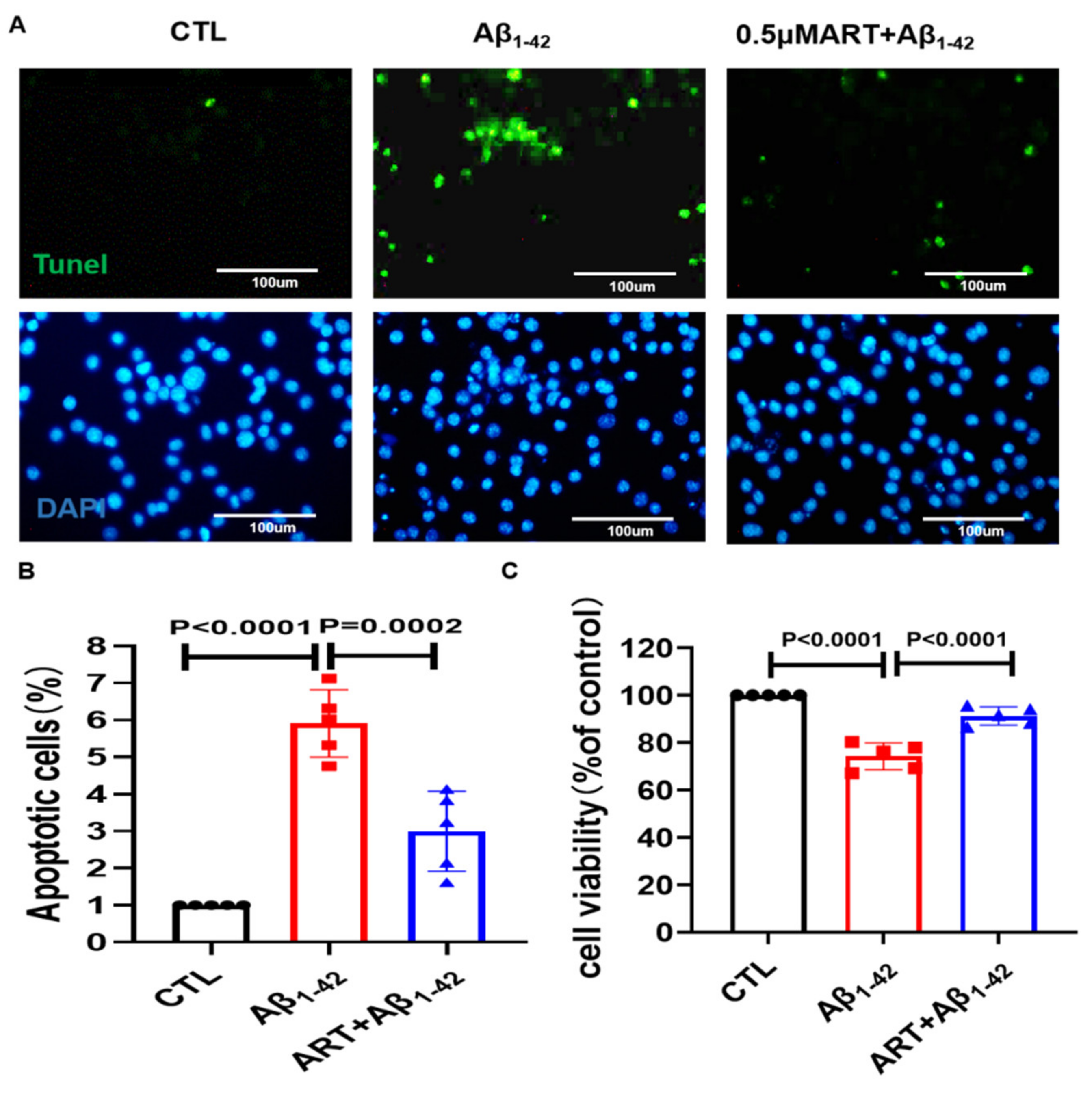
2.4. Microglia Conditioned Medium Regulates PC12 Cell Viability and Apoptosis
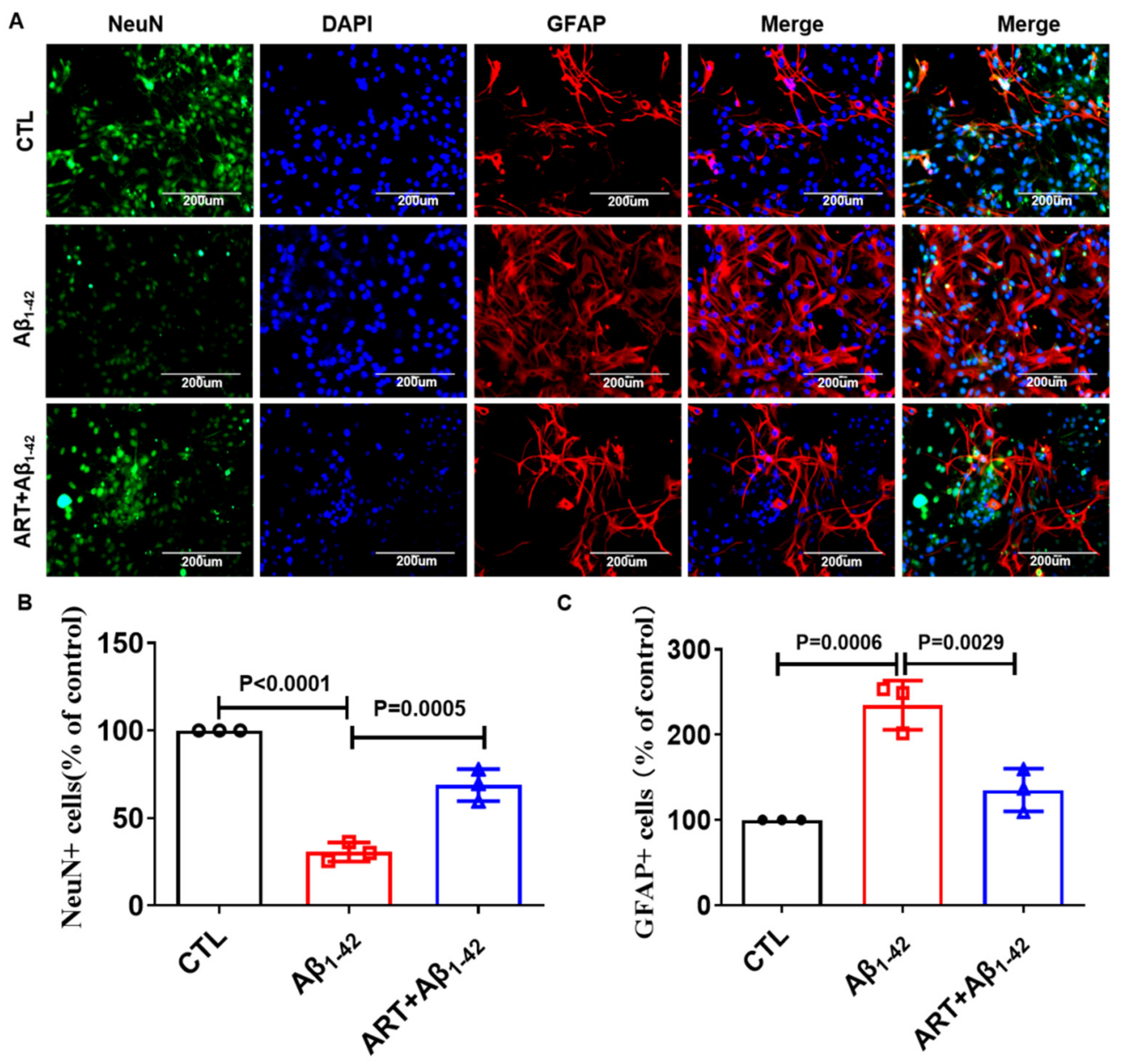
2.5. Artemisinin Reduced the Excessive Activation of Astrocytes in Primary Cultured Neurons
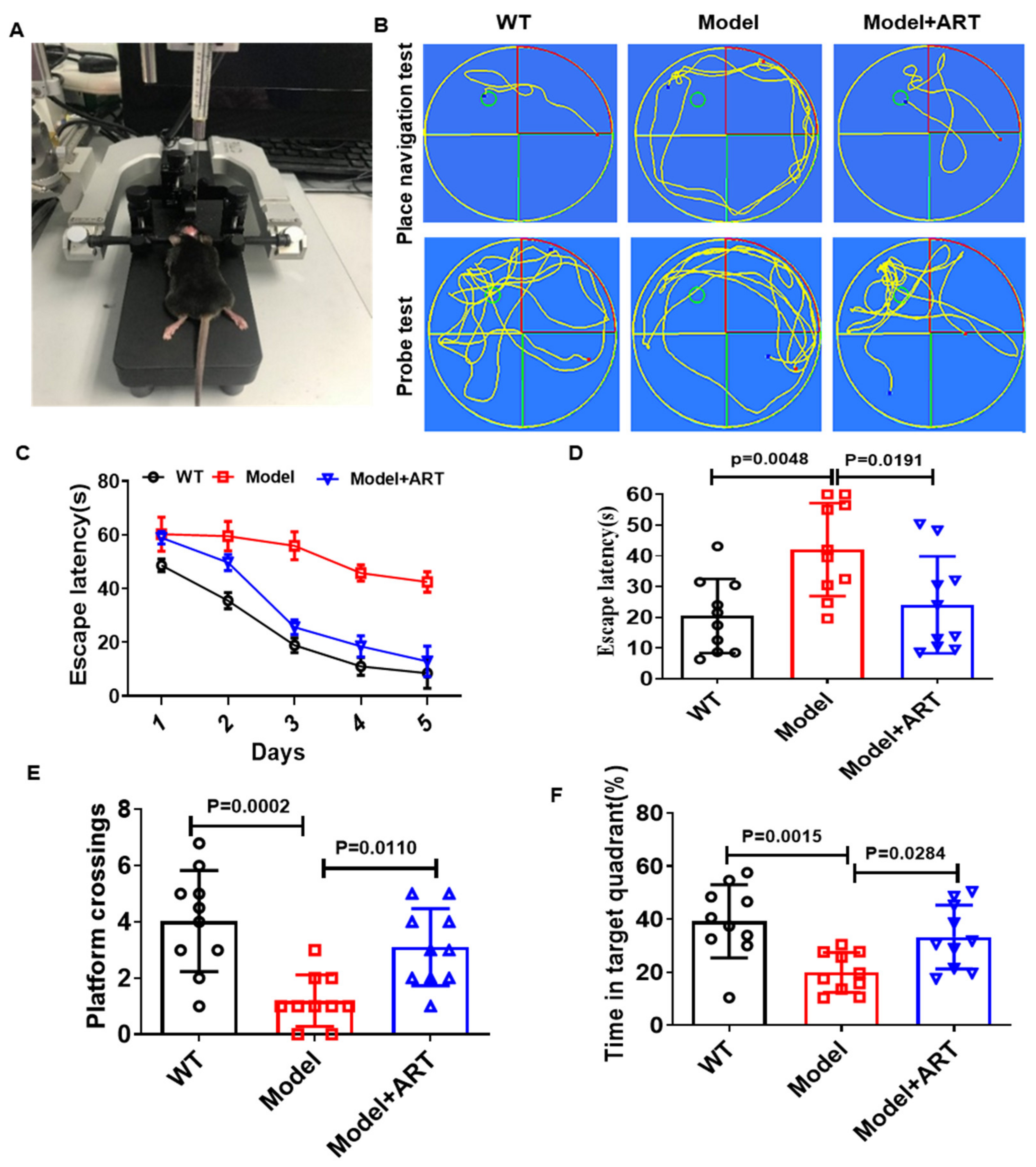
2.6. Artemisinin Improved the Cognitive Impairment of AD Model Mice
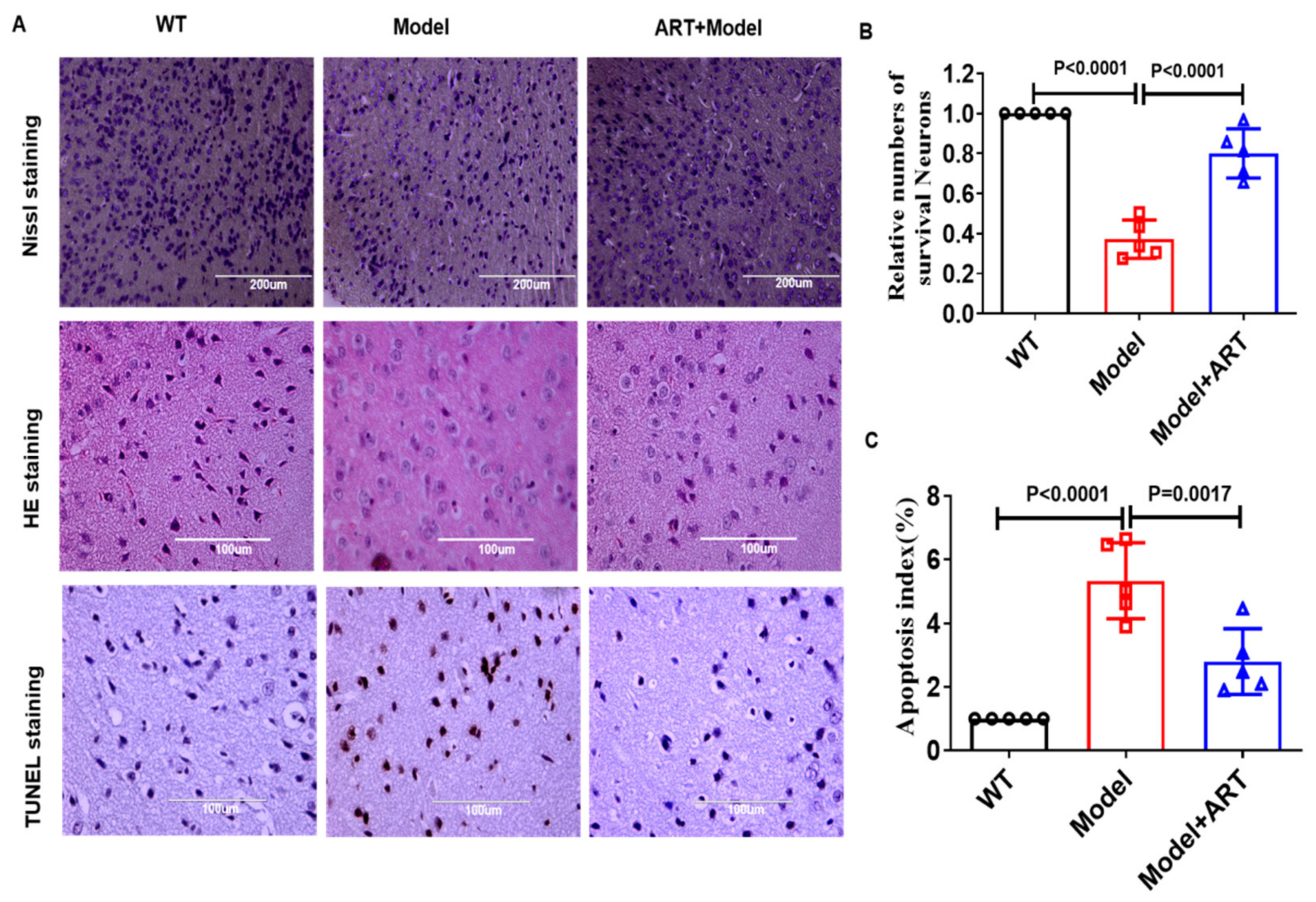
2.7. Artemisinin Reduced Neuronal Cell Damage in the AD Model Mice
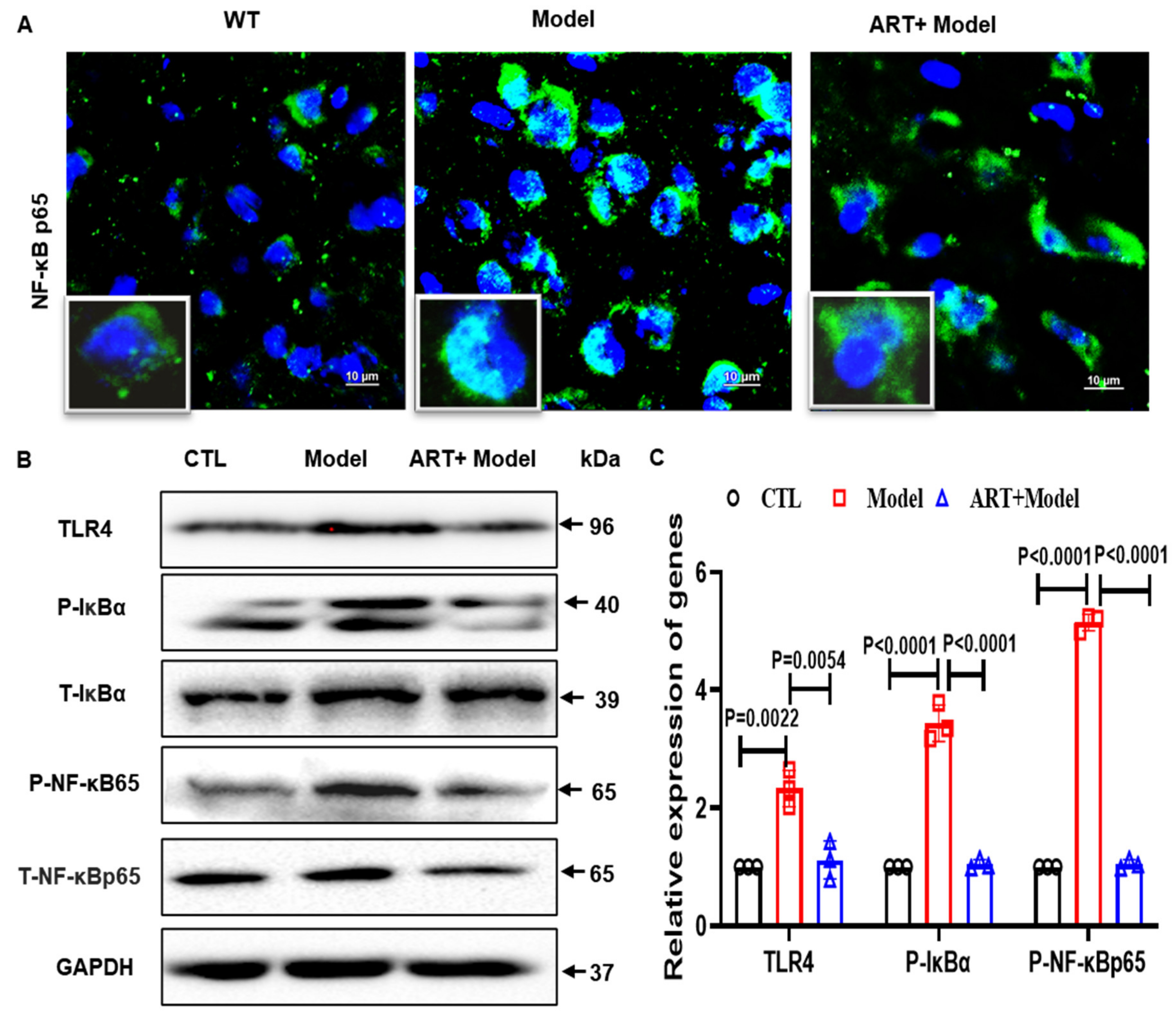
2.8. Artemisinin Reduced Neuroinflammation and Inhibited NF-κB Signaling in the AD Model Mice
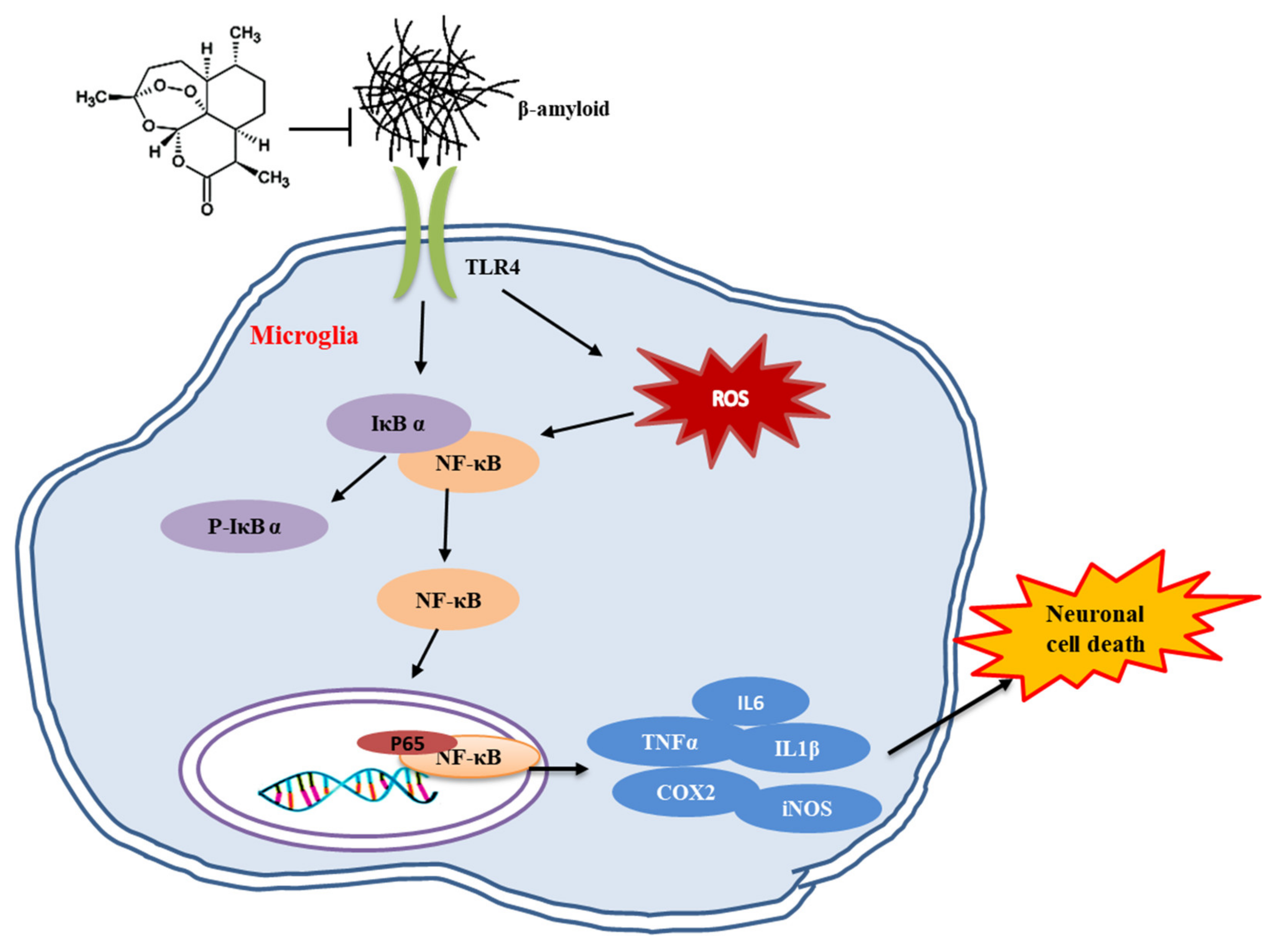
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Animal and Treatment
4.3. Stereotaxic Injection
4.4. Morris Water Maze (MWM)
4.5. Tissue Preparation
4.6. Immunohistochemistry and Immunofluorescence
4.7. Nissl Staining
4.8. BV2 Cell Line Culture
4.9. Primary Neuronal Cells
4.10. TUNEL Assay
4.11. Measurement of Intracellular ROS Levels
4.12. ELISA
4.13. Transwell Assay
4.14. Western Blot
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Li, L.; Chen, Z.; Zhong, X.; Huang, J.; Yu, Q.; Carlsson, C.; Okonkwo, O. Sweetening the process of biomarker discovery in Alzheimer’s disease: Development of improved chemical strategies for probing glycosylation patterns in AD. In Abstracts of Papers of The American Chemical Society; American Chemical Society: Washington, DC, USA, 2019. [Google Scholar]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243. [Google Scholar]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflamm. 2012, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [Green Version]
- Bates, T.W.; Kahle, K.M.; Stulz, R.M. Why do US firms hold so much more cash than they used to? J. Financ. 2009, 64, 1985–2021. [Google Scholar] [CrossRef] [Green Version]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1β, TNF-α and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflamm. 2010, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-F.; Niu, D.-B.; Xue, B.; Li, F.-Q.; Liu, X.-Y.; He, Q.-H.; Wang, X.-H.; Wang, X.-M. Triptolide inhibits TNF-α, IL-1β and NO production in primary microglial cultures. Neuroreport 2003, 14, 1091–1095. [Google Scholar]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Gan, L.; Ye, S.; Chu, A.; Anton, K.; Yi, S.; Vincent, V.A.; Von Schack, D.; Chin, D.; Murray, J.; Lohr, S. Identification of cathepsin B as a mediator of neuronal death induced by Aβ-activated microglial cells using a functional genomics approach. J. Biol. Chem. 2004, 279, 5565–5572. [Google Scholar] [CrossRef] [Green Version]
- Su, X.-Z.; Miller, L.H. The Discovery of Artemisinin and the Nobel Prize in Physiology or Medicine; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Chaturvedi, D.; Goswami, A.; Saikia, P.P.; Barua, N.C.; Rao, P.G. Artemisinin and its derivatives: A novel class of anti-malarial and anti-cancer agents. Chem. Soc. Rev. 2010, 39, 435–454. [Google Scholar] [CrossRef]
- Shi, C.; Li, H.; Yang, Y.; Hou, L. Anti-inflammatory and immunoregulatory functions of artemisinin and its derivatives. Mediat. Inflamm. 2015, 2015, 435713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Cheng, B.; Qian, H.; Ma, S.; Chen, Q. Identification of HSP90 as a direct target of artemisinin for its anti-inflammatory activity via quantitative chemical proteomics. Org. Biomol. Chem. 2019, 17, 6854–6859. [Google Scholar] [CrossRef]
- Zeng, Z.; Xu, J.; Zheng, W. Artemisinin protects PC12 cells against β-amyloid-induced apoptosis through activation of the ERK1/2 signaling pathway. Redox Biol. 2017, 12, 625–633. [Google Scholar] [CrossRef]
- Zhao, X.; Fang, J.; Li, S.; Gaur, U.; Xing, X.; Wang, H.; Zheng, W. Artemisinin attenuated hydrogen peroxide (H2O2)-induced oxidative injury in SH-SY5Y and hippocampal neurons via the activation of AMPK pathway. Int. J. Mol. Sci. 2019, 20, 2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Dong, H.; Li, N.; Zhang, S.; Sun, J.; Zhang, S.; Qian, Y. Activated brain mast cells contribute to postoperative cognitive dysfunction by evoking microglia activation and neuronal apoptosis. J. Neuroinflamm. 2016, 13, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schüffer, W.; et al. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer’s Disease. Cell. Physiol. Biochem. 2015, 20, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Filippis, L.D.; Peri, F. The Role of TLR4 in Neural Stem Cells–Mediated Neurogenesis and Neuroinflammation. In The Role of Toll-like Receptor 4 in Infectious and Non Infectious Inflammation; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J. Neuroinflamm. 2019, 16, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Song, X.; Li, M.; Wang, X.; Tao, Y.; Xiya, X.; Liu, H.; Zhao, Y.; Chang, D.; Sha, Q. The role of TLR4/NF-κB signaling pathway in activated microglia of rats with chronic high intraocular pressure and vitro scratch injury-induced microglia. Int. Immunopharmacol. 2020, 83, 106395. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, J.; Ruan, Z.; Tian, S.; Ma, Y.; Zhu, J.; Li, G. Intrahippocampal injection of Aβ1-42 inhibits neurogenesis and down-regulates IFN-γ and NF-κB expression in hippocampus of adult mouse brain. Amyloid 2013, 20, 13–20. [Google Scholar] [CrossRef]
- Lin, T.; Liu, G.A.; Perez, E.; Rainer, R.D.; Febo, M.; Cruz-Almeida, Y.; Ebner, N.C. Systemic inflammation mediates age-related cognitive deficits. Front. Aging Neurosci. 2018, 10, 236. [Google Scholar] [CrossRef] [Green Version]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol mitigates lipopolysaccharide-and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Buch, P.R.; Desai, I.; Balakrishnan, S. COX-2 activity and expression pattern during regenerative wound healing of tail in lizard Hemidactylus flaviviridis. Prostaglandins Other Lipid Mediat. 2018, 135, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Nakajima, K.; Kikuchi, Y.; Honda, S.; Ishikawa, M. Functional activation of cultured microglia by neurotrophins. J. Neurochem. 1997, 69, S4. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Imai, F.; Kanno, T.; Sawada, M. Preservation of neurotrophin expression in microglia that migrate into the gerbil’s brain across the blood–brain barrier. Neurosci. Lett. 2001, 312, 95–98. [Google Scholar] [CrossRef]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Hong, J.-S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wyss-Coray, T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat. Med. 2006, 12, 1005–1015. [Google Scholar]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, M.; Pandey, N.; Shukla, A.; Singh, S.; Singh, S.K. Multidimensional Roles of Microglial Cells in Neuroviral Infections. In The Biology of Glial Cells: Recent Advances; Springer: Singapore, 2022. [Google Scholar]
- Giacomo, T.; Leonardo, I.; Paola, C.S.; Luca, P.; Roberto, S.; Sandro, I.; Giuseppe, M.; Daniela, P. The combined effects of microglia activation and brain glucose hypometabolism in early-onset Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 50. [Google Scholar]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord.-Drug Targets Former. Curr. Drug Targets-CNS Neurol. Disord. 2010, 9, 156–167. [Google Scholar]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Lorne, E.; Dupont, H.; Abraham, E. Non-Septic Acute Lung Injury and Inflammation: Role of TLR4; Intensive Care Medicine. In Yearbook of Intensive Care and Emergency Medicine; Springer: Cham, Switzerland, 2009. [Google Scholar]
- Verstak, B.; Stack, J.; Ve, T.; Mangan, M.; Hjerrild, K.; Jeon, J.; Stahl, R.; Latz, E.; Gay, N.; Kobe, B. The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J. Leukoc. Biol. 2014, 96, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Guo, X.; You, Y.; Fu, R. Farrerol attenuates MPP + -induced inflammatory response by TLR4 signaling in a microglia cell line: Farrerol inhibits inflammation in microglia cells. Phytother. Res. 2019, 33, 1134–1141. [Google Scholar] [CrossRef]
- Chen, S.N.; Tan, Y.; Xiao, X.C.; Li, Q.; Wu, Q.; Peng, Y.Y.; Ren, J.; Dong, M.L. Deletion of TLR4 attenuates lipopolysaccharide-induced acute liver injury by inhibiting inflammation and apoptosis. Acta Pharmacol. Sin. 2021, 42, 1610–1619. [Google Scholar] [CrossRef]
- Bombardieri, M.; Barone, F.; Pittoni, V. Increased circulating levels and salivary gland expression of interleukin-18 in patients with Sjgren’s syndrome: Relationship with autoantibody production and lymphoid organization of the periductal inflammatory infiltrate. Arthritis Res. Ther 2004, 6, R447. [Google Scholar]
- Yang, Z.; Liang, Y.; Zhu, Y.; Li, C.; Zhang, L.; Zeng, X.; Zhong, R. Increased expression of Toll-like receptor 4 in peripheral blood leucocytes and serum levels of some cytokines in patients with ankylosing spondylitis. Clin. Exp. Immunol. 2007, 149, 48–55. [Google Scholar] [CrossRef]
- Yang, H.Z.; Wang, J.P.; Su, M.; Liu, H.Z.; Cui, B.; Yan, H.M.; Yan, J.; Zhe, L.; Hong, L.; Hua, F. TLR4 Activity Is Required in the Resolution of Pulmonary Inflammation and Fibrosis after Acute and Chronic Lung Injury. Am. J. Pathol. 2012, 180, 275–292. [Google Scholar] [CrossRef]
- Wajant, H. TRAIL and NFkappaB signaling—A complex relationship. Vitam. Horm. 2004, 67, 101. [Google Scholar]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jothishankar, B.; He, P.; Staufenbiel, M.; Li, R. Amyloid Precursor Protein Mutation Disrupts Reproductive Experience-Enhanced Normal Cognitive Development in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2013, 49, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohutnicka, M.; Czlonkowski, A.; Czlonkowska, A. Involvement of the immunological and inflammatory response in the pathogenesis of neurodegenerative and ischemic diseases. Med. Sci. Monit. 1998, 20, 42–47. [Google Scholar]
- Rossi, F.; Bianchini, E. Synergistic Induction of Nitric Oxide by β-Amyloid and Cytokines in Astrocytes. Biochem. Biophys. Res. Commun. 1996, 225, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Fan, Y.-C.; Wang, M.; Wang, D.; Li, X.-H. Atorvastatin attenuates the production of IL-1β, IL-6, and TNF-α in the hippocampus of an amyloid β1-42-induced rat model of Alzheimer’s disease. Clin. Interv. Aging 2013, 8, 103. [Google Scholar]
- White, J.A.; Manelli, A.M.; Holmberg, K.H.; Van Eldik, L.J.; LaDu, M.J. Differential effects of oligomeric and fibrillar amyloid-β1–42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005, 18, 459–465. [Google Scholar] [CrossRef]
- Bodegraven, E.; Sluijs, J.A.; Tan, K.A.; Robe, P.; Hol, E.M. New GFAP splice isoform (GFAPμ) differentially expressed in glioma translates into 21 kDa N-terminal GFAP protein. Cold Spring Harb. Lab. 2020, 35, e21389. [Google Scholar]
- Yokel, R.A.; O’Callaghan, J. An aluminum-induced increase in GFAP is attenuated by some chelators. Neurotoxicol. Teratol. 1998, 20, 55–60. [Google Scholar] [CrossRef]
- Facchinetti, R.; Bronzuoli, M.R.; Scuderi, C. An animal model of Alzheimer disease based on the intrahippocampal injection of amyloid β-peptide (1–42). In Neurotrophic Factors; Springer: Berlin/Heidelberg, Germany, 2018; pp. 343–352. [Google Scholar]
- Vorhees, C.; Williams, M. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Callaghan, D.; Wodzinska, J.; Xu, J.; Premyslova, M.; Liu, Q.Y.; Connelly, J.; Zhang, W. Biochemical and behavioral characterization of the double transgenic mouse model (APPswe/PS1dE9) of Alzheimer’s disease. Neurosci. Bull. 2011, 27, 221. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, S.; Gaur, U.; Zheng, W. Artemisinin Improved Neuronal Functions in Alzheimer’s Disease Animal Model 3xtg Mice and Neuronal Cells via Stimulating the ERK/CREB Signaling Pathway. Aging Dis. 2020, 11, 801. [Google Scholar] [CrossRef] [PubMed]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Zhao, X.; Zeng, Z.; Gaur, U.; Fang, J.; Peng, T.; Li, S.; Zheng, W. Metformin protects PC12 cells and hippocampal neurons from H2O2-induced oxidative damage through activation of AMPK pathway. J. Cell. Physiol. 2019, 234, 16619–16629. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Cat. NO | Company | Dilution |
|---|---|---|---|
| TLR4 | 35463 | SAB | WB:1:1000 |
| NF-κB p65 | 4764 | CST | WB:1:1000 |
| P-NF-κB p65 | 3033 | CST | WB:1:1000 |
| IκBα | 4814 | CST | WB:1:1000 |
| P- IκBα | 2859 | CST | WB:1:1000 |
| GFAP (GA5) | 3670 | CST | WB:1:1000/IF:1:200 |
| TNF-α | 41504 | SAB | WB:1:1000 |
| IL-6 | 32064 | SAB | WB:1:1000 |
| IL-1β (3A6) | 12242s | CST | WB:1:1000 |
| Cox2 | 21679 | SAB | WB:1:1000 |
| iNOS | 13120 | CST | WB:1:1000 |
| Iba1 | 49668 | SAB | WB:1:500/IF:1:1000 |
| Anti-rabbit IgG HRP | 7074 | CST | WB:1:2000 |
| Alexa Fluor® 594 | 8889 | CST | IF:1:500 |
| Alexa Fluor® 488 | 4412 | CST | IF:1:500 |
| Sequence (Three-Letter Code) | H-Asp-Ala-Glu-Phe-Arg-His-Asp-Ser-Gly-Tyr-Glu-Val-His-His-Gln-Lys-Leu-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met-Val-Gly-Gly-Val-Val-Ile-Ala-OH |
| One Letter Code | DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Huang, X.; Yang, C.; Jiang, Y.; Zhou, W.; Zheng, W. Artemisinin Attenuates Amyloid-Induced Brain Inflammation and Memory Impairments by Modulating TLR4/NF-κB Signaling. Int. J. Mol. Sci. 2022, 23, 6354. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116354
Zhao X, Huang X, Yang C, Jiang Y, Zhou W, Zheng W. Artemisinin Attenuates Amyloid-Induced Brain Inflammation and Memory Impairments by Modulating TLR4/NF-κB Signaling. International Journal of Molecular Sciences. 2022; 23(11):6354. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116354
Chicago/Turabian StyleZhao, Xia, Xiaosu Huang, Chao Yang, Yizhou Jiang, Wenshu Zhou, and Wenhua Zheng. 2022. "Artemisinin Attenuates Amyloid-Induced Brain Inflammation and Memory Impairments by Modulating TLR4/NF-κB Signaling" International Journal of Molecular Sciences 23, no. 11: 6354. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116354