
Accumulation of Deleterious Effects in Gastric Epithelial Cells and Vascular Endothelial Cells In Vitro in the Milieu of Helicobacter pylori Components, 7-Ketocholesterol and Acetylsalicylic Acid

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Upregulation of ROS Production in AGS or HUVEC Cell Cultures Carried out in the Presence of H. pylori Components Alone or Simultaneously with ASA and 7-kCh
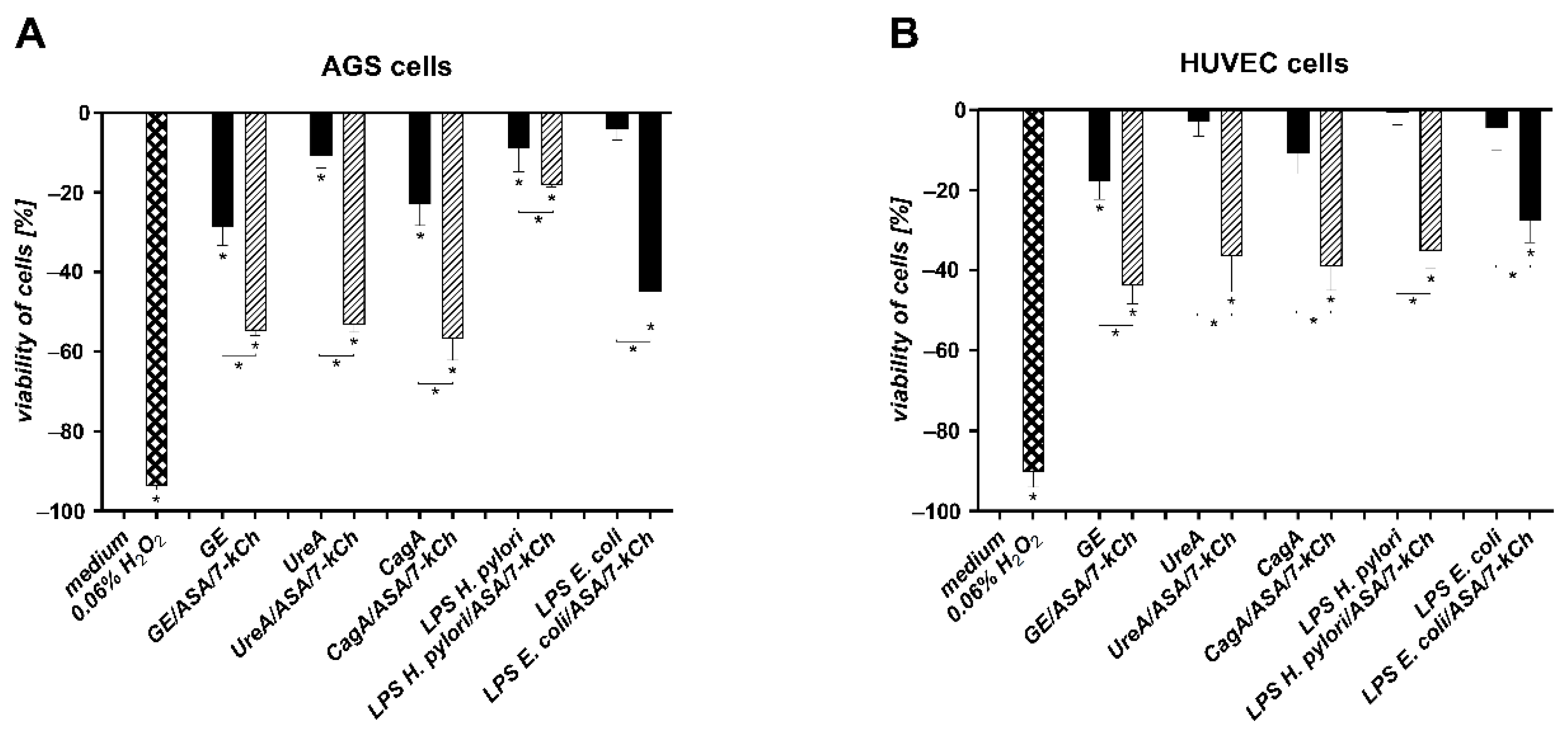
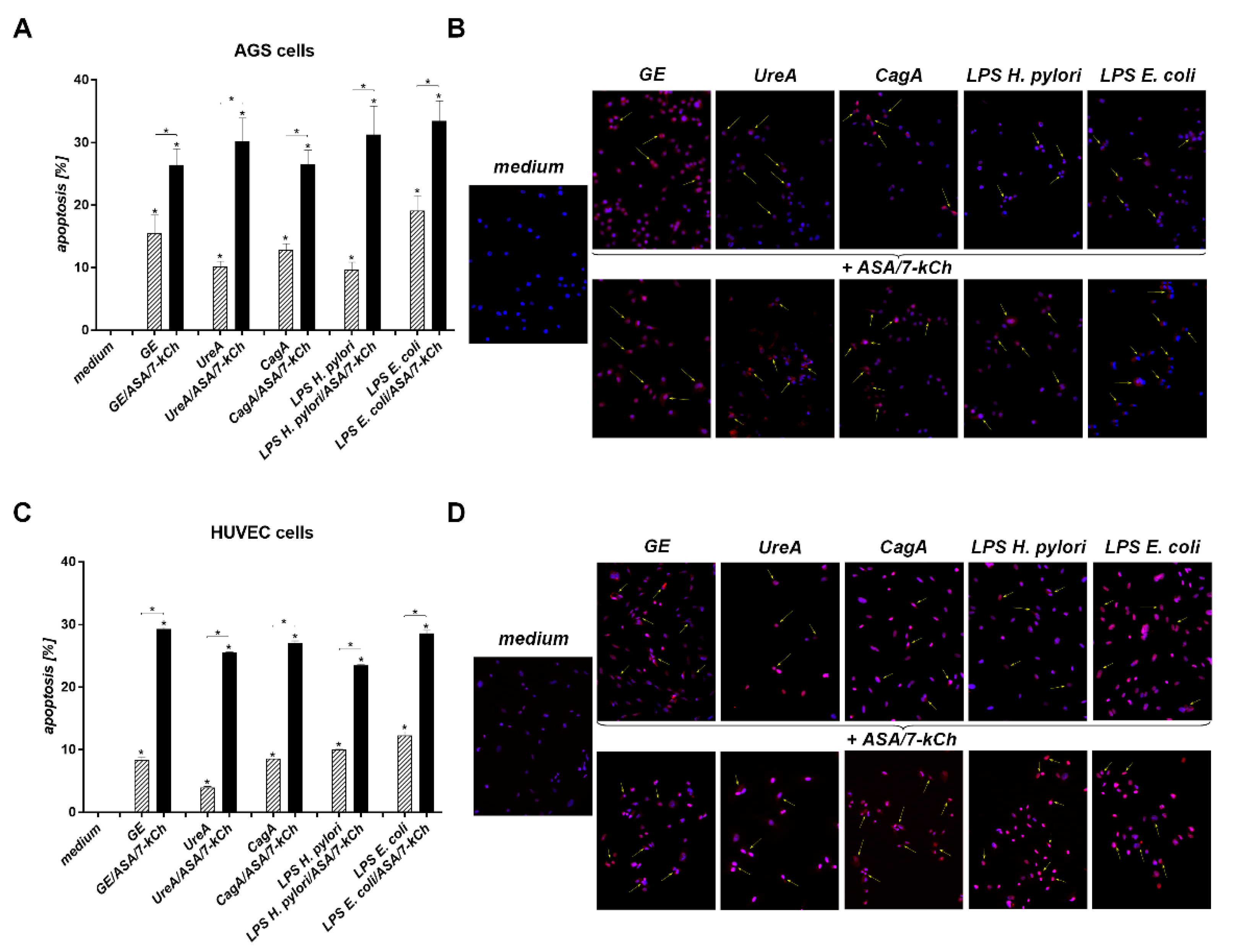
2.2. The Correlation between Diminished Viability of AGS or HUVEC Cells and an Increased Number of Cells Undergoing Apoptosis in Cell Cultures Carried out with H. pylori Components Alone or in the Presence of ASA and 7-kCh
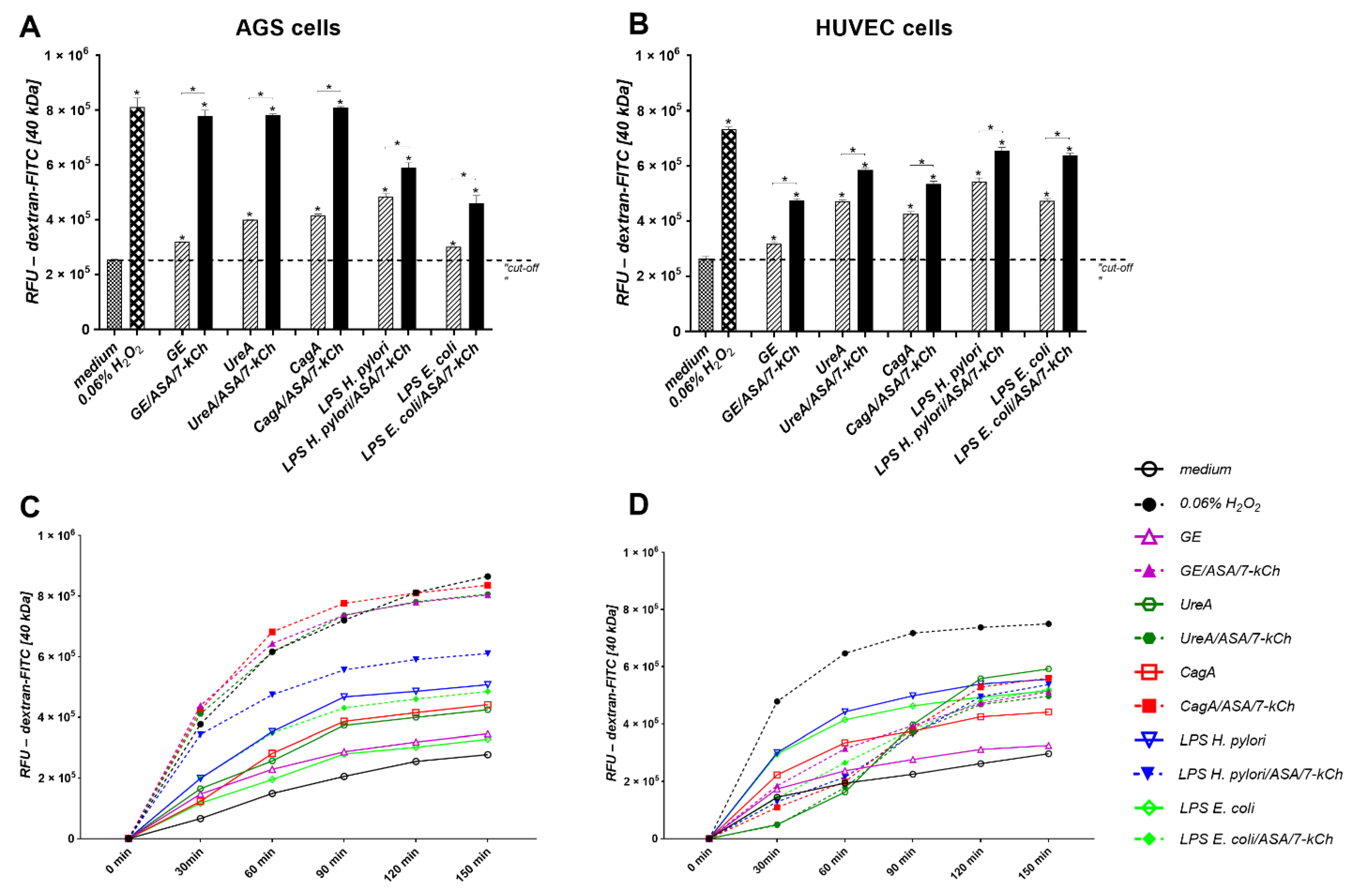
2.3. Disintegration of AGS or HUVEC Cell Monolayers Treated with H. pylori Components Alone or Simultaneously with ASA and 7-kCh
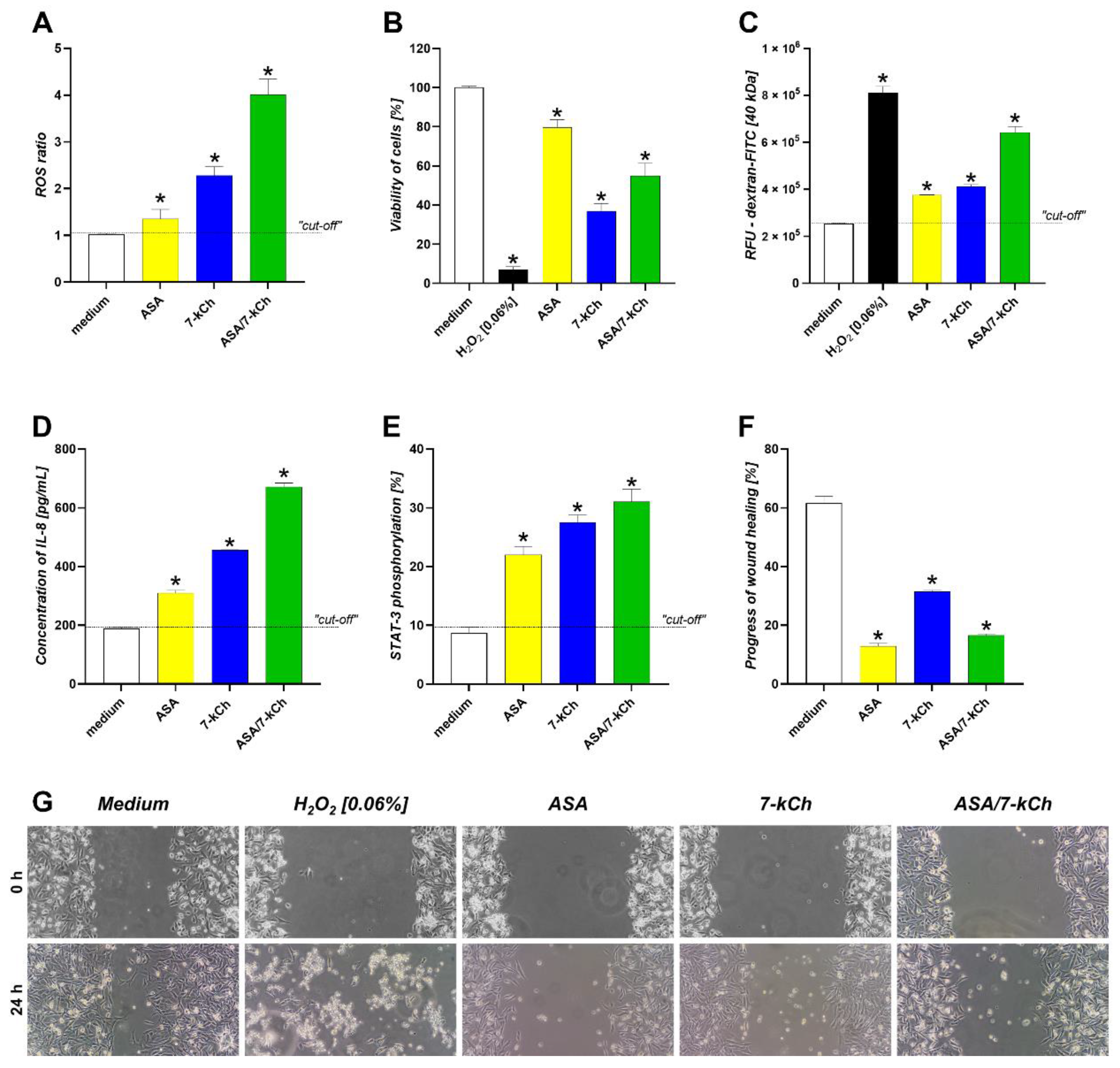
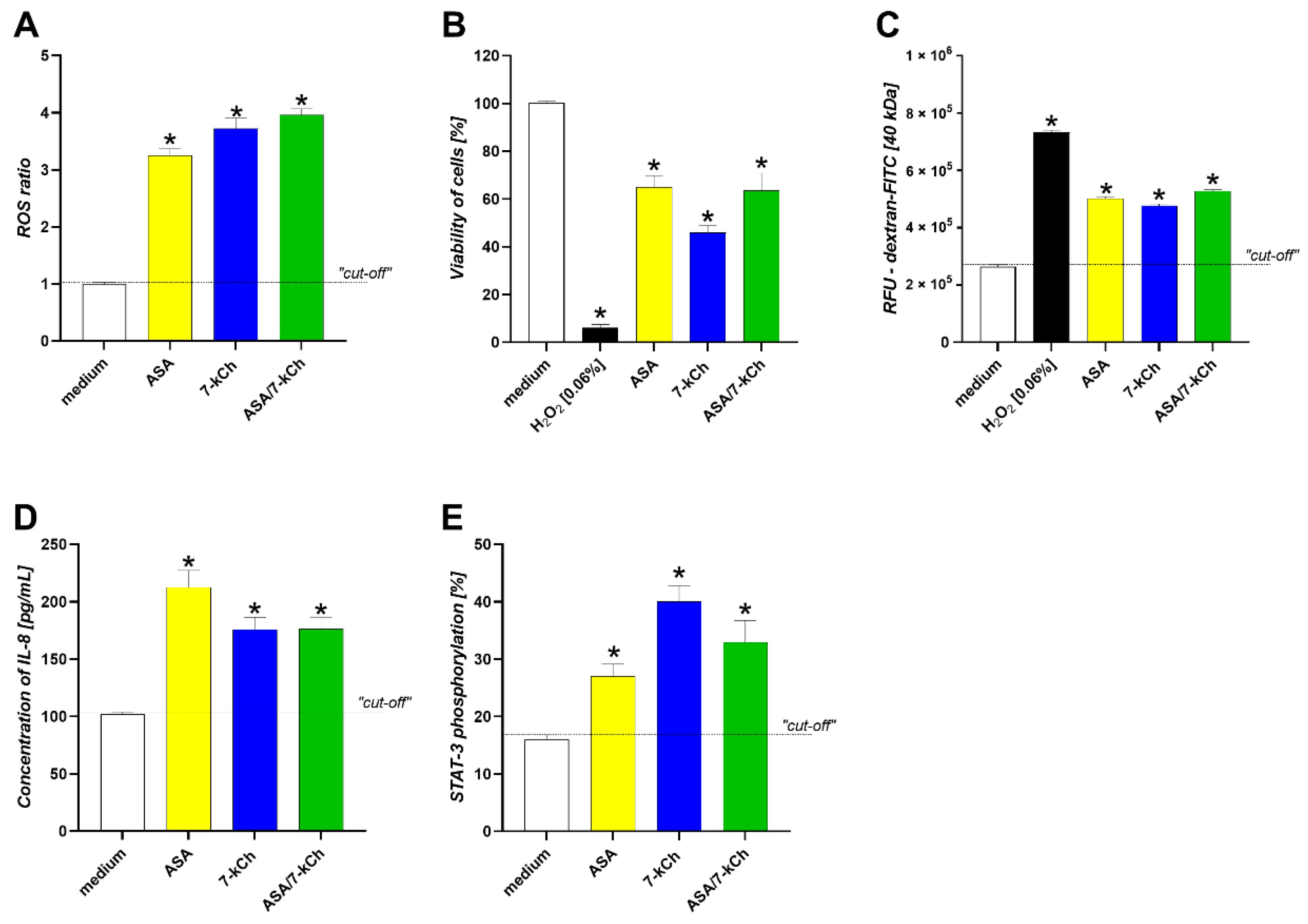
2.4. The Infuence of ASA or 7-kCh Alone on ROS Production, Cell Viability and Integrity, IL-8 Secretion, STAT3 Activation and Wound Healing in AGS or HUVEC Cell Models
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Cell Stimulation
4.3. Assessment of Reactive Oxygen Species (ROS)
4.4. Cell Viability Assay
4.5. Apoptosis
4.6. Cell Barrier Integrity—Paracelullar Flux Assay
4.7. Wound Healing Assay
4.8. STAT3 Activation
4.9. IL-8 Production
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGS | gastric adenocarcinoma epithelial cells |
| ASA | acetylsalicylic acid |
| ATCC | American Type Culture Collection |
| Bax | BCL2 associated x, apoptosis regulator |
| CagA | cytotoxin associated gene A antigen |
| CCUG | Culture Collection University of Gothenburg |
| CHD | coronary heart disease |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide salt |
| c-Myc | Myc-proto-oncogene protein |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DHE | dihydroetidine |
| EDTA | ethylenediaminetetraacetic acid |
| EGM-2 | endothelial growth medium-2 |
| EU | European Unit |
| FBS | fetal bovine serum |
| FITC | fluorescein isothiocyanate |
| GE | glycine acid extract |
| GMP | guanosine monophosphate |
| HspB | heat shock protein B |
| HUVEC | human umbilical vein endothelial cells |
| 7-kCh | 7-ketocholesterol |
| LDL | low density lipoprotein |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MMP-9 | metalloproteinase 9 |
| NFκB | nuclear factor kappa B |
| OipA | outer inflammatory protein A |
| oxLDL | oxidized low density lipoprotein |
| PARP | poly (ADP-ribose) polymerase |
| PBS | phosphate buffered saline |
| PCR | polymerase chain reaction |
| rCagA | recombinant cytotoxin associated gene A antigen |
| RFU | relative fluorescein unit |
| RPMI | Rosswell Park Memory Institute cell culture medium |
| ROS | reactive oxygen species |
| SD | standard deviation |
| SDS-PAGE | sodium dodecyl sulphate polyacrylamide gel electrophoresis |
| STAT3 | signal transducer and activator of transcription 3 |
| TLR-4 | Toll-like receptor 4 |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labelling |
| UreA | subunit A of urease |
| VacA | vacuolating cytotoxin A |
References
- Caladrini, C.A.; Ribeiro, A.C.; Gonnelli, A.C.; Ota-Tsuzuki, C.; Rangel, L.P.; Saba-Chujfi, E.; Mayer, M.P. Microbial composition of atherosclerotic plaques. Oral Dis. 2014, 20, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, J.S. Multiple infectious agents and the origins of atherosclerotic coronary artery disease. Front. Cardiovasc. Med. 2016, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Mendall, M.A.; Goggin, P.M.; Molineaux, N.; Levy, J.; Toosy, T.; Strachan, D.; Camm, A.J.; Northfield, T.C. Relation of Helicobacter pylori infection and coronary heart disease. Br. Heart J. 1994, 71, 437–439. [Google Scholar] [CrossRef] [PubMed]
- Longo-Mbenza, B.; Nsenga, J.N.; Mokondjimobe, E.; Gombet, T.; Assori, I.N.; Ibara, J.R.; Ellenga-Mbolla, B.; Vangu, D.N.; Fuele, S.M. Helicobacter pylori infection is identified as a cardiovascular risk factor in Central Africans. Vasc. Health Risk Manag. 2012, 8, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Chmiela, M.; Gajewski, A.; Rudnicka, K. Helicobacter pylori vs coronary heart disease—Searching for connections. World J Cardiol. 2015, 7, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Testerman, T.L.; Semino-Mora, C.; Cann, J.A.; Qiang, B.; Peña, E.A.; Liu, H.; Olsen, C.H.; Chen, H.; Appt, S.E.; Kaplan, J.R.; et al. Both diet and Helicobacter pylori infection contribute to atherosclerosis in pre- and postmenopausal cynomolgus monkeys. PLoS ONE 2019, 14, e0222001. [Google Scholar] [CrossRef]
- Krupa, A.; Gonciarz, W.; Rusek-Wala, P.; Rechciński, T.; Gajewski, A.; Samsel, Z.; Dziuba, A.; Śmiech, A.; Chmiela, M. Helicobacter pylori infection acts synergistically with a high-fat diet in the development of a proinflammatory and potentially proatherogenic endothelial cell environment in an experimental model. Int. J. Mol. Sci. 2021, 22, 3394. [Google Scholar] [CrossRef]
- Caron, T.J.; Scott, K.E.; Fox, J.G.; Hagen, S.J. Tight junction disruption: Helicobacter pylori and dysregulation of the gastric mucosal barrier. World J. Gastroenterol. 2015, 21, 11411–11427. [Google Scholar] [CrossRef] [PubMed]
- Mnich, E.; Kowalewicz-Kulbat, M.; Sicińska, P.; Hinc, K.; Obuchowski, M.; Gajewski, A.; Moran, A.P.; Chmiela, M. Impact of Helicobacter pylori on the healing process of the gastric barrier. World J. Gastroenterol. 2016, 22, 7536–7558. [Google Scholar] [CrossRef]
- Gonciarz, W.; Krupa, A.; Hinc, K.; Obuchowski, M.; Moran, A.P.; Gajewski, A.; Chmiela, M. The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts. PLoS ONE 2019, 14, e0220636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentino, M.; Ding, H.; Blanchard, T.G.; Czinn, S.J.; Sztein, M.B.; Fasano, A. Helicobacter pylori-induced disruption of monolayer permeability and proinflammatory cytokine secretion in polarized human gastric epithelial cells. Infect. Immun. 2013, 81, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Bravo, D.; Hoare, A.; Soto, C.; Valenzuela, M.A.; Quest, A.F. Helicobacter pylori in human health and disease: Mechanisms for local gastric and systemic effects. World J. Gastroenterol. 2018, 24, 3071–3089. [Google Scholar] [CrossRef]
- Rudnicka, W.; Czkwianianc, E.; Płaneta-Małecka, I.; Jurkiewicz, M.; Wiśniewska, M.; Cieslikowski, T.; Rózalska, B.; Wadström, T.; Chmiela, M. A potential double role of anti-LewisX antibodies in Helicobacter pylori-associated gastroduodenal diseases. FEMS Immunol. Med. Microbiol. 2001, 30, 121–125. [Google Scholar] [CrossRef]
- Matusiak, A.; Chałubiński, M.; Broncel, M.; Rechciński, T.; Rudnicka, K.; Miszczyk, E.; Walencka, M.; Strapagiel, D.; Gajewski, A.; Chmiela, M. Putative consequences of exposure to Helicobacter pylori infection in patients with coronary heart disease in terms of humoral immune response and inflammation. Arch. Med. Sci. 2016, 12, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonciarz, W.; Matusiak, A.; Rudnicka, K.; Rechcinski, T.; Chałubiński, M.; Czkwianuianc, E.; Broncel, M.; Gajewski, A.; Chmiela, M. Autoantibodies to a specific peptide epitope of human Hsp60 (ATVLA) with homology to Helicobacter pylori HspB in H. pylori-related patients. APMIS 2019, 127, 139–149. [Google Scholar] [CrossRef]
- Kutuk, O.; Basaga, H. Inflammation meets oxidation: NK-κB as mediator of initial lesion development in atherosclerosis. Trends Mol. Med. 2003, 9, 549–557. [Google Scholar] [CrossRef]
- Brown, A.J.; Leong, S.I.; Dean, R.T.; Jessup, W. 7-hydroperoxycholesterol and its products in oxidized low density lipoprotein and human atherosclerotic plaque. J. Lipid Res. 1997, 38, 1730–1745. [Google Scholar] [CrossRef]
- Tani, M.; Kamata, Y.; Deushi, M.; Osaka, M.; Yoshida, M. 7-ketocholesterol enhances leukocyte adhesion to endothelial cells via p38MAPK pathway. PLoS ONE 2018, 13, e0200499. [Google Scholar] [CrossRef]
- Lara-Guzmán, O.J.; Gil-Izquierdo, A.; Medina, S.; Osorio, E.; Álvarez-Quintero, R.; Zuluaga, N.; Oger, C.; Galano, J.M.; Durand, T.; Muñoz-Durango, K. Oxidized LDL triggers changes in oxidative stress and inflammatory biomarkers in human macrophages. Redox Biol. 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Yen, M.H.; Yen, C.H.; Lau, Y.T. Oxidized low density lipoprotein induces apoptosis via generation of reactive oxygen species in vascular smooth muscle cells. Cardivasc. Res. 2001, 49, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthe´le´my, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef]
- Wilterdink, J.; Bendixen, B.; Adams, H.P., Jr.; Woolson, R.F.; Clarke, W.R.; Hansen, M.D. Effect of prior aspirin use on stroke severity in the trial of Org 10172 in acute stroke treatment (TOAST). Stroke 2001, 32, 2836–2840. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.; Pignone, M.; Phillips, C.; Mulrow, C. Aspirin for the primary prevention of cardiovascular events: A summary of the evidence for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2002, 136, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, A.; Chang, D.K.; Ricciardiello, L.; Gasche, C.; Boland, C.R. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin. Cancer Res. 2003, 9, 383–390. [Google Scholar]
- Raza, H.; John, A.; Benedict, S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. Eur. J. Pharmacol. 2011, 668, 15–24. [Google Scholar] [CrossRef]
- Tadayuki, O.; Hiroto, M.; Takashi, J. Aspirin induces gastric epithelial barrier dysfunction by activating p38 MAPK via claudin-7. Am. J. Physiol. Cell Physiol. 2008, 295, C800–C806. [Google Scholar] [CrossRef] [Green Version]
- Castellsague, J.; Riera-Guardia, N.; Calingaert, B.; Varas-Lorenzo, C.; Fourrier-Reglat, A.; Nicotra, F.; Sturkenboom, M.; Perez-Gutthann, S. Individual NSAIDs and upper gastrointestinal complications: A systematic review and meta-analysis of observational studies (the SOS project). Drug Safety 2012, 35, 1127–1146. [Google Scholar] [CrossRef]
- Rechcinski, T.; Chmiela, M.; Małecka-Panas, E.; Płaneta-Małecka, I.; Rudnicka, W. Serological indicators of Helicobacter pylori in adult dyspeptic patients and healthy blood donors. Microbiol. Immuno. 1997, 41, 387–393. [Google Scholar] [CrossRef]
- Hinc, K.; Isticato, R.; Dembek, M.; Karczewska, J.; Iwanicki, A.; Peszyńska-Sularz, G.; De Felice, M.; Obuchowski, M.; Ricca, E. Expression and display of UreA of Helicobacter acinonychis on the surface of Bacillus subtilis spores. Microb. Cell Factories 2010, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petrcca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N.; et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 579–595. [Google Scholar] [CrossRef] [Green Version]
- Moran, A.P.; Helander, I.M.; Kosunen, T.U. Compositional analysis of Helicobacter pylori rough-form lipopolysaccharides. J. Bacteriol. 1992, 174, 1370–1377. [Google Scholar] [CrossRef] [Green Version]
- Wojtala, A.; Bonora, M.; Malinska, D.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Methods to monitor ROS production by fluorescence microscopy and fluorymetry. Meth. Enzymol. 2014, 524, 243–262. [Google Scholar] [CrossRef]
- Kowalski, M.; Konturek, P.C.; Pieniazek, P.; Karczewska, E.; Kluczka, A.; Grove, R.; Kranig, W.; Nasseri, R.; Thale, J.; Hahn, E.G.; et al. Prevalence of Helicobacter pylori infection in coronary lumen reduction after percutaneous coronary angioplasty. Dig. Liver Dis. 2001, 33, 222–229. [Google Scholar] [CrossRef]
- Valgimigli, M.; Frigoli, E.; Heg, D.; Tijssen, J.; Jüni, P.; Vranckx, P.; Ozaki, Y.; Morice, M.C.; Chevalier, B.; Onuma, Y.; et al. Dual Antiplatelet Therapy after PCI in Patients at High Bleeding Risk. N. Engl. J. Med. 2021, 385, 1643–1655. [Google Scholar] [CrossRef]
- Kelly, P.J.; Morrow, J.D.; Ning, M.M.; Koroshetz, W.; Lo, E.H.; Terry, E.; Milne, G.L.; Hubbard, J.; Lee, H.; Stevenson, E.; et al. Oxidative stress and matrix metalloproteinase-9 in acute ischemic stroke: The Biomarker Evaluation for Antioxidant Therapies in Stroke (BEAT-Stroke) study. Stroke 2008, 39, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Fadeel, B.; Xue, D.; Kagan, V. Programmed cell clearance: Molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem. Biophys. Res. Commun. 2010, 396, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging 2012, 4, 330–349. [Google Scholar] [CrossRef]
- Teymournejad, O.; Mobarez, A.; Hassan, Z.; Abadi, T.B. Binding of the Helicobacter pylori OipA causes apoptosis of host cells via modulation of Bax/Bcl-2 level. Sci. Rep. 2017, 7, 8036–8044. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Tian, Z.; Yao, S.; Yu, Y.; Zhang, C.; Li, X.; Mao, T.; Jing, X.; Ding, X.; Yang, R. High-fat diet-induced obesity upregulates the expression of lymphoid chemokines and promotes the formation of gastric lymphoid follicles after Helicobacter suis infection. Pathog. Dis. 2017, 30, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Kaiserling, E.; Heinle, H.; Itabe, H.; Takano, T.; Remmele, W. Lipid islands in human gastric mucosa: Morphological and immunohistochemical findings. Gastroenterology 1996, 110, 369–374. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Redox signaling in cardiovascular health and disease. Free Radic Biol Med. 2013, 61, 5323–5344. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Xia, Y.; Luo, X.; Chen, S.; Li, B.; Ye, Z.; Chen, S.; Mao, L.; Jin, H.; Li, Y.; et al. Exosomal CagA derived from Helicobacter pylori-infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis. J. Mol. Cell Cardiol. 2019, 135, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Kiss, L.; Chen, M.; Gero, D.; Módis, K.; Lacza, Z.; Szabó, C. Effects of 7-ketocholesterol on the activity of endothelial poly(ADP-ribose) polymerase and on endothelium-dependent relaxant function. Int. J. Mol. Med. 2006, 18, 1113–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbas, H.S.; Suleymanlar, I.; Kemaloglu, D.; Koc, S.; Davran, F.; Demir, I.; Suleymanlar, G. The assessment of carotid intima media thickness and serum paraoxygenase-1 activity in Helicobacter pylori positive subjects. Lipds Health Dis. 2010, 9, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.A.; Lu, W.; Ogasawara, M.A.; Rivera-Del Valle, N.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Kacprzak, D.; Pawliczak, R. Does aspirin-induced oxidative stress cause asthma exacerbation? Arch. Med. Sci. 2015, 3, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escaned, J.; Cao, D.; Baber, U.; Nicolas, J.; Sartori, S.; Zhang, Z.; Dangas, G.; Angiolillo, D.J.; Briguori, C.; Cohen, D.J.; et al. Ticagrelor monotherapy in patients at high bleeding risk undergoing percutaneous coronary intervention: TWILIGHT-HBR. Eur. Heart J. 2021, 42, 4624–4634. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liao, Z.; Li, Y.; Zhao, X.; Ma, S.; Bao, D.; Qiu, M.; Deng, J.; Wang, J.; Qu, P.; et al. Magnetically controlled capsule endoscopy for assessment of antiplatelet therapy-induced gastrointestinal injury. J. Am. Coll. Cardiol. 2022, 79, 116–128. [Google Scholar] [CrossRef]
- Mladenova, I. Helicobacter pylori and cardiovascular disease: Update 2019. Minerva. Cardioangiol. 2019, 65, 425–432. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajewski, A.Ł.; Gawrysiak, M.; Krupa, A.; Rechciński, T.; Chałubiński, M.; Gonciarz, W.; Chmiela, M. Accumulation of Deleterious Effects in Gastric Epithelial Cells and Vascular Endothelial Cells In Vitro in the Milieu of Helicobacter pylori Components, 7-Ketocholesterol and Acetylsalicylic Acid. Int. J. Mol. Sci. 2022, 23, 6355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116355
Gajewski AŁ, Gawrysiak M, Krupa A, Rechciński T, Chałubiński M, Gonciarz W, Chmiela M. Accumulation of Deleterious Effects in Gastric Epithelial Cells and Vascular Endothelial Cells In Vitro in the Milieu of Helicobacter pylori Components, 7-Ketocholesterol and Acetylsalicylic Acid. International Journal of Molecular Sciences. 2022; 23(11):6355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116355
Chicago/Turabian StyleGajewski, Adrian Ł., Mateusz Gawrysiak, Agnieszka Krupa, Tomasz Rechciński, Maciej Chałubiński, Weronika Gonciarz, and Magdalena Chmiela. 2022. "Accumulation of Deleterious Effects in Gastric Epithelial Cells and Vascular Endothelial Cells In Vitro in the Milieu of Helicobacter pylori Components, 7-Ketocholesterol and Acetylsalicylic Acid" International Journal of Molecular Sciences 23, no. 11: 6355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116355