1. Introduction
Cyclic guanosine monophosphate (cGMP) is a second messenger involved in the regulation of many physiological processes including cardiovascular homeostasis, smooth muscle tone, blood pressure, platelet aggregation, memory, learning, and sensory transduction [
1,
2,
3]. cGMP is synthesised from guanosine triphosphate (GTP) by two forms of guanylate cyclase (GC): soluble GCs (sGCs), which are cytosolic enzymes activated by nitric oxide (NO) and carbon monoxide [
4]; and particulate GCs (pGCs), which are membrane receptors activated by natriuretic peptides and some intestinal peptides [
5]. cGMP signals via protein kinase G (PKG) and cyclic nucleotide-gated ion channels, and also modulates the activity of some phosphodiesterases (PDEs) [
6]. cGMP is degraded by PDE-mediated hydrolysis [
6].
Modulation of cGMP activity has therapeutic value for several disorders. For example, NO donors such as glyceryl trinitrate are used to treat angina [
7], the sGC stimulator riociguat is used for treatment of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension [
8], PDE5 inhibitors such as sildenafil and vardenafil are used to treat erectile dysfunction [
9], and agonists for GC-C (a type of pGC) such as linaclotide and plecanatide are used to treat irritable bowel syndrome with constipation [
10]. Natriuretic peptides are also under investigation for the treatment of cardiovascular disorders such as heart failure [
11], and drug discovery targeting cGMP signalling cascades is under continued investigation for the treatment of a variety of other disorders and diseases [
12,
13,
14,
15,
16].
cGMP is usually measured in populations of cells in a plate-based format using traditional end point signalling assays that involve the detection of cGMP in cell lysates. However, although such assays may be sensitive, they are laborious, as cells must be lysed at individual time points. Furthermore, end point cGMP assays usually require the addition of PDE inhibitors to stop the breakdown of cGMP, leading to the accumulation and measurement of total cGMP, which can produce a type of “observational bias”, whereby the dynamics of cGMP are not easily detected, leading to an incomplete or misleading idea of the signalling patterns induced by different ligands [
17]. Conversely, assays that can detect cGMP activity in real time will help remove the confounding effects of observational bias and will also allow more efficient examination of the kinetic aspects of signalling.
Genetically encoded biosensors based on Förster/fluorescence resonance energy transfer (FRET) are sometimes used to image cGMP activity in real time in single cells [
18,
19,
20], where FRET refers to the transfer of energy from an excited donor fluorophore to an appropriate acceptor fluorophore when they are in close proximity [
21]. These FRET-based biosensors for cGMP consist of a “sensor” domain that binds cGMP, which is sandwiched by an appropriate FRET donor/acceptor fluorophore pair such as cyan fluorescent protein (CFP)/yellow fluorescent protein (YFP). When cGMP binds to the sensor, the FRET pair moves either closer together or farther apart, producing a change in FRET that can be monitored in real time. However, although FRET-based biosensors are valuable for cellular microscopy and imaging of single cells with high spatial and temporal resolution, they are less suited for the detection of activity from populations of cells in plate-based assays, due to the higher background noise and photobleaching of the donor caused by laser excitation [
22]. In contrast, bioluminescence resonance energy transfer (BRET), which uses a luciferase rather than a fluorescent protein as the donor, is a more sensitive technique for plate-based assays, as luciferases do not require laser excitation, but rather emit light after oxidation of a substrate molecule. As a result, BRET assays do not induce photobleaching of the donor, and show a lower background noise and a higher signal-to-noise ratio than FRET assays [
22].
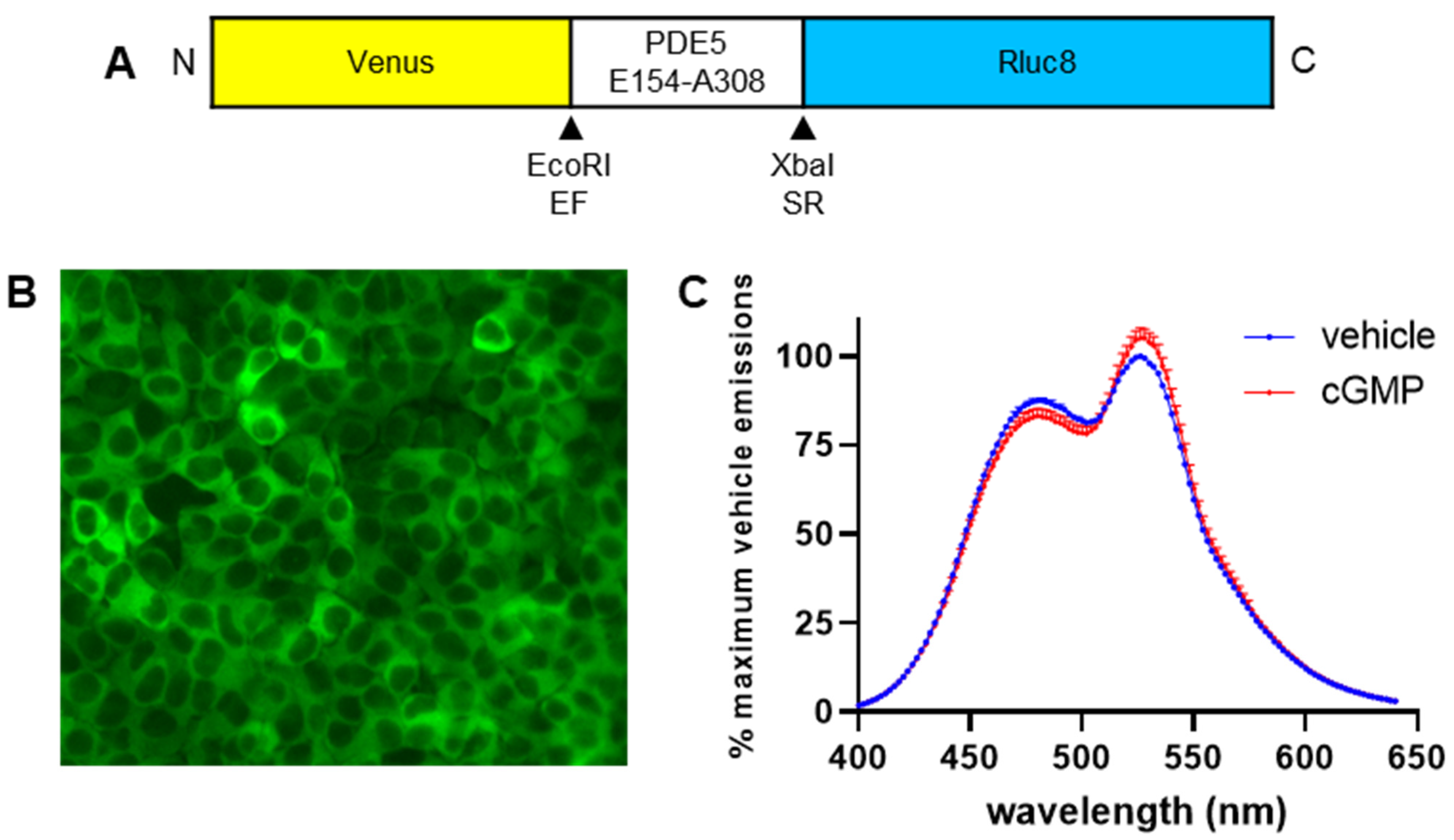
A BRET-based assay for cGMP activity would allow for high sensitivity, real-time detection of cGMP activity in live cells in a plate-based assay, which is more suited to ligand screening and pharmacological characterisation than imaging of single cells. Therefore, we report here the conversion of the cGES-DE5 FRET-based cGMP biosensor [
19] to a BRET-based biosensor (cyclic GMP sensor using YFP-PDE5-Rluc8; CYGYEL), cloning of the novel biosensor into a lentiviral vector for stable expression in mammalian cells, validation and characterisation of the biosensor in live cells in a real-time multi-well plate-based format, and investigation of the applicability of the sensor for the detection of sGC- and pGC-mediated cGMP activity in human primary vascular cells.
3. Discussion
Traditional end point signalling assays for cGMP do not easily measure the temporal aspects of signalling as they require lysing cells at individual time points and usually employ PDE inhibitors to allow the accumulation of cGMP to detectable levels. The ability of BRET-based biosensors to detect signalling in real time in plate-based cellular assays prompted us to develop a BRET-based biosensor for cGMP activity. The FRET-based biosensor cGES-DE5 was converted to BRET due to its high selectivity for cGMP over cAMP and high temporal resolution compared with other available biosensors [
19]. This was accomplished by substituting the region coding for ECFP with Rluc8, as well as swapping the region coding for EYFP to Venus. The resulting biosensor, which was named CYGYEL, was cloned into a lentiviral vector and subsequently tested across several cell types including human primary vascular cells, where it successfully detected real-time changes in cGMP activity, and in some cases showed differences in temporal patterns between ligands and cell types.
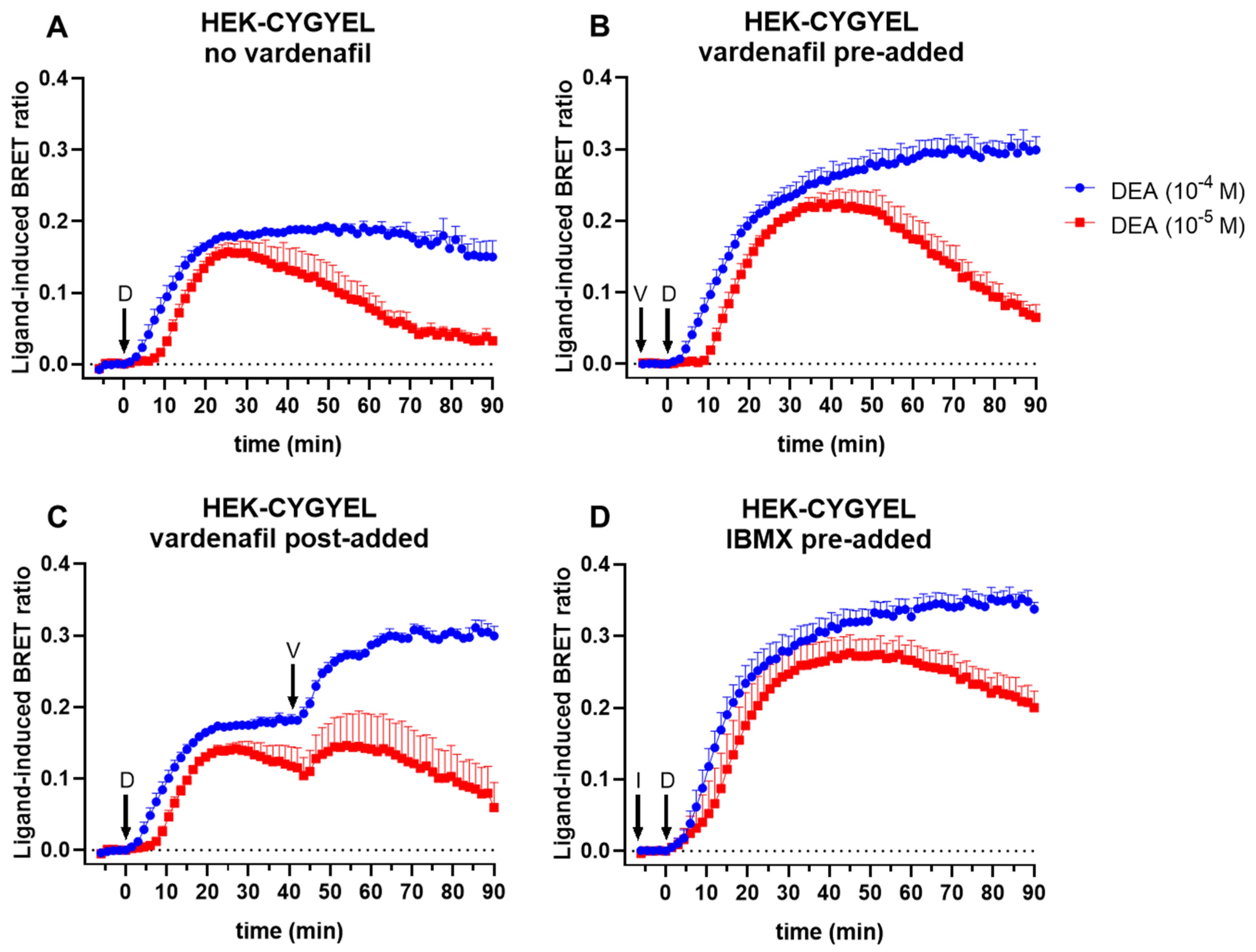
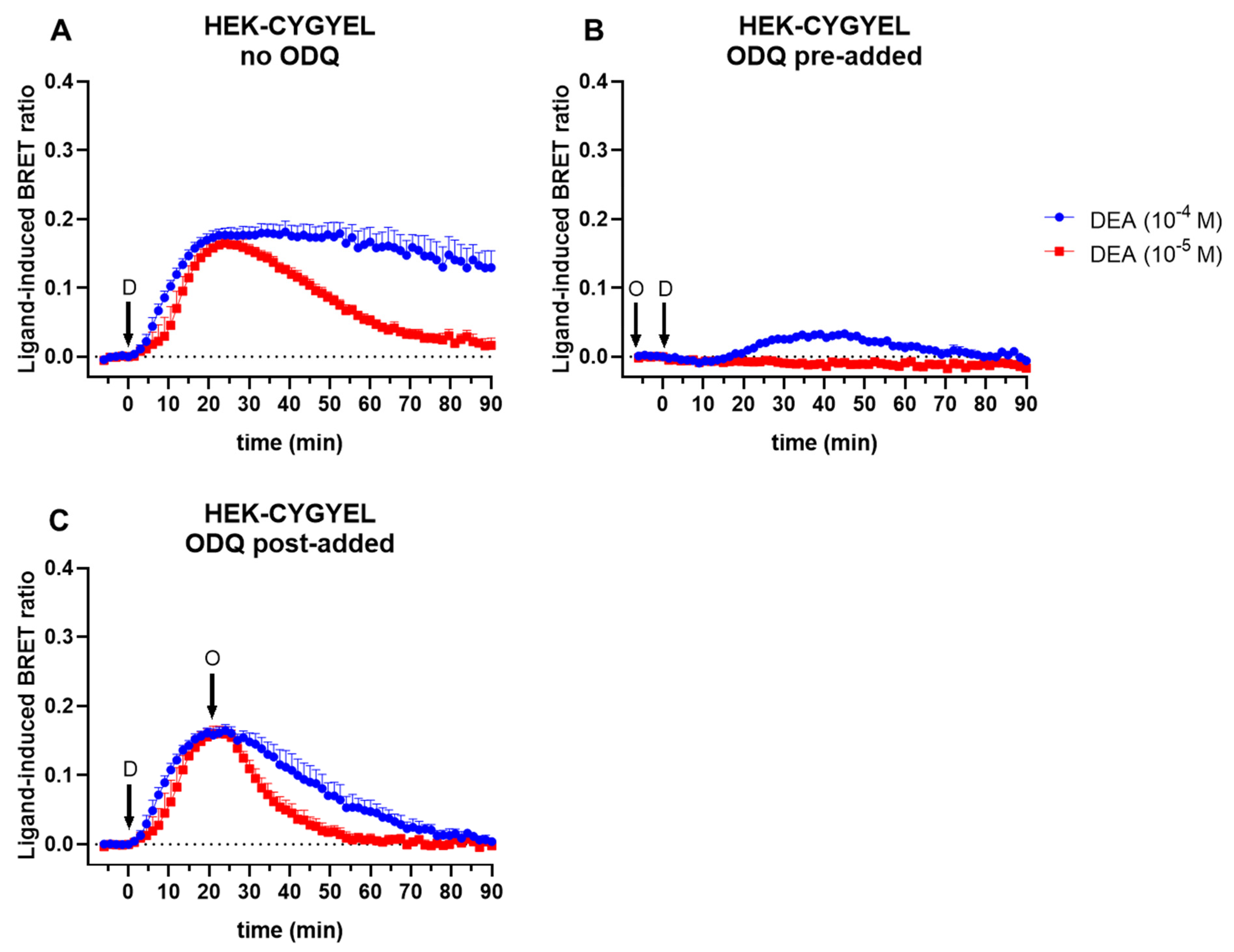
First, HEK293T cells were transduced and sorted by FACS for basic characterisation of the sensor. The imaging of HEK-CYGYEL cells using a fluorescence microscope demonstrated that CYGYEL was expressed throughout the cytoplasm, suggesting that the detection of cGMP throughout the cell would be possible. Real-time assays in these cells showed that CYGYEL detects robust increases and decreases in sGC-mediated cGMP activity for extended periods of time (at least 90 min) after stimulation with a NO donor. The assay was sensitive enough to detect cGMP activity in the absence of any PDE inhibitors, but the addition of the cGMP-specific PDE5 inhibitor vardenafil or the nonspecific PDE inhibitor IBMX did increase the BRET ratio and prolong the response. Furthermore, preincubation with the sGC inhibitor ODQ diminished or abolished the increases in BRET ratio, depending on the DEA concentration, further confirming the assay was specific for cGMP activity.
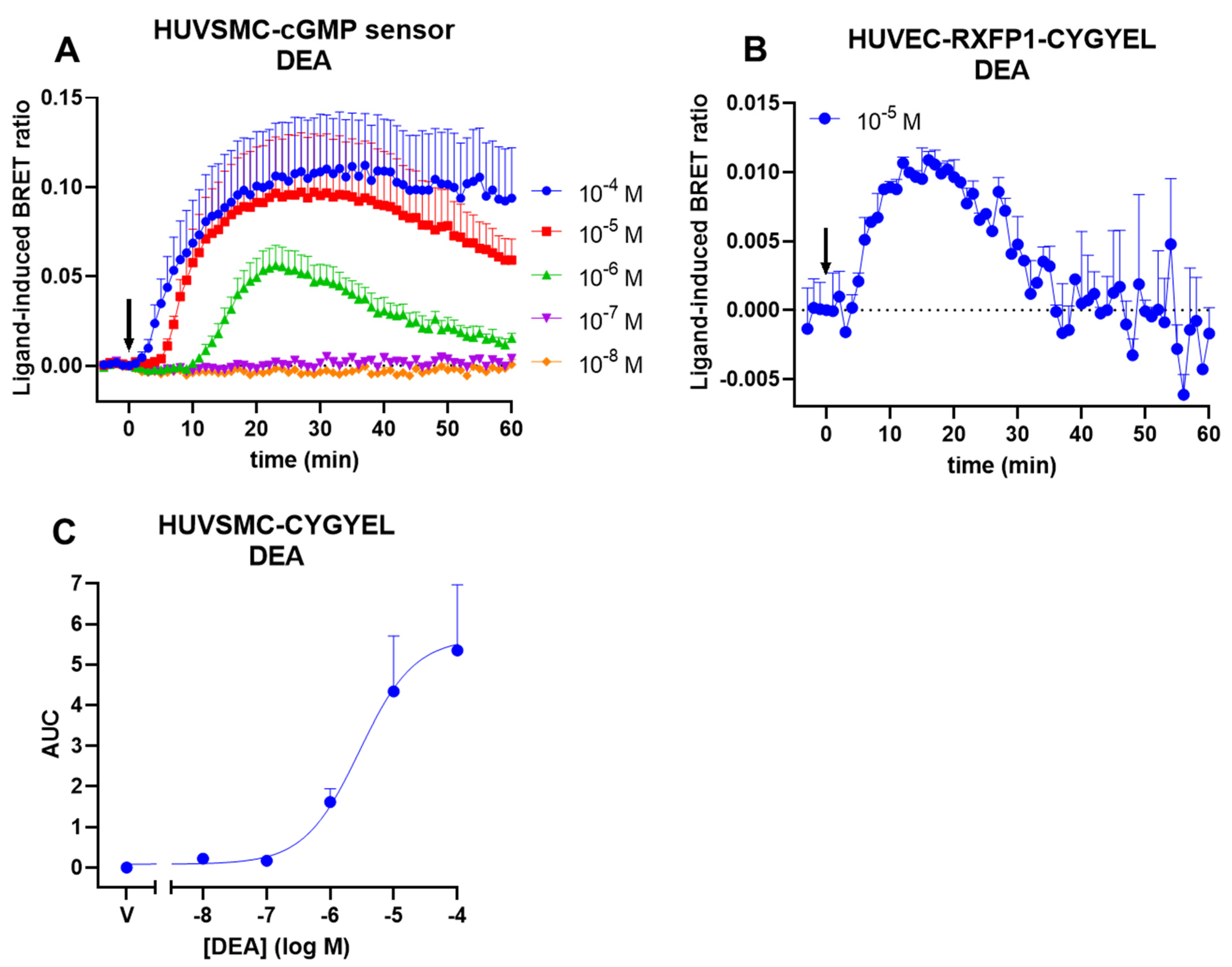
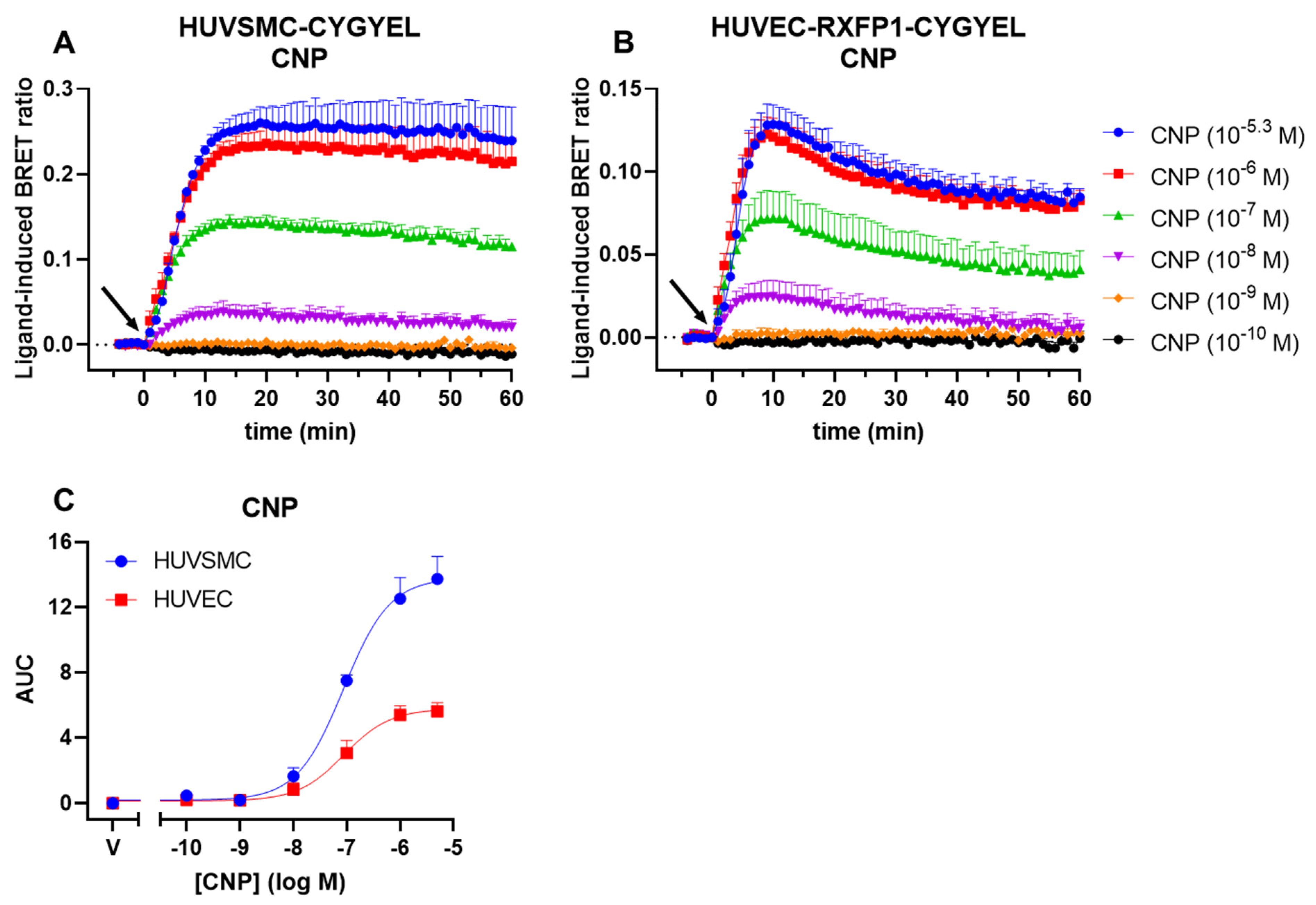
Following this, we demonstrated that CYGYEL can detect cGMP activity mediated through both sGC and pGC in human primary vascular cells, specifically HUVECs and HUVSMCs. Interestingly, we observed differences in cGMP signalling both between cell types and between ligands. Both cell types showed robust, concentration-dependent responses to CNP, but cGMP activity was more sustained in HUVSMCs than in HUVECs. Notably, however, the potency of CNP was almost identical between cell types. In contrast, DEA responses were weak and difficult to detect in HUVECs, whereas there were robust, concentration-dependent DEA-mediated cGMP increases in HUVSMCs, which were also substantially less potent than the CNP responses. Furthermore, CNP showed faster increases in cGMP compared with DEA, consistent with the role of CNP as a membrane receptor agonist, compared with DEA, which is a NO donor. Additionally, the weak responses to DEA observed in HUVECs, compared with strong responses to CNP observed in the same cell type, are consistent with findings from other studies showing that natriuretic peptides induced stronger cGMP responses in HUVECs compared with NO donors [
26,
27]. Finally, as robust cGMP activity was detected in both types of vascular cells tested, it is expected that the lentiviral vector, which uses a constitutive human promoter, will also aid in the detection of cGMP across a variety of other cell types.
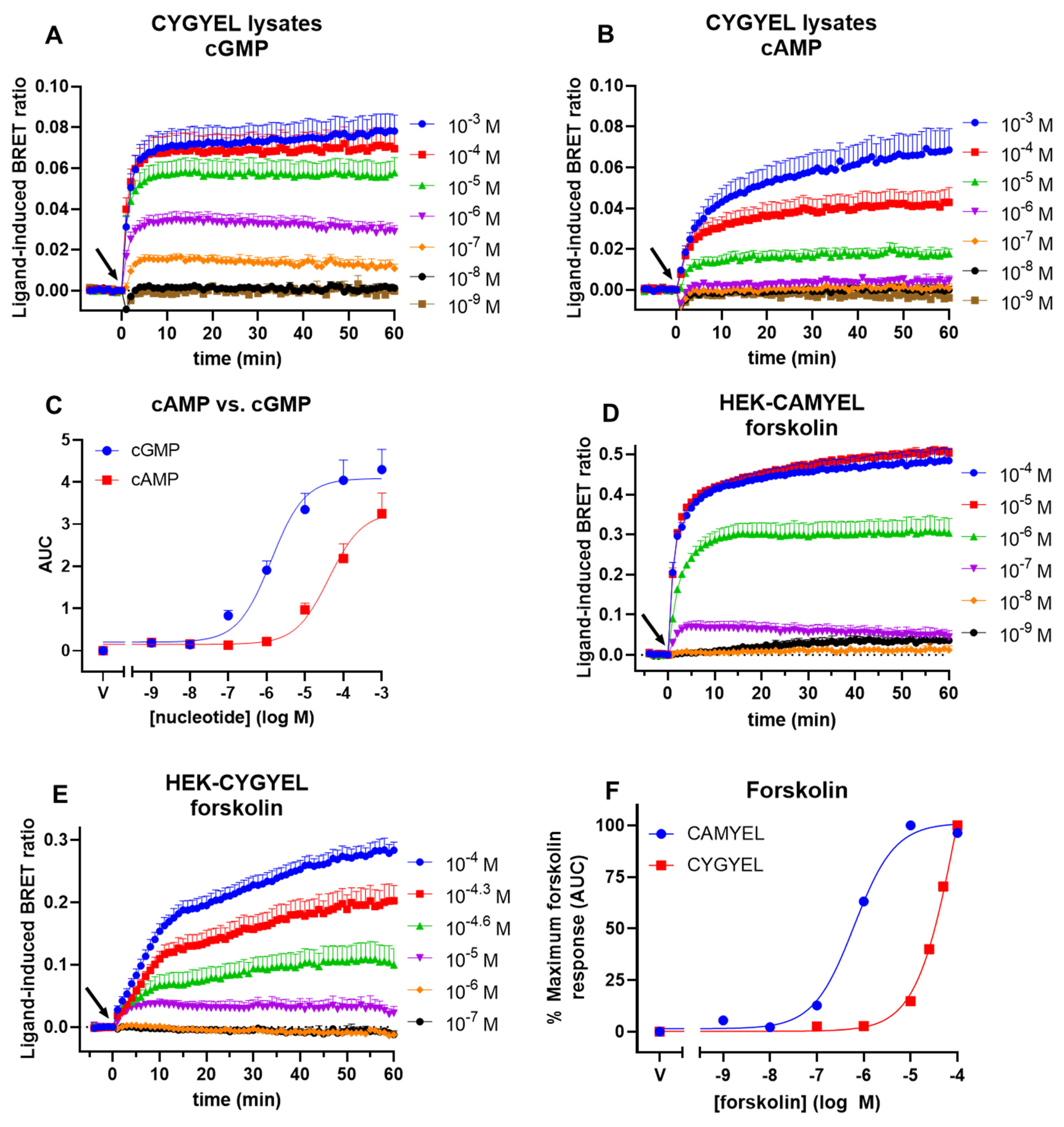
One potential concern for the use of genetically encoded cGMP sensors is that cAMP can also bind to cGMP-binding domains, including the binding domain from PDE5, albeit with a lower affinity than cGMP. As noted, the low crosstalk from cAMP observed for cGES-DE5 was one of the primary reasons this sensor was chosen as the basis to generate CYGYEL. In lysates from TsA201 cells, cGES-DE5 was activated by cGMP with a potency of about 1.5 μM and by cAMP with a potency of about 630 μM [
19]. Here, we demonstrated that CYGYEL was activated by cGMP with a potency (EC
50) of about 1.3 μM and by cAMP with a potency of about 42 μM—a selectivity of about 32-fold. Thus, although the EC
50 of cGMP is comparable between the two variants of the biosensor, the EC
50 of cAMP is considerably lower for the activation of CYGYEL than cGES-DE5. As noted, however, results from such biochemical assays in cell lysates will not always reflect what occurs in live cells due to a variety of factors such as localised cyclic nucleotide and PDE activity. Therefore, we also tested the ability of cAMP to activate CYGYEL in live cell assays by stimulating HEK-CYGYEL cells with the adenylate cyclase activator forskolin and compared these results to those observed for the same assay conducted in HEK-CAMYEL cells. These findings showed that in contrast to the activation of CAMYEL, relatively high concentrations of forskolin were required to induce a robust activation of CYGYEL, and that there was a 160-fold difference in forskolin potencies between the activation of the two sensors. However, it should also be noted that cAMP-mediated activation of CYGYEL may depend on the cell type being tested due to differential expression of adenylate cyclases and PDEs, for example, so users of this biosensor should carefully consider this. On the other hand, it is also important to note that for many applications of CYGYEL, such as screening of NO donors and pGC agonists as conducted here, the crosstalk from cAMP may not be an important factor. Examples of situations where crosstalk may be important include situations when using a ligand induces the activation of both cGMP and cAMP, or when using cell types that have high background levels of cAMP. Furthermore, other characteristics of a sensor, such as its temporal resolution, are also equally important to consider in the choice of a biosensor. As noted, for example, the superior temporal resolution of cGES-DE5 reported by Nikolaev et al. [
19], compared with other available FRET-based biosensors, was the other key reason that we chose cGES-DE5 as the basis for our BRET variant.
We also note the development of another BRET-based biosensor for cGMP, known as GFP
2-GAFa-Rluc [
28]. This biosensor is based on the BRET
2 methodology [
29], using the combination of Rluc, GFP
2, and DeepBlueC as the substrate, compared with CYGYEL which is based on BRET
1, using the combination of Rluc8, Venus, and coelenterazine
h. GFP
2-GAFa-Rluc uses a longer segment of PDE5A than CYGYEL, which is sandwiched between the Rluc donor and GFP
2 acceptor. In lysates from transfected cells, cGMP had a surprisingly low EC
50 of approximately 30 nM for the activation of GFP
2-GAFa-Rluc, and cAMP did not appear to activate the sensor, although it was only tested up to 1 µM in lysates [
28]. This finding is intriguing as it raises the possibility that this truncation of PDE5 may be more optimal than the one described by Nikolaev et al. [
19], which was incorporated into CYGYEL. However, two caveats must be noted. First, GFP
2-GAFa-Rluc may act as a “sink” that sequesters cGMP in the cell, suggesting that cGMP may have a slow off-rate and remains bound, preventing its degradation by PDEs, thus making it difficult to detect the kinetics of signalling [
28]. Second, our own results have demonstrated that CYGYEL is dynamic and reversible, able to detect the kinetics of cGMP activity over extended periods of time; yet similar kinetic assays were not conducted using GFP
2-GAFa-Rluc [
28]. This may be in part related to the use of the BRET
2 methodology, which does not typically allow extended time courses due to weak emissions and a rapid loss of signal [
30]. In contrast, BRET
1 maintains a high luminescence for a longer duration and allows for a higher throughput. Due to these factors, we cannot directly compare the activity of the two biosensors and are unsure of the temporal resolution of GFP
2-GAFa-Rluc. Future studies could convert GFP
2-GAFa-Rluc to BRET
1 in order to directly compare the temporal resolution and reversibility of the two different truncations of PDE5, in order to determine the utility of each truncation as a sensor domain.
Similarly, future studies could also aim to increase the affinity of cGMP for CYGYEL or other cGMP biosensors while at the same time decreasing the affinity of cAMP, with the caveat that very high affinities might lead to sequestration cGMP [
28]. Interestingly, a variant of cGES-DE5 that swapped the ECFP/EYFP FRET pair to a T-sapphire/dimer2 FRET pair showed substantial increases in both affinity for cGMP (~40 nM) and selectivity for cGMP over cAMP [
31], allowing the detection of cGMP in adult cardiomyocytes [
32]. However, as we have shown, changing the ECFP/EYFP pair to Rluc8/Venus did not have the same effect on selectivity. Additionally, the recently developed FRET-based cGMP sensor
PfPKG, which was based on PKG (rather than PDE5) from
Plasmodium falciparum, showed a high cGMP affinity of about 23 nM, but this was coupled with a high cAMP affinity of about 4.6 µM, suggesting that it may be difficult to decouple the affinities of the two cyclic nucleotides for cGMP-binding domains [
33]. In fact, the authors even mutated the cGMP-binding domain from PKG in an attempt to reduce the affinity of cAMP, but were unsuccessful [
33].
The compartmentalisation of cyclic nucleotides is a current research topic in cell biology [
34], and although cGMP compartmentalisation is less well understood than that of cAMP, it does appear that there are spatially segregated pools of cGMP within the cell [
35,
36], which is not surprising given that sGC and pGC are already separately localised. Importantly, microdomains of cGMP may have different functional roles in cardiovascular physiology, which is still under investigation [
37]. The ability of a biosensor to detect cGMP, and its selectivity for cGMP over cAMP, may differ depending on whether lysates or live cells are stimulated, and may also differ based on cell type, due to the differential expression and compartmentalisation of enzymes that regulate cyclic nucleotide levels within the cell. Notably, CYGYEL showed diffuse cytoplasmic expression when cells expressing CYGYEL were imaged using a fluorescence microscope, which was consistent with its ability to detect cGMP synthesised by either sGC or pGC in live cells. Although BRET is not typically used to detect localised signalling, it may be possible to do this by targeting the biosensor to particular locations of interest, such as the membrane [
38]. Alternatively, cGES-DE5 may be used as a complementary biosensor to detect localised signalling via sGC versus pGC in single cells, for example, which is one of the primary advantages of FRET over BRET. Additionally, the recent development of enhanced Nano-lanterns [
39] enabled the generation of a dual FRET/BRET-based cAMP biosensor [
40], and it would be interesting to see whether a similar approach is viable for the generation of a single cGMP sensor that can be used for plate-based assays in addition to cellular microscopy.
In summary, we have generated a genetically encoded, BRET-based biosensor for cGMP activity that streamlines the process of detecting cGMP compared to traditional end point assays. CYGYEL can detect changes in sGC- and pGC-mediated cGMP activity over time in the plate-based format, and the lentiviral vector allows for the generation of human cell lines stably expressing the biosensor, allowing the detection of cGMP across physiologically relevant cell types.
4. Methods
4.1. Materials and Reagents
1H-[1,2,4]Oxadiazolo[4,3-a]quinoxaline-1-one (ODQ), 3-isobutyl-1-methylxanthine (IBMX), adenosine 3′,5′-cyclic monophosphate sodium salt monohydrate, C-type natriuretic peptide (CNP), diethylamine NONOate diethylammonium salt (DEA), forskolin, guanosine 3′,5′-cyclic monophosphate sodium salt, Polybrene, Tris base, and vardenafil, were from Sigma-Aldrich (Burlington, MA, USA). Dulbecco’s modified Eagle medium (DMEM), Medium 199 (M199), L-glutamine, and penicillin–streptomycin (P/S) were from Gibco (Waltham, MA, USA). Foetal bovine serum (FBS) was from Scientifix (Melbourne, VIC, Australia). EGM-2 endothelial cell growth medium-2 BulletKit was from Lonza (Basel, Switzerland). Smooth muscle cell growth supplement was from ScienCell (Carlsbad, CA, USA). Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) was from ChemSupply (Adelaide, SA, Australia). Coelenterazine h was from Nanolight (Pinetop, AZ, USA).
4.2. Design of a BRET-Based Biosensor for cGMP Activity
The BRET-based biosensor CYGYEL was created based on the cGES-DE5 FRET-based biosensor [
19]. cGES-DE5 consists of the cGMP-binding domain (Glu154 to Ala308) from human PDE5A1 (GenBank accession number NM_001083) inserted between enhanced yellow fluorescent protein (EYFP) and enhanced cyan fluorescent protein (ECFP) using the
EcoRI and
XbaI restriction sites, respectively. The sequence for EYFP was replaced with the sequence for Venus, and the sequence for ECFP was replaced with the sequence for Rluc8. A Kozak sequence (GCCACC) was placed before the start codon, and the gene for the biosensor was synthesised by GenScript (Piscataway, NJ, USA) into pcDNA3.1/Zeo
(+) between the
NheI and
ApaI restriction sites.
4.3. Cell Culture
All cells were cultured at 37 °C with 5% CO2. HEK293T cells (CRL-3216) were cultured in DMEM supplemented with 10% FBS, 1% L-glutamine, and 1% P/S. Human umbilical vein smooth muscle cells (HUVSMCs) were grown in M199 supplemented with 5% heat-inactivated FBS, 1% P/S, and smooth muscle cell growth supplement. Human umbilical vein endothelial cells (HUVECs) were grown in EGM-2 endothelial cell growth medium-2 (including 2% FBS) supplemented with 1% P/S. These are henceforth referred to as “complete media”. HUVSMCs were purchased from ScienCell.
4.4. Isolation of Human Umbilical Vein Endothelial Cells
This study was approved by the Monash Health Human Research and Ethics Committee (HREC: 01067B). Healthy term umbilical cord from singleton pregnancies were collected, with informed written consent, at the time of elective caesarean section. Exclusion criteria included multiple pregnancy, maternal medical conditions, any history of preeclampsia and/or foetal growth restriction, smoking, alcohol or drug use during pregnancy. Exclusions were made for the following medications: antihypertensive, aspirin, nonsteroidal anti-inflammatory drugs, or thyroid medications. To isolate HUVECs, the vein of the umbilical cord (~15 cm) was rinsed with 1× Hank’s Buffered Saline Solution (HBSS) until all blood was removed. Luer-lok fittings were placed at both ends of the vein and 10 mL of collagenase type II (0.5 mg/mL in HBSS) syringed into the vein. The cord was then incubated for 10 min at 37 °C in a humidified 95% air/5% CO2 incubator. The collagenase was collected by a syringe and placed in a 50 mL falcon tube. The vein was washed twice with 10 mL EBMTM-2 endothelial cell growth media (Lonza), which was collected and added into the falcon tube. The tube was spun at 500× g for 5 min at room temperature to pellet the cells. Cells were resuspended in 5 mL EBMTM-2 media and cultured in a T25 flask at 37 °C in a humidified incubator at 95% air/5% CO2. Cells were passaged to P2 before slowly freezing in 90% FBS and 10% DMSO (1 million cells/mL) and stored in liquid nitrogen.
4.5. Cloning CYGYEL into a Lentiviral Vector
CYGYEL was cloned into a lentiviral vector under the control of the constitutive human Ef1α promoter using Gateway technology (Invitrogen), according to the manufacturer’s instructions. The CYGYEL coding sequence from the pcDNA3.1/Zeo
(+) vector was flanked with appropriate
attB5 and
attB2 sequences and amplified by polymerase chain reaction (PCR) using the forward primer 5′ GGGG ACA ACT TTG TAT ACA AAA GTT GGC CAC CAT GGT GAG CAA GGG C 3′ and reverse primer 5′ GGGG ACC ACT TTG TAC AAG AAA GCT GGG TAT TA CTG CTC GTT CTT CAG CAC 3′. The PCR product of the correct size was extracted, gel-purified, and cloned into the pDONR 221 P5-P2 vector using BP Clonase II. The resulting pENTR L5-L2 entry clone was confirmed to have the correct sequence using Sanger sequencing. Our pENTR L1-R5 Ef1α entry clone was cloned along with the pENTR L5-L2 CYGYEL clone into the pLenti X1 Zeo DEST vector [
41] using LR Clonase II, to generate the pLenti X1 Ef1α CYGYEL expression clone.
4.6. Generation of HEK-CAMYEL and HEK-CYGYEL Cell Lines via Lentiviral Transduction
HEK293T stably expressing the cAMP sensor CAMYEL [
25] or CYGYEL were generated using crude lentivirus. HEK293T cells were plated at 3.5 million cells on a 10 cm dish. The next day, cells were transfected with 8 µg of either pLenti X1 Ef1α CAMYEL [
42] or pLenti X1 Ef1α CYGYEL, along with the lentiviral packaging and envelope plasmids pMDL, pRSV-Rev, and pCMV-VSV-G, using Lipofectamine 2000. The following day, HEK293T cells to be transduced were plated on a 10 cm dish at 400,000 cells. The next day, media containing lentivirus from transfected cells were harvested and filtered through a 0.45 µm syringe filter. The newly plated HEK293T cells were transduced by replacing the cell culture media with the media containing lentivirus, mixed with 4 μg/mL hexadimethrine bromide (Polybrene) to increase transduction efficiency. Three rounds of transduction were performed over 32 h, before allowing cells to recover for three days in complete DMEM.
4.7. Purification of Lentivirus and Transduction of Primary Vascular Cells
CYGYEL lentivirus was produced in HEK293T cells and purified for transduction of primary cells. HEK293T cells were plated out on five 20 cm dishes at 11 million cells per dish. The next day, cells were transfected with the pLenti X1 Ef1α CYGYEL expression clone, pMDL/pRRE, pRSV-Rev, and pCMV-VSV-G, using Lipofectamine 2000. Two days later, media containing secreted lentivirus was harvested and spun at 2000 rpm for five minutes to pellet any cellular debris. The supernatant was then filtered through a 0.45 µm filter and spun in a Hitachi CP100MX ultracentrifuge at approximately 72,000× g at 4 °C for two and a half hours to pellet the virus. The supernatant was then discarded, and 50 µL of cold phosphate-buffered saline (PBS) was added per tube (for a total of 150 µL per viral preparation), which was stored at 4 °C overnight. Some of the crude lentivirus-containing media were also stored at 4 °C overnight. The next day, the virus was resuspended in the PBS, pooled, then stored in aliquots at −80 °C. Moreover, 1 µL of the virus was also used for a visual titration on HEK293T cells, along with some of the crude lentivirus, to ensure that it was of high enough titre to transduce cells in vitro.
HUVECs and HUVSMCs were transduced with the purified virus in a T175 flask at approximately 70% confluency. Then, 5 μL of purified lentivirus was resuspended in cell culture media along with 4 μg/mL hexadimethrine bromide (Polybrene). Cell culture media in the flask were replaced with virus-containing media, which were left on cells overnight before replacing it with fresh cell culture media.
4.8. Flow Cytometry and Fluorescence-Activated Cell Sorting
Transduced cells were sorted from non-transduced cells using fluorescence-activated cell sorting (FACS), using a Becton Dickinson FACSAria III. Cells were isolated by gating after plotting the forward scatter area (FSC-A) against the side scatter area. Single cells were isolated after plotting the side scatter height against the side scatter width and the forward scatter height (FSC-H) against the forward scatter width. Dead cells stained with DAPI were removed after laser excitation at 405 nm and gating FSC-A against emissions at 450/40 nm. From the population of single, live cells, cells showing higher YFP emissions than control cells, as detected after laser excitation at 488 nm and emissions at 530/30 nm, were sorted into separate populations, as noted. Cells were subsequently grown to confluency in a T175 flask and cryopreserved. For HUVECs, the cells were already expressing “high” levels of a relaxin family peptide receptor 1 (RXFP1)–internal ribosome entry site (IRES)–green fluorescent protein (GFP) construct, from a previous transduction and sort (
Figure S1); however, emissions were separated from YFP emissions by using separate emission filters for GFP (510/20 nm) and YFP (530/30 nm) and applying compensation. These cells had subsequently been cryopreserved. For analytical flow cytometry, cells were isolated as above, but with the following exceptions: a Beckman Coulter CytoFLEX S was used, single cells were isolated after plotting only for FSC-A versus FSC-H, and YFP was detected using a 525/40 nm emission filter.
4.9. Fluorescence Microscopy
HEK293T cells that were stably expressing “high” levels of CYGYEL were imaged using a Leica DM IL LED microscope with DFC450 C camera, EL6000 external light source, and GFP filter, at 20× magnification.
4.10. BRET Assays in Live Cells
HEK293T cells, HUVSMCs, or HUVECs stably expressing either CYGYEL or CAMYEL were seeded onto 96-well CulturPlate microplates (PerkinElmer, Waltham, MA, USA) and were incubated overnight. At the time of the assay, HEK293T or HUVSMC cell culture media were replaced with phenol red free complete media containing 25 mM HEPES and 5 µM coelenterazine h, whereas HUVEC cell culture media, EGM-2, were replaced with complete EGM-2 containing 25 mM HEPES and 5 µM coelenterazine h. Emissions were detected simultaneously from Rluc (CAMYEL) or Rluc8 (CYGYEL) (475/30 nm) and citrine (CAMYEL) or Venus (CYGYEL) (525/30 nm) using a PHERAstar FSX plate reader (BMG Labtech, Ortenberg, Germany) at 37 °C. Emissions were read for five minutes to establish a baseline before addition of a vehicle or ligand.
4.11. BRET Assays in Cell Lysates
HEK-CYGYEL cells were washed and resuspended in DPBS, and then centrifuged in order to pellet the cells. DPBS was aspirated and cells were resuspended in buffer containing 5 mM Tris and 2 mM EDTA (pH 7.3), then sonicated on low power for three 10 s cycles at 4 °C. Lysates were then spun down at 21,000× g for 20 min at 4 °C, after which the supernatant was obtained and stored at −80 °C or used for BRET assays. During the assay, lysates were mixed with coelenterazine h (5 µM), distributed into a white, 384-well OptiPlate (PerkinElmer) with lysates corresponding to the equivalent of approximately 5000 cells per well, and then assayed as above using a PHERAstar FSX plate reader. Cell lysates were also used to generate spectral scans, using a CLARIOstar plate reader (BMG Labtech).
4.12. Presentation of BRET Data
The ligand-induced BRET ratio was calculated by subtracting the ratio of acceptor channel emissions (525/30 nm) to donor channel emissions (475/30 nm) for vehicle wells from the same ratio for treated wells:
For time courses, the ligand-induced BRET ratio was plotted against time, with the last prereading before ligand addition displayed as the zero time point (time of vehicle/ligand addition). For CAMYEL cAMP data only, decreases in ligand-induced BRET ratio represent increases in cAMP, so all values were subtracted from zero. For spectral scan data, the wavelength (nm) was plotted against emissions. Concentration–response curves were fit to the data by applying three-parameter nonlinear regressions to the area under the curve data, using Prism 9 from GraphPad (San Diego, CA, USA).


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}