
Oxidative DNA Damage and Cisplatin Neurotoxicity Is Exacerbated by Inhibition of OGG1 Glycosylase Activity and APE1 Endonuclease Activity in Sensory Neurons

 , and
, and 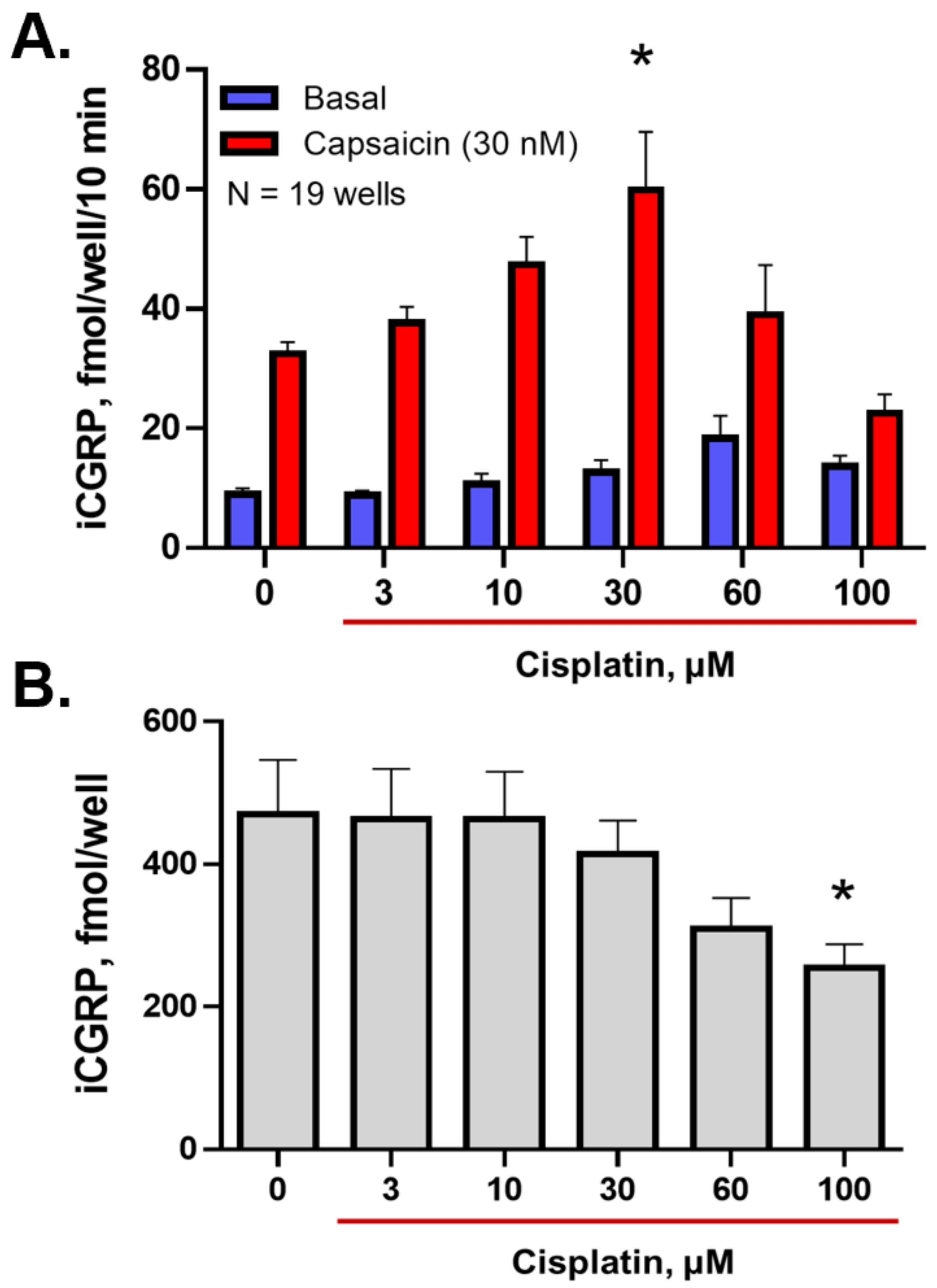{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
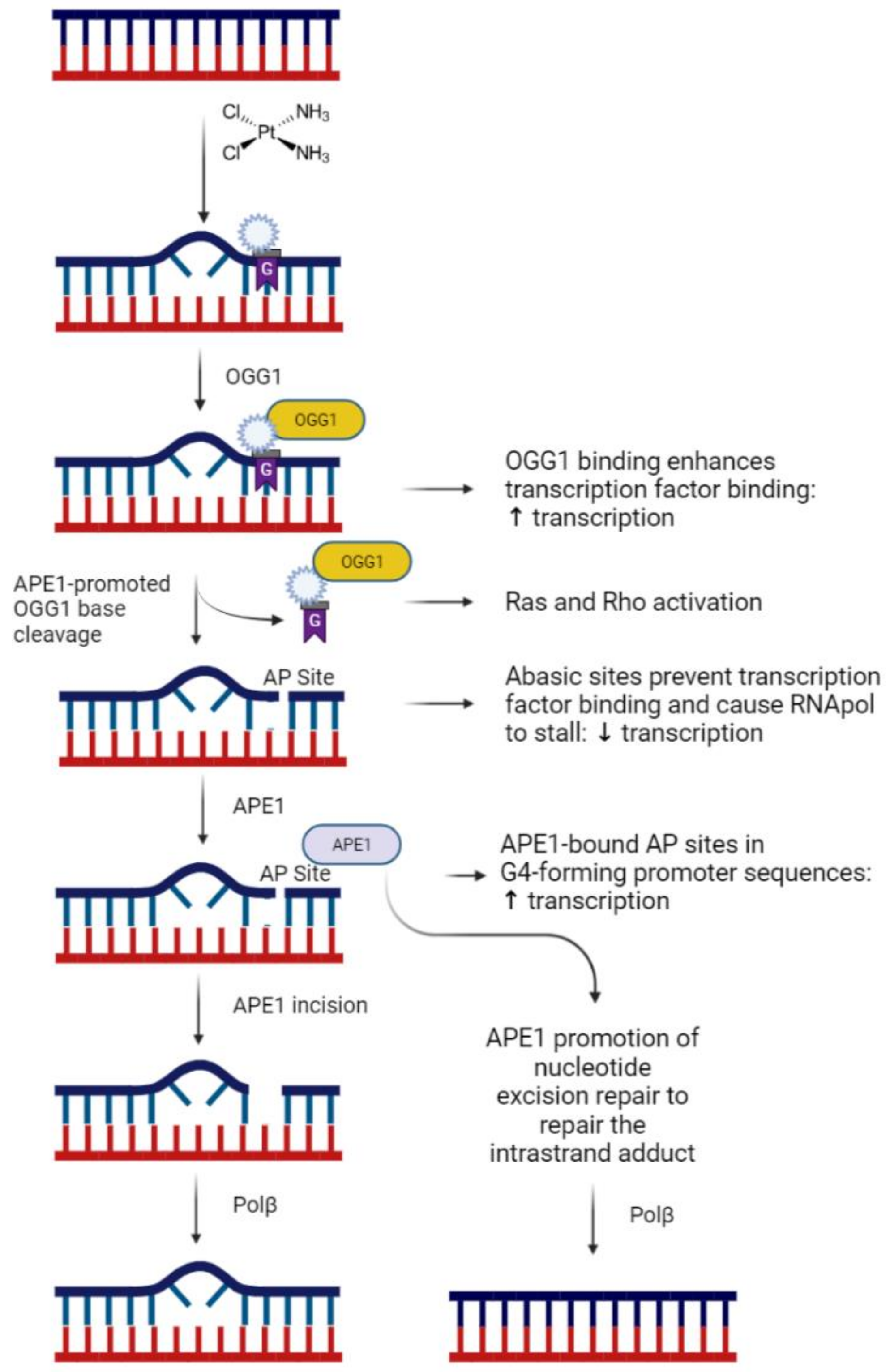{kind=link}
Abstract
:1. Introduction
2. Results
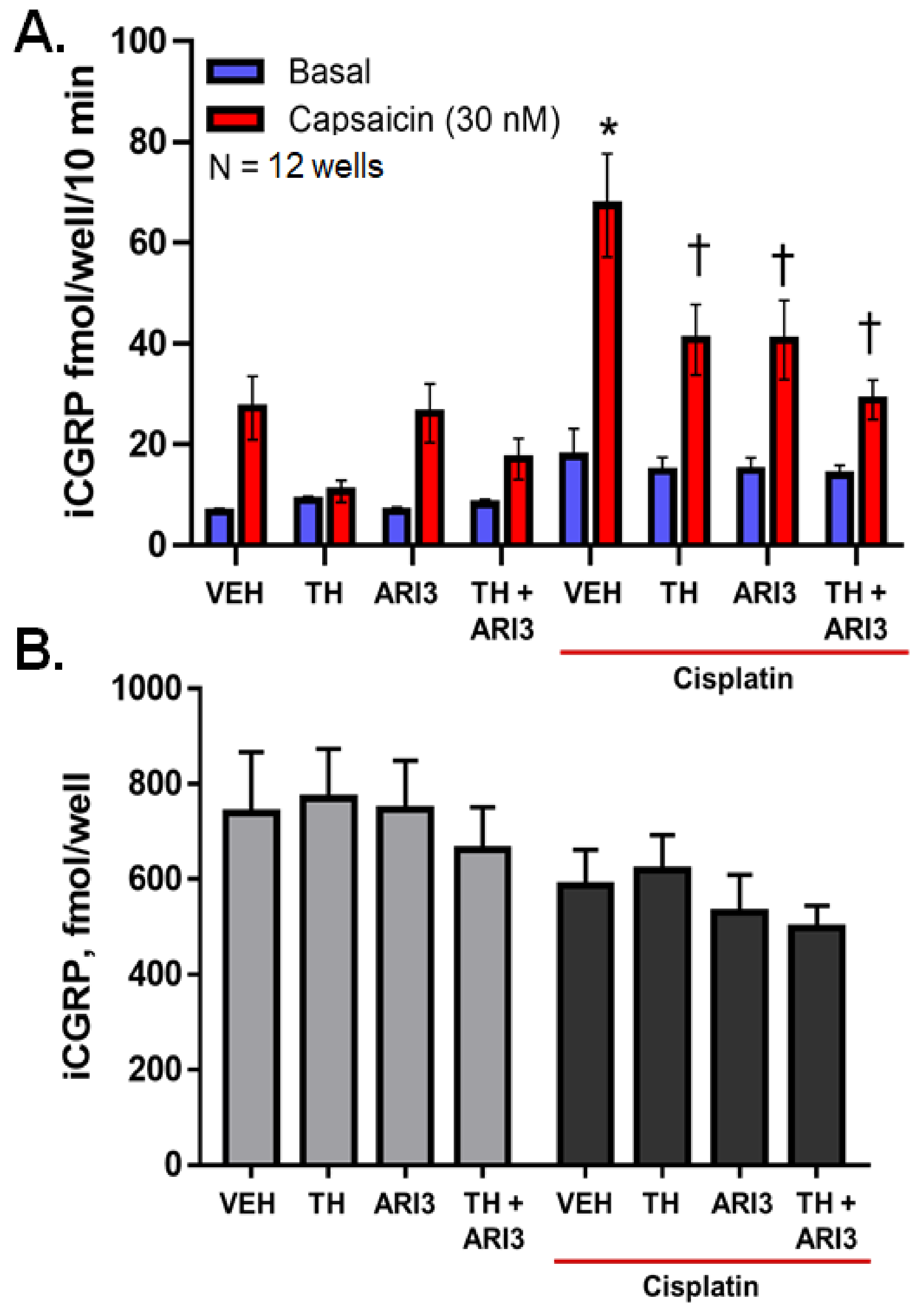
2.1. Inhibition of Either OGG1 Glycosylase Activity or APE1 DNA Repair Activity Diminishes the Sensitizing Effects of Cisplatin on Neuropeptide Release
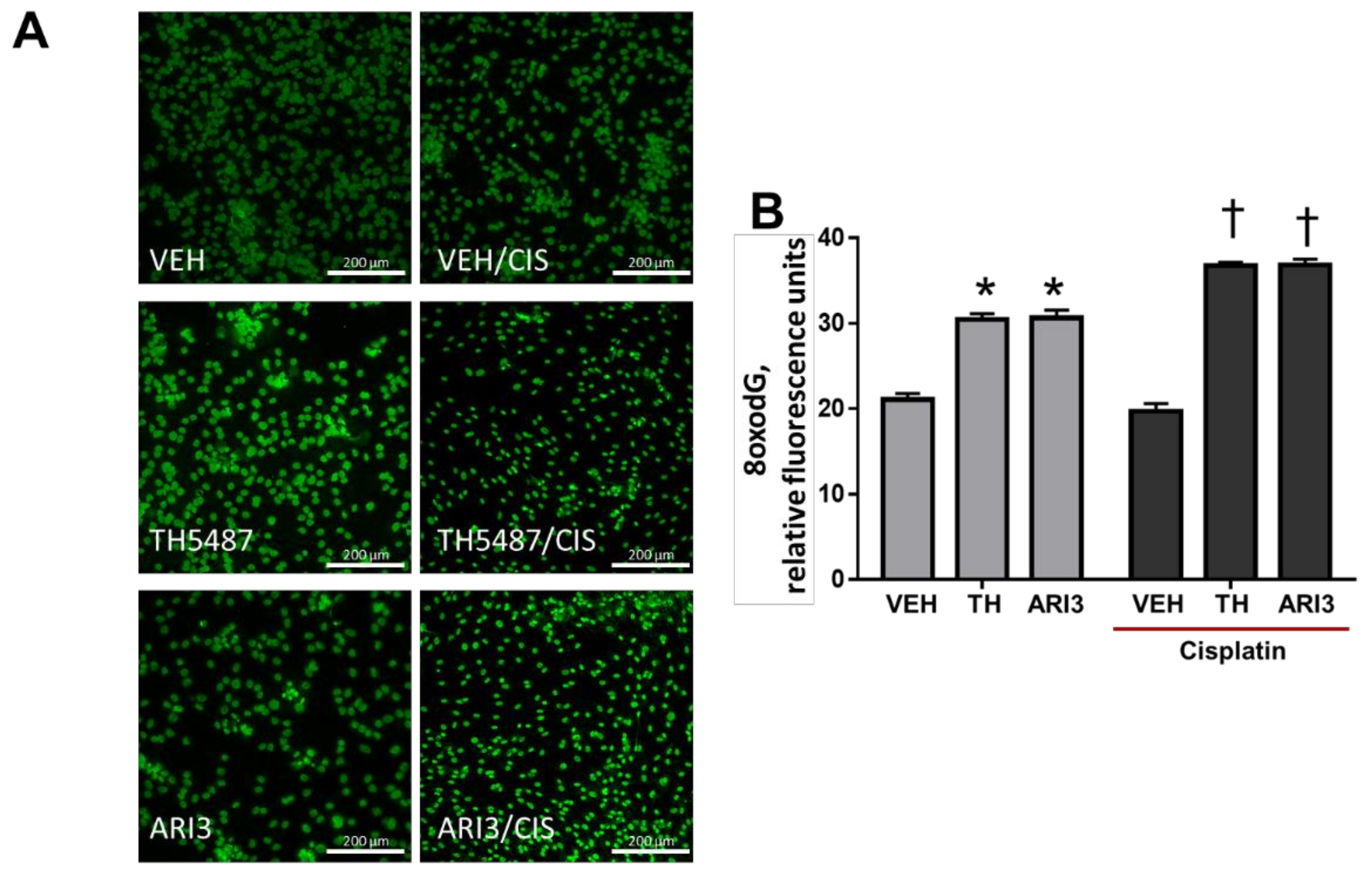
2.2. Inhibition of Either OGG1 Glycosylase Activity or APE1 DNA Repair Activity Enhances Basal Levels of 8oxo-dG Lesions and This Effect Is Exacerbated by Cisplatin Treatment
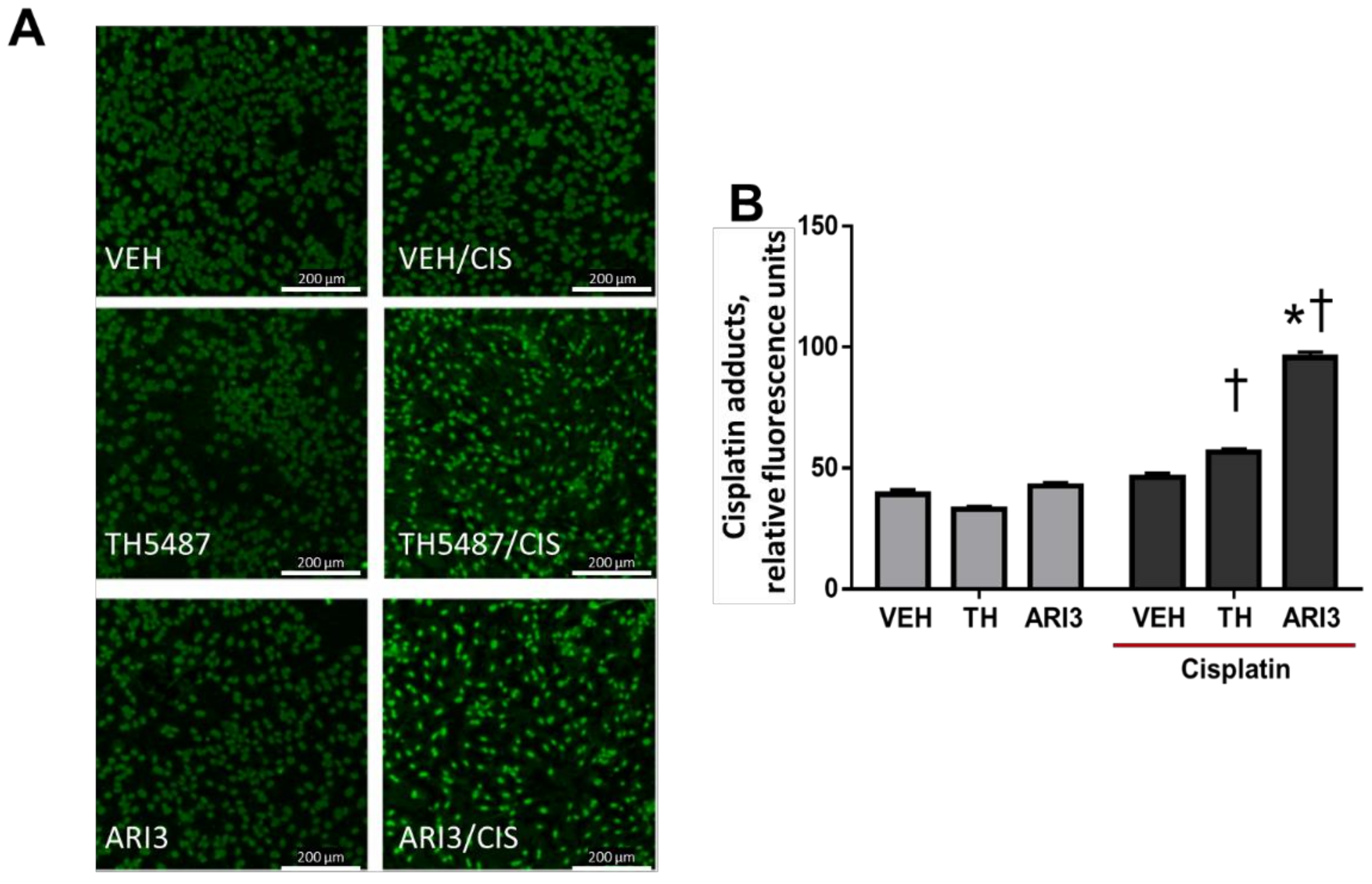
2.3. Inhibition of APE1 DNA Repair Activity Enhances Cisplatin Adduct Levels in Neuronal Cultures
2.4. Cisplatin Elicits a Decrease in Neurite Outgrowth in Cultures of Sensory Neurons, and This Is Partially Attenuated by Inhibition of OGG1 Glycosylase Activity
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation of Primary Sensory Neuron Cultures
4.3. Calcitonin Gene-Related Peptide Release
4.4. Neurite Length Assessment
4.5. Staining and Quantification of 7,8-Dihydro-8-oxo-2′-deoxyguanosine (8-oxodG) and Cisplatin Adducts
4.6. Reagents
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Podratz, J.L.; Lee, H.; Knorr, P.; Koehler, S.; Forsythe, S.; Lambrecht, K.; Arias, S.; Schmidt, K.; Steinhoff, G.; Yudintsev, G.; et al. Cisplatin induces mitochondrial deficits in Drosophila larval segmental nerve. Neurobiol. Dis. 2017, 97, 60–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobylev, I.; Joshi, A.R.; Barham, M.; Neiss, W.F.; Lehmann, H.C. Depletion of Mitofusin-2 Causes Mitochondrial Damage in Cisplatin-Induced Neuropathy. Mol. Neurobiol. 2018, 55, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.; Schmitt, L.I.; Kusterarent, P.; Kutritz, A.; Rassaf, T.; Kleinschnitz, C.; Hendgen-Cotta, U.B.; Hagenacker, T. Platinum-Based Drugs Cause Mitochondrial Dysfunction in Cultured Dorsal Root Ganglion Neurons. Int. J. Mol. Sci. 2020, 21, 8636. [Google Scholar] [CrossRef]
- Kelley, M.R.; Jiang, Y.; Guo, C.; Reed, A.; Meng, H.; Vasko, M.R. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS ONE 2014, 9, e106485. [Google Scholar] [CrossRef]
- Dzagnidze, A.; Katsarava, Z.; Makhalova, J.; Liedert, B.; Yoon, M.S.; Kaube, H.; Limmroth, V.; Thomale, J. Repair capacity for platinum-DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J. Neurosci. 2007, 27, 9451–9457. [Google Scholar] [CrossRef] [Green Version]
- Gorgun, M.F.; Zhuo, M.; Englander, E.W. Cisplatin Toxicity in Dorsal Root Ganglion Neurons Is Relieved by Meclizine via Diminution of Mitochondrial Compromise and Improved Clearance of DNA Damage. Mol. Neurobiol. 2017, 54, 7883–7895. [Google Scholar] [CrossRef]
- Jirsova, K.; Mandys, V. Differences in the inhibition of neuritic outgrowth in organotypic cultures of rat foetal dorsal root ganglia treated with cisplatin and carboplatin: A comparative study. Folia Histochem. Cytobiol. 1997, 35, 215–219. [Google Scholar]
- Donaldson, K.L.; Goolsby, G.L.; Wahl, A.F. Cytotoxicity of the anticancer agents cisplatin and taxol during cell proliferation and the cell cycle. Int. J. Cancer 1994, 57, 847–855. [Google Scholar] [CrossRef]
- Kim, Y.K.; Jung, J.S.; Lee, S.H.; Kim, Y.W. Effects of antioxidants and Ca2+ in cisplatin-induced cell injury in rabbit renal cortical slices. Toxicol. Appl. Pharm. 1997, 146, 261–269. [Google Scholar] [CrossRef]
- Siomek, A.; Tujakowski, J.; Gackowski, D.; Rozalski, R.; Foksinski, M.; Dziaman, T.; Roszkowski, K.; Olinski, R. Severe oxidatively damaged DNA after cisplatin treatment of cancer patients. Int. J. Cancer 2006, 119, 2228–2230. [Google Scholar] [CrossRef]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mutat. Res. 2007, 614, 24–36. [Google Scholar] [CrossRef]
- Yan, F.; Liu, J.J.; Ip, V.; Jamieson, S.M.; McKeage, M.J. Role of platinum DNA damage-induced transcriptional inhibition in chemotherapy-induced neuronal atrophy and peripheral neurotoxicity. J. Neurochem. 2015, 135, 1099–1112. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.; Douki, T.; Ravanat, J.L. One-electron oxidation of DNA and inflammation processes. Nat. Chem. Biol. 2006, 2, 348–349. [Google Scholar] [CrossRef]
- Margolin, Y.; Cloutier, J.F.; Shafirovich, V.; Geacintov, N.E.; Dedon, P.C. Paradoxical hotspots for guanine oxidation by a chemical mediator of inflammation. Nat. Chem. Biol. 2006, 2, 365–366. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Monden, Y.; Arai, T.; Asano, M.; Ohtsuka, E.; Aburatani, H.; Nishimura, S. Human MMH (OGG1) type 1a protein is a major enzyme for repair of 8-hydroxyguanine lesions in human cells. Biochem. Biophys. Res. Commun. 1999, 258, 605–610. [Google Scholar] [CrossRef]
- Singhal, R.K.; Prasad, R.; Wilson, S.H. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J. Biol. Chem. 1995, 270, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Vasko, M.R.; Guo, C.; Thompson, E.L.; Kelley, M.R. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA Repair 2011, 10, 942–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Guo, C.; Vasko, M.R.; Kelley, M.R. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008, 68, 6425–6434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, A.; Floyd, A.M.; Dangeti, M.; Lei, W.; Sobol, R.W.; Patrick, S.M. Differential role of base excision repair proteins in mediating cisplatin cytotoxicity. DNA Repair 2017, 51, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailer-Morrison, M.K.; Kotler, J.M.; Martin, B.D.; Sugden, K.D. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2′-deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry 2003, 42, 9761–9770. [Google Scholar] [CrossRef]
- Ramon, O.; Sauvaigo, S.; Gasparutto, D.; Faure, P.; Favier, A.; Cadet, J. Effects of 8-oxo-7,8-dihydro-2′-deoxyguanosine on the binding of the transcription factor Sp1 to its cognate target DNA sequence (GC box). Free Radic. Res. 1999, 31, 217–229. [Google Scholar] [CrossRef]
- Moore, S.P.; Toomire, K.J.; Strauss, P.R. DNA modifications repaired by base excision repair are epigenetic. DNA Repair 2013, 12, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Maeda, L.S.; Kolodner, R.D.; Hanawalt, P.C. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair 2004, 3, 483–494. [Google Scholar] [CrossRef]
- Visnes, T.; Cazares-Korner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Roychoudhury, S.; Pramanik, S.; Harris, H.L.; Tarpley, M.; Sarkar, A.; Spagnol, G.; Sorgen, P.L.; Chowdhury, D.; Band, V.; Klinkebiel, D.; et al. Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc. Natl. Acad. Sci. USA 2020, 117, 11409–11420. [Google Scholar] [CrossRef]
- Feghali, J.G.; Liu, W.; Van De Water, T.R. L-n-acetyl-cysteine protection against cisplatin-induced auditory neuronal and hair cell toxicity. Laryngoscope 2001, 111, 1147–1155. [Google Scholar] [CrossRef]
- Van den Berg, J.H.; Beijnen, J.H.; Balm, A.J.; Schellens, J.H. Future opportunities in preventing cisplatin induced ototoxicity. Cancer Treat. Rev. 2006, 32, 390–397. [Google Scholar] [CrossRef]
- Haihong, Z.; Takatsugu, M.; Jaime, C.-T.; Liwen, L.; Akihiro, N.; Horace, J.S.; Paul, M.S.; Bruce, R.S.; Amanda, J.W.; Geoffrey, P.M.; et al. Targeting human 8-oxoguanine DNA glycosylase (hOGG1) to mitochondria enhances cisplatin cytotoxicity in hepatoma cells. Carcinogenesis 2007, 28, 1629–1637. [Google Scholar]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittman, S.K.; Gracias, N.G.; Vasko, M.R.; Fehrenbacher, J.C. Paclitaxel alters the evoked release of calcitonin gene-related peptide from rat sensory neurons in culture. Exp. Neurol. 2014, 253, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Meijer, C.; de Vries, E.G.; Marmiroli, P.; Tredici, G.; Frattola, L.; Cavaletti, G. Cisplatin-induced DNA-platination in experimental dorsal root ganglia neuronopathy. Neurotoxicology 1999, 20, 883–887. [Google Scholar] [PubMed]
- McDonald, E.S.; Randon, K.R.; Knight, A.; Windebank, A.J. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: A potential mechanism for neurotoxicity. Neurobiol. Dis. 2005, 18, 305–313. [Google Scholar] [CrossRef]
- Kim, H.S.; Guo, C.; Thompson, E.L.; Jiang, Y.; Kelley, M.R.; Vasko, M.R.; Lee, S.H. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat. Res. 2015, 779, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Boyette-Davis, J.A.; Cata, J.P.; Zhang, H.; Driver, L.C.; Wendelschafer-Crabb, G.; Kennedy, W.R.; Dougherty, P.M. Follow-up psychophysical studies in bortezomib-related chemoneuropathy patients. J. Pain 2011, 12, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggioni, D.; Nicolini, G.; Chiorazzi, A.; Meregalli, C.; Cavaletti, G.; Tredici, G. Different effects of erythropoietin in cisplatin- and docetaxel-induced neurotoxicity: An in vitro study. J. Neurosci. Res. 2010, 88, 3171–3179. [Google Scholar] [CrossRef]
- Malgrange, B.; Delree, P.; Rigo, J.M.; Baron, H.; Moonen, G. Image analysis of neuritic regeneration by adult rat dorsal root ganglion neurons in culture: Quantification of the neurotoxicity of anticancer agents and of its prevention by nerve growth factor or basic fibroblast growth factor but not brain-derived neurotrophic factor or neurotrophin-3. J. Neurosci. Methods 1994, 53, 111–122. [Google Scholar]
- Pittman, S.K.; Gracias, N.G.; Fehrenbacher, J.C. Nerve growth factor alters microtubule targeting agent-induced neurotransmitter release but not MTA-induced neurite retraction in sensory neurons. Exp. Neurol. 2016, 279, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Ta, L.E.; Espeset, L.; Podratz, J.; Windebank, A.J. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. NeuroToxicology 2006, 27, 992–1002. [Google Scholar] [CrossRef]
- Wang, D.; Hara, R.; Singh, G.; Sancar, A.; Lippard, S.J. Nucleotide excision repair from site-specifically platinum-modified nucleosomes. Biochemistry 2003, 42, 6747–6753. [Google Scholar] [CrossRef]
- Clayton, D.A.; Doda, J.N.; Friedberg, E.C. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 1974, 71, 2777–2781. [Google Scholar] [CrossRef] [Green Version]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar]
- Maj, M.A.; Ma, J.; Krukowski, K.N.; Kavelaars, A.; Heijnen, C.J. Inhibition of Mitochondrial p53 Accumulation by PFT-mu Prevents Cisplatin-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 108. [Google Scholar] [CrossRef] [Green Version]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Preston, T.J.; Henderson, J.T.; McCallum, G.P.; Wells, P.G. Base excision repair of reactive oxygen species-initiated 7,8-dihydro-8-oxo-2′-deoxyguanosine inhibits the cytotoxicity of platinum anticancer drugs. Mol. Cancer 2009, 8, 2015–2026. [Google Scholar] [CrossRef] [Green Version]
- Allgayer, J.; Kitsera, N.; von der Lippen, C.; Epe, B.; Khobta, A. Modulation of base excision repair of 8-oxoguanine by the nucleotide sequence. Nucleic Acids Res. 2013, 41, 8559–8571. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized Guanine Base Lesions Function in 8-Oxoguanine DNA Glycosylase-1-mediated Epigenetic Regulation of Nuclear Factor kappaB-driven Gene Expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldogh, I.; Hajas, G.; Aguilera-Aguirre, L.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Sur, S.; Hazra, T.K.; Mitra, S. Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J. Biol. Chem. 2012, 287, 20769–20773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Hosoki, K.; Bacsi, A.; Radak, Z.; Hegde, M.L.; Sur, S.; Hazra, T.K.; Brasier, A.R.; Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase-1-mediated DNA repair is associated with Rho GTPase activation and alpha-smooth muscle actin polymerization. Free Radic. Biol. Med. 2014, 73, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell. Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Zhu, J.; Jara-Espejo, M.; Burrows, C.J. Cruciform DNA Sequences in Gene Promoters Can Impact Transcription upon Oxidative Modification of 2′-Deoxyguanosine. Biochemistry 2020, 59, 2616–2626. [Google Scholar] [CrossRef]
- Kim, N. The Interplay between G-quadruplex and Transcription. Curr. Med. Chem. 2019, 26, 2898–2917. [Google Scholar] [CrossRef]
- Mijit, M.; Caston, R.; Gampala, S.; Fishel, M.L.; Fehrenbacher, J.; Kelley, M.R. APE1/Ref-1—One Target with Multiple Indications: Emerging Aspects and New Directions. J. Cell Signal. 2021, 2, 151–161. [Google Scholar]
- Malfatti, M.C.; Antoniali, G.; Codrich, M.; Tell, G. Coping with RNA damage with a focus on APE1, a BER enzyme at the crossroad between DNA damage repair and RNA processing/decay. DNA Repair 2021, 104, 103133. [Google Scholar] [CrossRef]
- Tosolini, D.; Antoniali, G.; Dalla, E.; Tell, G. Role of phase partitioning in coordinating DNA damage response: Focus on the Apurinic Apyrimidinic Endonuclease 1 interactome. Biomol. Concepts 2020, 11, 209–220. [Google Scholar] [CrossRef]
- Kelley, M.R.; Wikel, J.H.; Guo, C.; Pollok, K.E.; Bailey, B.J.; Wireman, R.; Fishel, M.L.; Vasko, M.R. Identification and Characterization of New Chemical Entities Targeting Apurinic/Apyrimidinic Endonuclease 1 for the Prevention of Chemotherapy-Induced Peripheral Neuropathy. J. Pharm. Exp. 2016, 359, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Specificity of stimulation of human 8-oxoguanine-DNA glycosylase by AP endonuclease. Biochem. Biophys. Res. Commun. 2008, 368, 175–179. [Google Scholar] [CrossRef]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef]
- Kothandapani, A.; Dangeti, V.S.; Brown, A.R.; Banze, L.A.; Wang, X.H.; Sobol, R.W.; Patrick, S.M. Novel role of base excision repair in mediating cisplatin cytotoxicity. J. Biol. Chem. 2011, 286, 14564–14574. [Google Scholar] [CrossRef] [Green Version]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair 2017, 56, 51–64. [Google Scholar] [CrossRef]
- Kothandapani, A.; Sawant, A.; Dangeti, V.S.; Sobol, R.W.; Patrick, S.M. Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity. Nucleic Acids Res. 2013, 41, 7332–7343. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.K.; Muftuoglu, M.; Beck, G.; Imam, S.Z.; Bohr, V.A.; Wilson, D.M., 3rd. Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007, 35, 4103–4113. [Google Scholar] [CrossRef]
- Boukelmoune, N.; Laumet, G.; Tang, Y.; Ma, J.; Mahant, I.; Singh, S.K.; Nijboer, C.; Benders, M.; Kavelaars, A.; Heijnen, C.J. Nasal administration of mesenchymal stem cells reverses chemotherapy-induced peripheral neuropathy in mice. Brain Behav. Immun. 2021, 93, 43–54. [Google Scholar] [CrossRef]
- Tan, D.; Zhang, H.; Deng, J.; Liu, J.; Wen, J.; Li, L.; Wang, X.; Pan, M.; Hu, X.; Guo, J. RhoA-GTPase Modulates Neurite Outgrowth by Regulating the Expression of Spastin and p60-Katanin. Cells 2020, 9, 230. [Google Scholar] [CrossRef] [Green Version]
- Calls, A.; Torres-Espin, A.; Navarro, X.; Yuste, V.J.; Udina, E.; Bruna, J. Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro-Oncology 2021, 23, 88–99. [Google Scholar] [CrossRef]
- Acklin, S.; Zhang, M.; Du, W.; Zhao, X.; Plotkin, M.; Chang, J.; Campisi, J.; Zhou, D.; Xia, F. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci. Rep. 2020, 10, 14170. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Yang, X.; Lu, X.; Dai, N.; Zhang, S.; Cheng, Y.; Zhang, L.; Yang, Y.; Liu, Y.; Yang, Z.; et al. APE1 deficiency promotes cellular senescence and premature aging features. Nucleic Acids Res. 2018, 46, 5664–5677. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.M.F.; Helleday, T.; Mortusewicz, O. OGG1 Inhibitor TH5487 Alters OGG1 Chromatin Dynamics and Prevents Incisions. Biomolecules 2020, 10, 1483. [Google Scholar] [CrossRef]
- Rai, G.; Vyjayanti, V.N.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M., 3rd; Maloney, D.J. Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. J. Med. Chem. 2012, 55, 3101–3112. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behrouzi, A.; Xia, H.; Thompson, E.L.; Kelley, M.R.; Fehrenbacher, J.C. Oxidative DNA Damage and Cisplatin Neurotoxicity Is Exacerbated by Inhibition of OGG1 Glycosylase Activity and APE1 Endonuclease Activity in Sensory Neurons. Int. J. Mol. Sci. 2022, 23, 1909. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031909
Behrouzi A, Xia H, Thompson EL, Kelley MR, Fehrenbacher JC. Oxidative DNA Damage and Cisplatin Neurotoxicity Is Exacerbated by Inhibition of OGG1 Glycosylase Activity and APE1 Endonuclease Activity in Sensory Neurons. International Journal of Molecular Sciences. 2022; 23(3):1909. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031909
Chicago/Turabian StyleBehrouzi, Adib, Hanyu Xia, Eric L. Thompson, Mark R. Kelley, and Jill C. Fehrenbacher. 2022. "Oxidative DNA Damage and Cisplatin Neurotoxicity Is Exacerbated by Inhibition of OGG1 Glycosylase Activity and APE1 Endonuclease Activity in Sensory Neurons" International Journal of Molecular Sciences 23, no. 3: 1909. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031909