
DRG2 Depletion Promotes Endothelial Cell Senescence and Vascular Endothelial Dysfunction

 ,
, 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
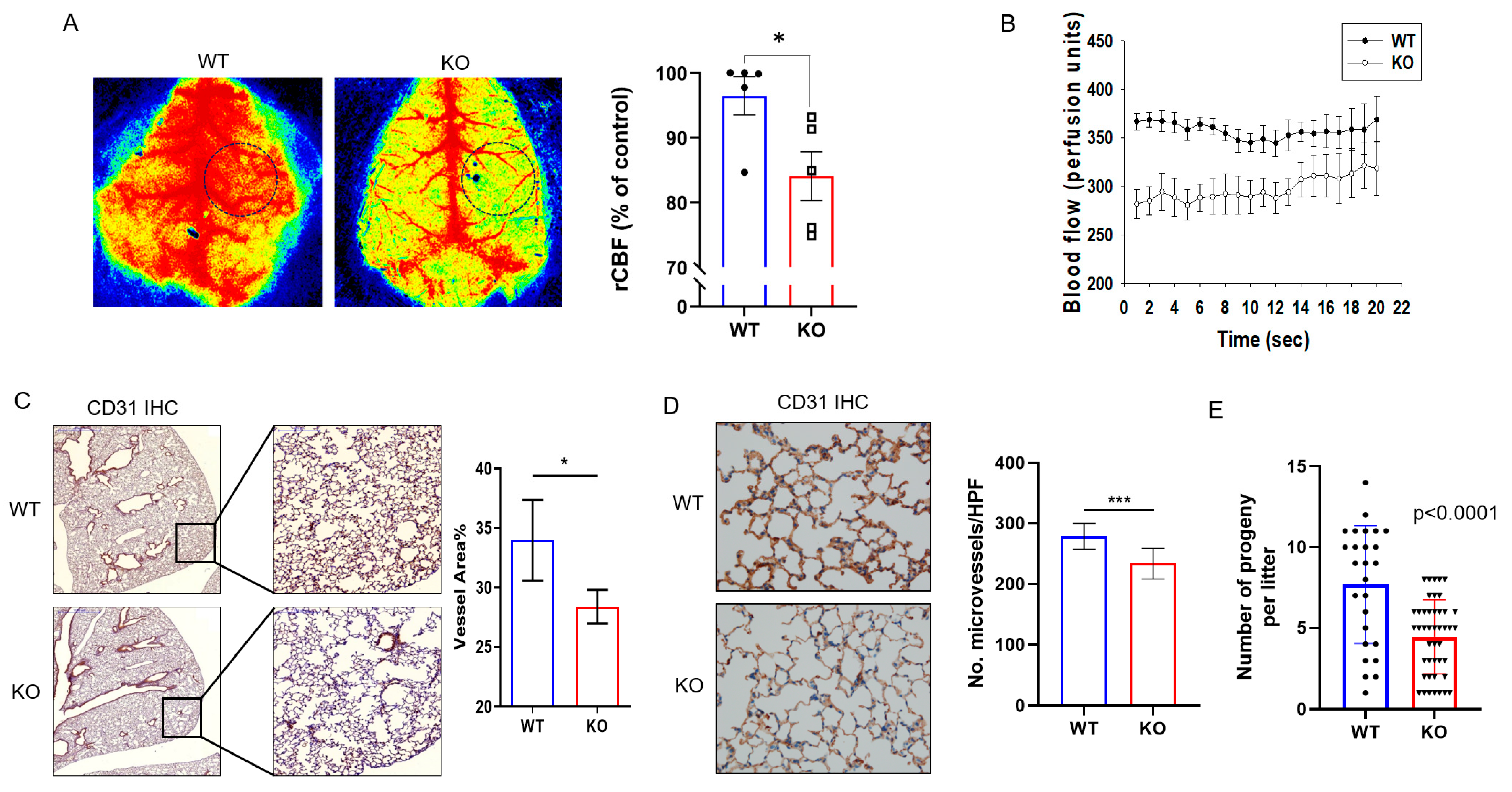
2.1. DRG2−/− Mice Exhibit Decreased Microvascular Circulation and Blood Vessel Density
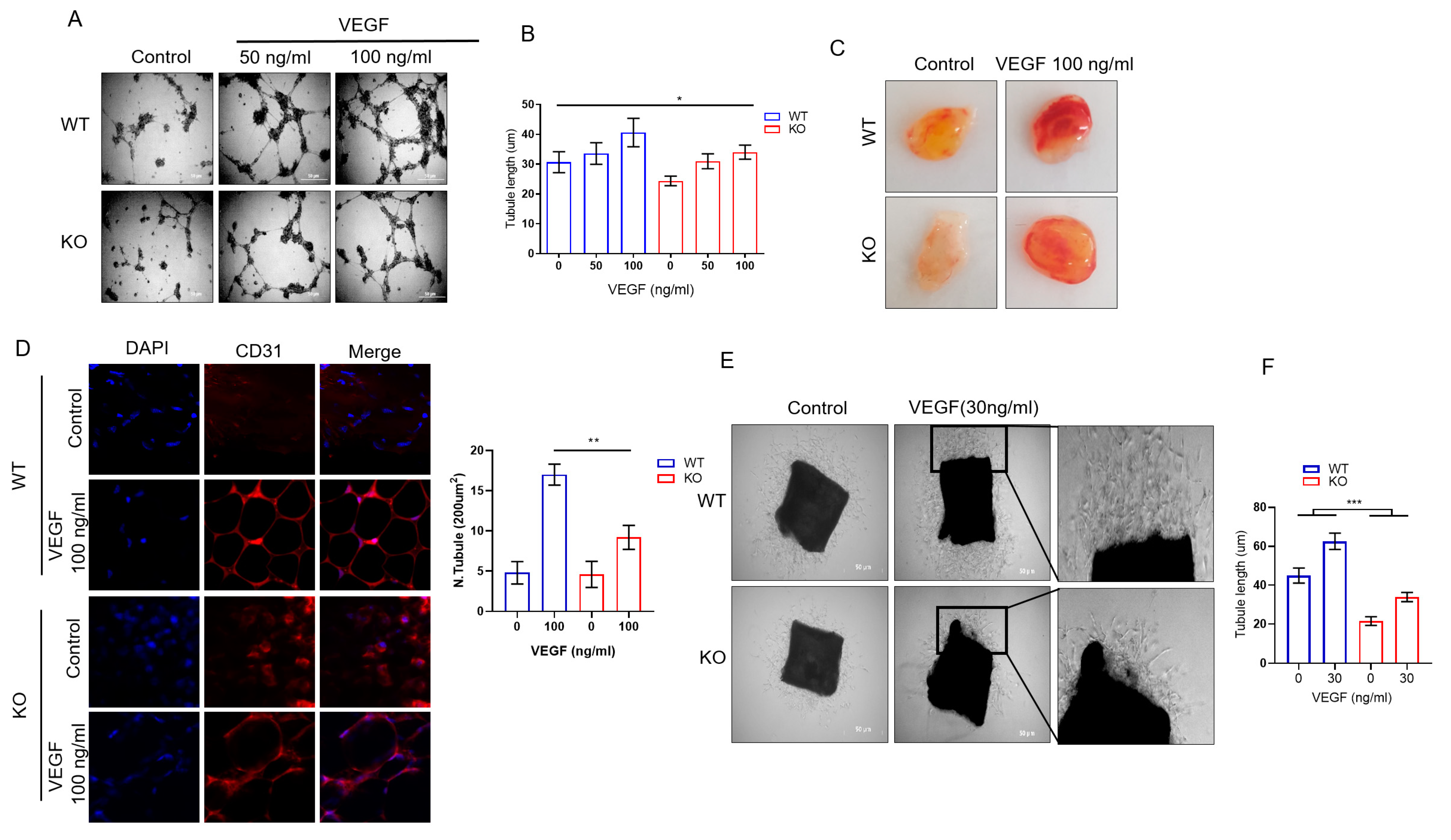
2.2. DRG2 Deficiency Inhibits Angiogenic Functions of Endothelial Cells
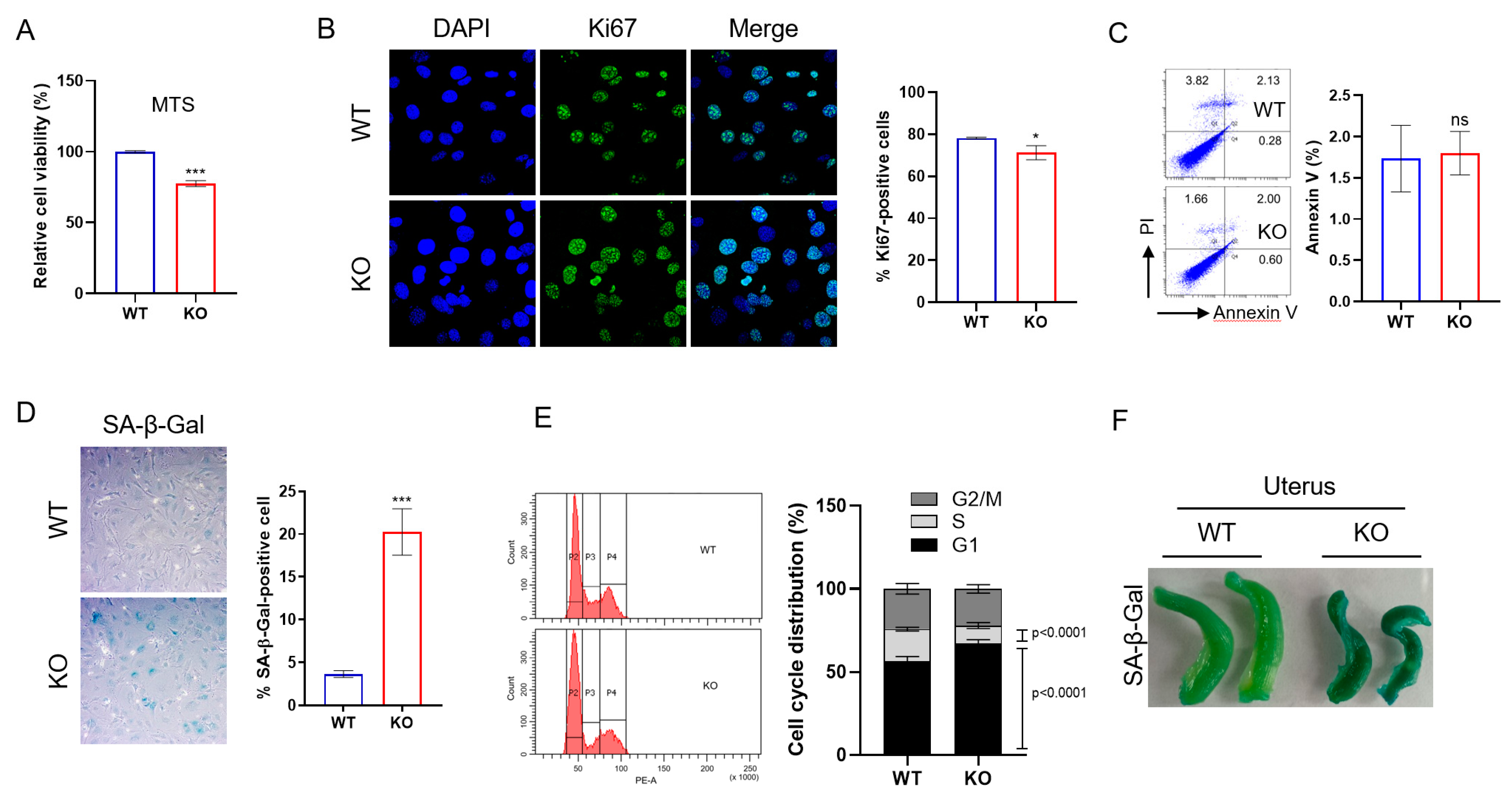
2.3. DRG2 Deficiency Decreases Proliferation and Enhances Senescence of Endothelial Cells
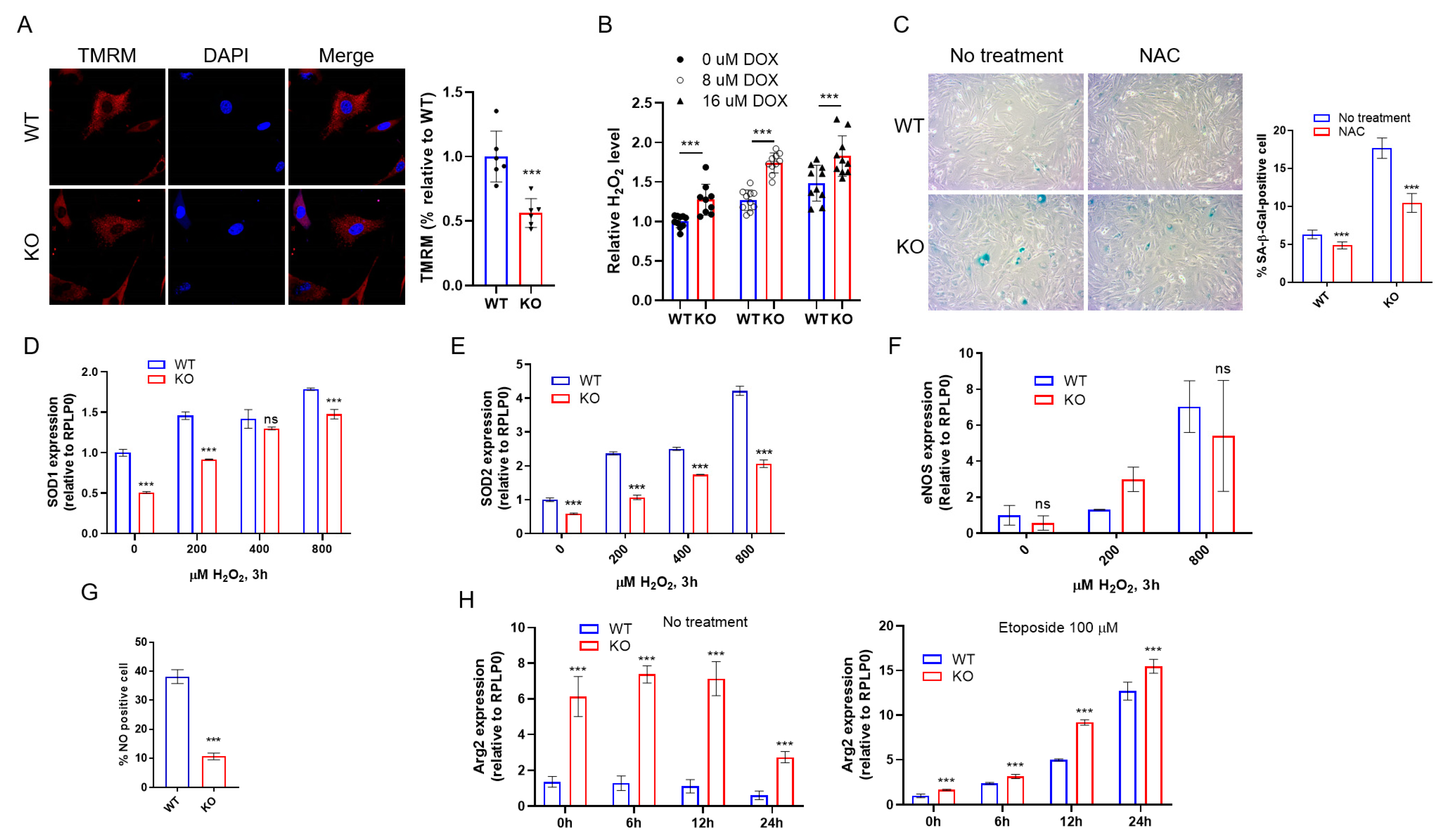
2.4. DRG2 Deficiency Enhances DNA Damage and Senescence Induced by Oxidative Stress
2.5. DRG2 Deficiency Affect the Expression of Antioxidant Genes
2.6. DRG2 Deficiency Decreases eNOS Expression and NO Production in Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mice and Animal Research Ethics
4.3. Cortical Cerebral Blood Flow Measurement
4.4. In Vivo Matrigel Plug Assays
4.5. Aortic Ring Assay
4.6. Immunohistochemistry and Quantification of Lung Angiogenesis
4.7. Endothelial Tube Formation In Vitro Assays
4.8. Colony-Forming Assay
4.9. Cell Cycle Analysis
4.10. Ki-67 Analysis
4.11. MTS Assay
4.12. Annexin-V Staining
4.13. Senescence-Associated β-Galactosidase (SA β-gal) Activity
4.14. Confocal Microscopy
4.15. SDS-PAGE Analysis and Immunoblotting
4.16. Quantitative Real-Time PCR and Semi-qRT-PCR
4.17. Measurement of NO Production
4.18. Measurement of ROS Levels
4.19. Statistics Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilken, H.M.; Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010, 22, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I. Vascular Cell Senescence. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef] [Green Version]
- Counter, C.M.; Avilion, A.A.; Le Feuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [CrossRef]
- Colavitti, R.; Finkel, T. Reactive Oxygen Species as Mediators of Cellular Senescence. IUBMB Life 2005, 57, 277–281. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative Med. Cell. Longev. 2016, 2016, e3565127. [Google Scholar] [CrossRef] [Green Version]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH Oxidases in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Kaley, G.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Mechanisms of Vascular Aging: New Perspectives. J. Gerontol. Ser. A 2010, 65A, 1028–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Förstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef]
- Ming, X.-F.; Barandier, C.; Viswambharan, H.; Kwak, B.R.; Mach, F.; Mazzolai, L.; Hayoz, D.; Ruffieux, J.; Rusconi, S.; Montani, J.-P.; et al. Thrombin Stimulates Human Endothelial Arginase Enzymatic Activity via RhoA/ROCK Pathway. Circulation 2004, 110, 3708–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luiking, Y.C.; Ten Have, G.A.M.; Wolfe, R.R.; Deutz, N.E.P. Arginine de novo and nitric oxide production in disease states. Am. J. Physiol. -Endocrinol. Metab. 2012, 303, E1177–E1189. [Google Scholar] [CrossRef] [Green Version]
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase Reciprocally Regulates Nitric Oxide Synthase Activity and Contributes to Endothelial Dysfunction in Aging Blood Vessels. Circulation 2003, 108, 2000–2006. [Google Scholar] [CrossRef] [Green Version]
- Steppan, J.; Ryoo, S.; Schuleri, K.H.; Gregg, C.; Hasan, R.K.; White, A.R.; Bugaj, L.J.; Khan, M.; Santhanam, L.; Nyhan, D.; et al. Arginase modulates myocardial contractility by a nitric oxide synthase 1-dependent mechanism. Proc. Natl. Acad. Sci. 2006, 103, 4759–4764. [Google Scholar] [CrossRef] [Green Version]
- Topal, G.; Brunet, A.; Walch, L.; Boucher, J.-L.; David-Dufilho, M. Mitochondrial Arginase II Modulates Nitric-Oxide Synthesis through Nonfreely Exchangeable l-Arginine Pools in Human Endothelial Cells. J. Pharmacol. Exp. Ther. 2006, 318, 1368–1374. [Google Scholar] [CrossRef] [Green Version]
- Schenker, T.; Lach, C.; Kessler, B.; Calderara, S.; Trueb, B. A novel GTP-binding protein which is selectively repressed in SV40 transformed fibroblasts. J. Biol. Chem. 1994, 269, 25447–25453. [Google Scholar] [CrossRef]
- Li, B.; Trueb, B. DRG represents a family of two closely related GTP-binding proteins. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1491, 196–204. [Google Scholar] [CrossRef]
- Ishikawa, K.; Azuma, S.; Ikawa, S.; Semba, K.; Inoue, J. Identification of DRG family regulatory proteins (DFRPs): Specific regulation of DRG1 and DRG2. Genes Cells Devoted Mol. Cell. Mech. 2005, 10, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.S.; Lee, U.H.; Kim, S.I.; Kim, H.J.; Park, J.J.; Cha, S.J.; Kim, S.B.; Song, H.; Chung, D.K.; Han, I.S.; et al. Overexpression of DRG2 suppresses the growth of Jurkat T cells but does not induce apoptosis. Arch. Biochem. Biophys. 2004, 422, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Song, H. Overexpression of DRG2 Increases G2/M Phase Cells and Decreases Sensitivity to Nocodazole-Induced Apoptosis. J. Biochem. 2004, 135, 331–335. [Google Scholar] [CrossRef]
- Ko, M.S.; Kim, H.J.; Kim, H.K.; Yoon, N.A.; Lee, U.H.; Lee, S.C.; Chung, D.K.; Lee, B.J.; Suh, J.H.; Cho, W.J.; et al. Developmentally regulated GTP-binding protein 2 ameliorates EAE by suppressing the development of TH17 cells. Clin. Immunol. 2014, 150, 225–235. [Google Scholar] [CrossRef]
- Mani, M.; Lee, U.H.; Yoon, N.A.; Kim, H.J.; Ko, M.S.; Seol, W.; Joe, Y.; Chung, H.T.; Lee, B.J.; Moon, C.H.; et al. Developmentally regulated GTP-binding protein 2 coordinates Rab5 activity and transferrin recycling. MBoC 2016, 27, 334–348. [Google Scholar] [CrossRef]
- Mani, M.; Thao, D.T.; Kim, B.C.; Lee, U.H.; Kim, D.J.; Jang, S.H.; Back, S.H.; Lee, B.J.; Cho, W.J.; Han, I.-S.; et al. DRG2 knockdown induces Golgi fragmentation via GSK3β phosphorylation and microtubule stabilization. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 1463–1474. [Google Scholar] [CrossRef]
- Vo, M.-T.; Ko, M.S.; Lee, U.H.; Yoon, E.H.; Lee, B.J.; Cho, W.J.; Ha, C.M.; Kim, K.; Park, J.W. Developmentally regulated GTP-binding protein 2 depletion leads to mitochondrial dysfunction through downregulation of dynamin-related protein 1. Biochem. Biophys. Res. Commun. 2017, 486, 1014–1020. [Google Scholar] [CrossRef]
- Mani, M.; Lee, U.H.; Yoon, N.A.; Yoon, E.H.; Lee, B.J.; Cho, W.J.; Park, J.W. Developmentally regulated GTP-binding protein 2 is required for stabilization of Rac1-positive membrane tubules. Biochem. Biophys. Res. Commun. 2017, 493, 758–764. [Google Scholar] [CrossRef]
- Yoon, N.A.; Jung, S.J.; Choi, S.H.; Ryu, J.H.; Mani, M.; Lee, U.H.; Vo, M.-T.; Jeon, D.Y.; Chung, S.W.; Ju Lee, B.; et al. DRG2 supports the growth of primary tumors and metastases of melanoma by enhancing VEGF-A expression. FEBS J. 2020, 287, 2070–2086. [Google Scholar] [CrossRef]
- Lim, H.R.; Vo, M.-T.; Kim, D.J.; Lee, U.H.; Yoon, J.H.; Kim, H.-J.; Kim, J.; Kim, S.R.; Lee, J.Y.; Yang, C.H.; et al. DRG2 Deficient Mice Exhibit Impaired Motor Behaviors with Reduced Striatal Dopamine Release. Int. J. Mol. Sci. 2020, 21, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox. Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. 1995, 92, 4337–4341. [Google Scholar] [CrossRef] [Green Version]
- Fatokun, A.A.; Stone, T.W.; Smith, R.A. Hydrogen peroxide-induced oxidative stress in MC3T3-E1 cells: The effects of glutamate and protection by purines. Bone 2006, 39, 542–551. [Google Scholar] [CrossRef]
- Gutiérrez-Uzquiza, Á.; Arechederra, M.; Bragado, P.; Aguirre-Ghiso, J.A.; Porras, A. p38α Mediates Cell Survival in Response to Oxidative Stress via Induction of Antioxidant Genes: Effect on the p70S6K pathway. J. Biol. Chem. 2012, 287, 2632–2642. [Google Scholar] [CrossRef] [Green Version]
- Ou, H.L.; Schumacher, B. DNA Damage Responses and P53 in the Aging Process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Lou, Z.; Chen, J. Cellular senescence and DNA repair. Exp. Cell Res. 2006, 312, 2641–2646. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLOS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.-F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. 2006, 103, 17018–17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Bugaj, L.J.; Oh, Y.J.; Bivalacqua, T.J.; Ryoo, S.; Soucy, K.G.; Santhanam, L.; Webb, A.; Camara, A.; Sikka, G.; et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J. Appl. Physiol. 2009, 107, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.; Berkowitz, D.E.; Ryoo, S. Increased arginase II activity contributes to endothelial dysfunction through endothelial nitric oxide synthase uncoupling in aged mice. Exp. Mol. Med. 2012, 44, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Ashby, J.W.; Mack, J.J. Endothelial Control of Cerebral Blood Flow. Am. J. Pathol. 2021, 191, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Finkel, T. Free radicals and senescence. Exp. Cell Res. 2008, 314, 1918–1922. [Google Scholar] [CrossRef]
- Rai, P.; Onder, T.T.; Young, J.J.; McFaline, J.L.; Pang, B.; Dedon, P.C.; Weinberg, R.A. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc. Natl. Acad. Sci. 2009, 106, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Bringold, F.; Serrano, M. Tumor suppressors and oncogenes in cellular senescence. Exp. Gerontol. 2000, 35, 317–329. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Y.; St. Clair, D.K. ROS and p53: A versatile partnership. Free. Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unemori, E.N.; Ferrara, N.; Bauer, E.A.; Amento, E.P. Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J. Cell Physiol. 1992, 153, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen Reduction by Nitric-oxide Synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Bachetti, T.; Comini, L.; Francolini, G.; Bastianon, D.; Valetti, B.; Cadei, M.; Grigolato, P.; Suzuki, H.; Finazzi, D.; Albertini, A.; et al. Arginase pathway in human endothelial cells in pathophysiological conditions. J. Mol. Cell. Cardiol. 2004, 37, 515–523. [Google Scholar] [CrossRef]
- Passaniti, A. Extracellular matrix-cell interactions: Matrigel and complex cellular pattern formation. Lab. Investig. 1992, 67, 804. [Google Scholar]
- Reynolds, L.E.; Wyder, L.; Lively, J.C.; Taverna, D.; Robinson, S.D.; Huang, X.; Sheppard, D.; Hynes, R.O.; Hodivala-Dilke, K.M. Enhanced pathological angiogenesis in mice lacking β3 integrin or β3 and β5 integrins. Nat. Med. 2002, 8, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Weidner, N.; Carroll, P.R.; Flax, J.; Blumenfeld, W.; Folkman, J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am. J. Pathol. 1993, 143, 401–409. [Google Scholar] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, A.-N.; Park, S.-S.; Le, M.-X.; Lee, U.H.; Ko, B.K.; Lim, H.R.; Yu, R.; Choi, S.H.; Lee, B.J.; Ham, S.-Y.; et al. DRG2 Depletion Promotes Endothelial Cell Senescence and Vascular Endothelial Dysfunction. Int. J. Mol. Sci. 2022, 23, 2877. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052877
Le A-N, Park S-S, Le M-X, Lee UH, Ko BK, Lim HR, Yu R, Choi SH, Lee BJ, Ham S-Y, et al. DRG2 Depletion Promotes Endothelial Cell Senescence and Vascular Endothelial Dysfunction. International Journal of Molecular Sciences. 2022; 23(5):2877. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052877
Chicago/Turabian StyleLe, Anh-Nhung, Seong-Soon Park, Minh-Xuan Le, Unn Hwa Lee, Byung Kyun Ko, Hye Ryeong Lim, Ri Yu, Seong Hee Choi, Byung Ju Lee, Soo-Youn Ham, and et al. 2022. "DRG2 Depletion Promotes Endothelial Cell Senescence and Vascular Endothelial Dysfunction" International Journal of Molecular Sciences 23, no. 5: 2877. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052877