
STAT3 Role in T-Cell Memory Formation

Department of Pathology and Laboratory Medicine, Center for Cellular Immunotherapies, University of Pennsylvania, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(5), 2878; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052878
Submission received: 22 October 2021
/
Revised: 28 February 2022
/
Accepted: 3 March 2022
/
Published: 7 March 2022
(This article belongs to the Special Issue Advances in Immuno-Oncology)
{kind=link}
{kind=link}
Abstract
:Along with the clinical success of immuno-oncology drugs and cellular therapies, T-cell biology has attracted considerable attention in the immunology community. Long-term immunity, traditionally analyzed in the context of infection, is increasingly studied in cancer. Many signaling pathways, transcription factors, and metabolic regulators have been shown to participate in the formation of memory T cells. There is increasing evidence that the signal transducer and activator of transcription-3 (STAT3) signaling pathway is crucial for the formation of long-term T-cell immunity capable of efficient recall responses. In this review, we summarize what is currently known about STAT3 role in the context of memory T-cell formation and antitumor immunity.
1. STAT3 Signaling Overview
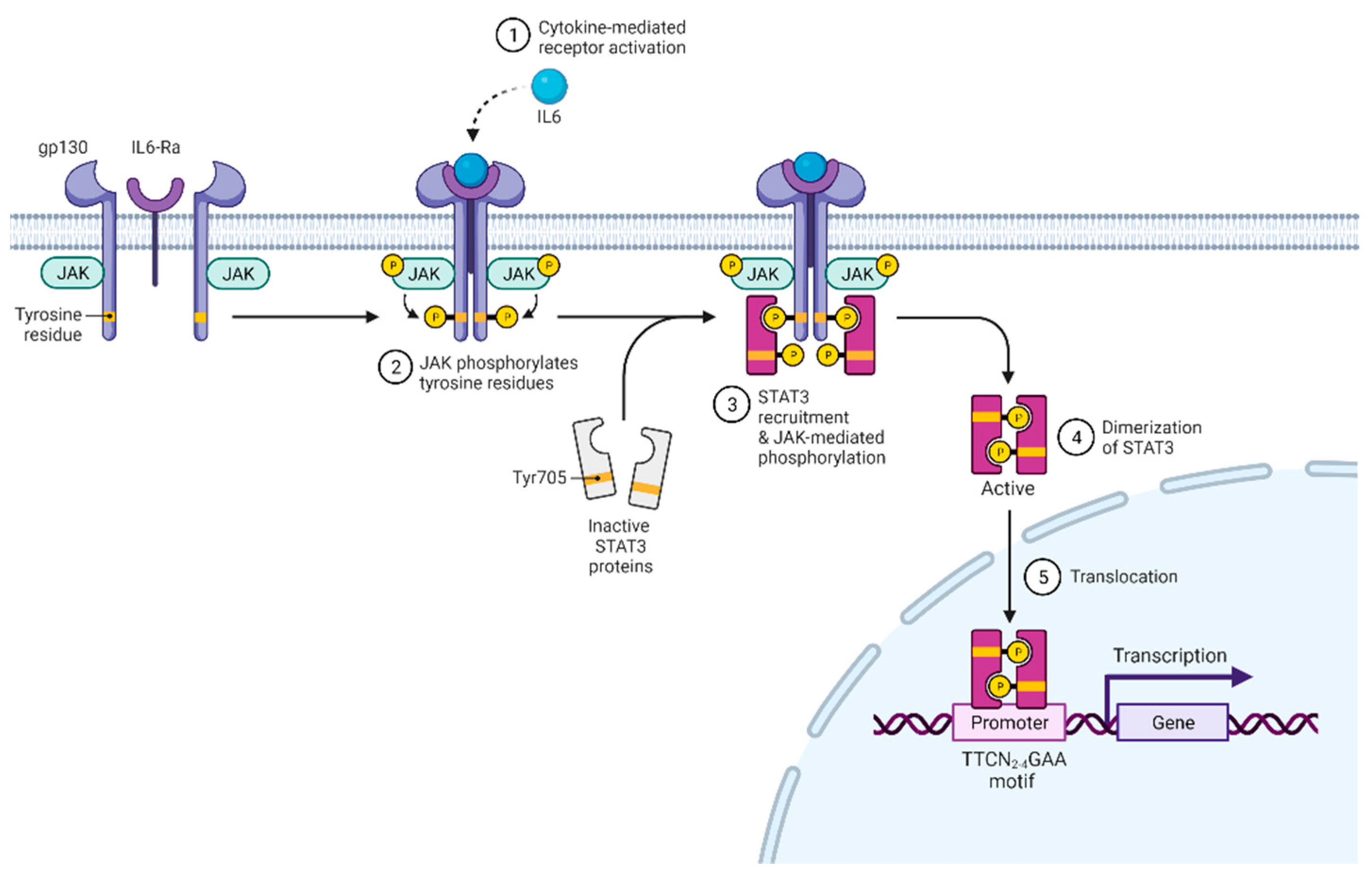
STAT3 was discovered in 1994 and initially shown to have transcription factor activity in response to IL-6 and EGF [1,2]. Subsequently, STAT3 was shown to be activated by various growth factors and cytokines acting through the gp130 subunit, common γ and β chain subunits, homodimeric cytokine receptors, type II receptors, receptor tyrosine kinases, and G-protein-coupled receptors [3]. After receptor activation and oligomerization (Figure 1), Janus kinases (JAK1, JAK2, JAK3, TYK2) bind to the receptor, phosphorylate it, and create binding sites for the SH2 domain of STAT3 [4]. STAT3 binds to phosphor-(p)YXXQ motifs on the receptor and is subsequently phosphorylated at Tyr705 by JAK [5,6]. This modification allows STAT3 to dimerize and initiate transcription regulation [7]. Besides Tyr705 residue, Ser727 can also be phosphorylated, which increases STAT3 transcriptional activity [8,9]. Besides being phosphorylated, STAT3 can also be methylated, acetylated, or exert transcriptional activity in an unphosphorylated form [10,11,12,13,14,15,16]. Future studies are required to understand how relevant these modifications are to T cells.
The primary role of STAT3 is transcriptional regulation, and it preferentially binds GAS elements with the consensus motif TTCN2-4GAA [17]. STAT3 target genes are numerous, and genome-wide ChIP-Seq analysis of STAT3 in CD4+ showed about 3000 targets [18]. Moreover, unphosphorylated STAT3 can also act as a transcriptional coregulator with collaboration with other proteins [12,15,19].
STAT3 also has important non-translational activities. It was shown to promote mitochondrial metabolism with an effect on other cellular functions [20,21,22]. During the activation of CD4+ T cells, STAT3 accumulates at mitochondria and sustains high mitochondrial membrane potential skewing differentiation toward the Th17 phenotype [23].
STAT3 is pleiotropic and was shown to play a role in physiology and development [24,25]. Its knockout leads to embryonic lethality [26], and STAT3 signaling is essential for cardiac function and maintaining the differentiation potential of stem cells [27,28]. In the blood compartment, STAT3 is required for the proper functioning of both B cells [29,30,31,32] and myeloid cells [33,34,35]. More importantly for the T-cell context, STAT3 signaling is required for hematopoiesis [36,37,38] and the proper development of the thymus [39,40,41] and can lead to abnormalities if perturbed. Moreover, there is increasing evidence that STAT3 signaling plays a role at the late stage of T-cell development [42,43]. For that reason, mouse models with a conditional knockout in immature T cells should be interpreted with caution as the observed phenotype in mature T cells can be affected by prior development abnormalities.
2. STAT3 Role in Th17 and Tfh Differentiation
STAT3 is crucial for the differentiation of specific CD4+ T cells subsets. Th17 is a proinflammatory CD4+ effector subset characterized by IL-17 secretion and its differentiation from naïve CD4+ T cells, which occurs in the presence of TGF-β and IL-6 [44,45]. Through STAT3, IL-6 induces the expression of lineage-defining transcription factor RORγt, and IL-21 and IL-23 receptors. IL-21 and IL-23 then create a positive feedback loop and further increase RORγt. STAT3 also induces IL-17 expression in collaboration with RORγt, and STAT3-targeted deletion impairs Th17 differentiation [46,47]. In the presence of IL-6 and/or IL-21 and the absence of TGF-β during priming, CD4+ T cells can differentiate into Tfh (follicular helper) subset [48]. This T-cell subset is characterized by CXCR5 expression and is necessary for B-cell activation and germinal center formation [49]. BCL-6 is the lineage-defining factor of Tfh, whose expression is also positively regulated by STAT3 [48,50]. It is important to note that, in these settings, lineage-defining factors such as STAT3 promote differentiation into specific CD4+ subtypes, in addition to restricting differentiation into alternative subtypes [51,52]. In support of this idea, Martinez-Fabregas et al. (2019) performed STAT3 ChIP-seq and RNA-seq in human Th1 cells and found that, out of 3400 STAT3 bound genes, only 23 were actually upregulated [53]. Th1 differentiation is driven by the STAT4–T-bet signaling axis and suppresses alternative CD4+ T-cell fates [54].
3. STAT3 Role in T-Cell Memory Formation
The crucial role of STAT3 in T-cell survival was initially demonstrated by the conditional deletion of this transcription factor in T-cell lineage [55]. In this system, T cells from STAT3-deficient mice were compared with T cells from wild-type mice in vitro. IL-6/STAT3 signaling was shown to have an antiapoptotic effect on T cells independent of Bcl-2 protein. This effect was abrogated in STAT3-deficient cells. IL-6/STAT3 signaling did not have any impact on T cell proliferation. Subsequently, the positive effect of STAT3 signaling on T-cell survival and its lack of proliferation was confirmed in another model [56]. They vaccinated mice with pigeon cytochrome C with or without concurrent IL-6 administration. IL-6 administration did not lead to more significant primary T-cell division, but instead reduced the apoptosis of antigen-specific T cells and increased their numbers 72 days after immunization. Although recall responses of memory CD4+ from both IL-6 treated and nontreated groups were similar, this study shows the STAT3 role in T-cell memory formation in vivo. The survival benefit of IL-6/STAT3 signaling in CD8+ T cells was demonstrated in the adoptive transfer mouse model [57]. After adoptive transfer, IL6Ra+ CD8+ T cells survived longer and had better recall responses than IL6Ra- counterparts. Durant et al. (2010) performed a ChIP-seq analysis of STAT3 in CD4+ and showed that STAT3 binds to Bcl-2 and activates its expression [18]. Moreover, the addition of STAT5-activating IL-2 could not rescue the viability of STAT3-/- suggesting nonoverlapping roles of STAT3 and STAT5 in CD4+ T-cell survival. Another group used conditional knockout of IL-6 receptor alpha chain in double-positive CD4+ CD8+ T-cell precursors and demonstrated that IL-6/STAT3 signaling is essential for proper CD4+ T-cell memory formation [58]. Wild-type or IL6raKO mice were immunized with OVA peptide with a second immunization 60 days later. Seven days after the second immunization, CD4+ T cells from IL6raT-KO hosts were analyzed and showed impaired proliferation and cytokine production in vitro. Interestingly, IL6ra and gp130 are downregulated upon TCR engagement in both CD8+ and CD4+ T cells, demonstrating the importance of IL-6 signaling timing [59]. Supporting previous findings, Oh et al. (2011) showed that STAT3 constrains T-cell proliferation but enhances survival. They used CD4-Cre STAT3 conditional deletion to demonstrate STAT3-driven expression of Bcl-2, FoxO1, and FoxO3a. STAT3 directly bound FoxO1 and FoxO3a promoters and STAT3 deficiency caused downregulation of FoxO target genes: p27Kip1 and IkB. The former (p27Kip1) is a cell-cycle inhibitor, and the latter (IkB) is inhibitor of NF-kB, the pathway that is required for the production of mitogenic IL-2.
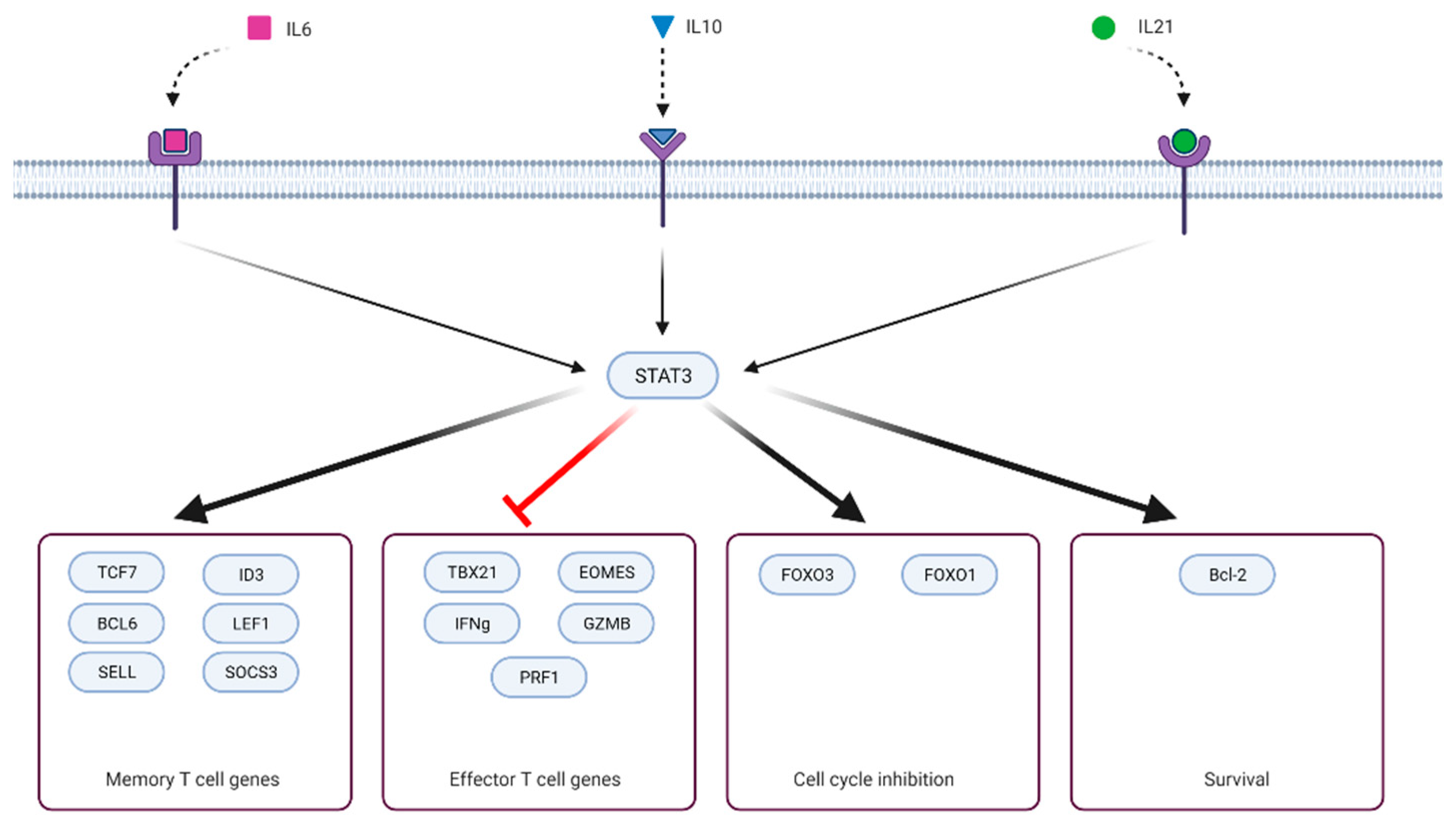
The role of STAT3 in driving expression of T-cell memory-related BCL6 and SOCS3 was revealed in the acute LCMV mouse model with conditional knockout of STAT3 in CD8+ T cells driven by granzyme B promoter [60]. IL-21 and IL-10 activate STAT3, and the IL-21/IL-10/STAT3/BCL6/SOCS3 axis was shown to be essential for the formation of memory precursors CD8+ T cells (KLRG1- IL7R+). Removal of any component from this axis led to an impaired accumulation of memory precursors T cells. As a result, significantly fewer memory CD8+ T cells formed in the long term. Mechanistically, SOCS3 reduces IL-12-driven phosphorylation of STAT4, which is known to promote short-lived effector (KLRG1+ IL7R-) and suppress memory precursor differentiation [61]. At least in CD4+ T cells, the SOCS3 locus is directly activated by STAT3 binding [18]. In another study, this group immunized mice with dendritic cells in the presence of different adjuvants [62]. They showed that LPS as adjuvant increased serum levels of IL-6 and IL-10 and resulted in more T-cell memory precursors and better secondary immune response. CpG adjuvant, on the other hand, did not induce the expression of either cytokine and, instead, led to inferior memory T-cell formation. The role of IL-6/STAT3 in memory T-cell formation was also indirectly demonstrated when antigen-specific mouse T cells were cultured with APC in the presence of the cytokine [63]. In this system, IL-6 induced expression of ID3, a transcription factor that is essential for proper differentiation of memory T cells. Interestingly, IL-21, which also activates STAT3 and drives memory formation, did not induce ID3 expression. If anything, IL-21 upregulated expression of ID3-antagonizing transcription factor ID2, likely through STAT4 activation. The role of IL-10 in T-cell memory formation was demonstrated earlier in the Listeria infection model [64]. IL-10 deficient mice had a better primary response but fewer memory T cells and suboptimal secondary response.
It is essential to realize that T-cell memory formation negatively correlates with effector function (Figure 2). Key transcription factors (that also initiate epigenetic reprogramming) drive memory differentiation simultaneously blocking terminal effector differentiation [63,65,66,67,68,69]. Depending on the presence, timing, and strength of signals (antigen, costimulatory/inhibitory molecules, cytokines or other signaling molecules, small molecules, ions) that initiate transcriptional reprogramming, T cells can acquire some or all attributes of memory or effector cell lineage [70,71,72,73,74,75,76]. STAT3 signaling is part of the memory formation program and was shown to downregulate T-cell cytotoxic effector functions. Ciucci et al. (2017) cultured CD8+ under Th17 polarizing conditions (IL-6, TGF-b, antibodies against IFNγ, IL-4, and IL-12) and showed that T cells expressing wild-type STAT3 repressed cytotoxic gene expression (GZMB, Pfr1, IFNγ, Tbx21, Eomes), whereas STAT3-/- failed to do so. This effect was mediated, in part, through the STAT3 target gene RORγt [77]. Besides IL-21–IL-10–STAT3–BCL6–SOCS3 axis, there is another mechanistic link between STAT3 and restriction of terminal effector differentiation. STAT3 upregulates expression of FoxO1, which, in turn, represses expression of T-bet, a master regulator of terminal differentiation [78,79].
On the other hand, IL-2/STAT5 signaling is one of the pathways that drive T-cell terminal differentiation and suppress memory formation [68,80,81]. There is evidence that STAT5 and STAT3 act as mutual antagonists. Kalie et al. (2010) demonstrated that on day 3.5 of acute LCMV infection primed T cells can be divided into CD25low and CD25high, which preferentially differentiate into memory precursors (KLRG1- IL7R+) and short-lived effector cells (KLRG1+ IL7R-), respectively [82]. CD25 is the IL-2 receptor alpha-chain and is required for IL-2 signal transduction [83]. CD25low cells had decreased IL-2/STAT5 signaling but were enriched with the IL-6 signaling pathway that acts through STAT3. In another study, the authors compared murine CD8+ T cell ex vivo expansion in the presence of IL-2, IL-15, or IL-21 [84]. Unlike IL-2 and IL-15, which utilize STAT5 for downstream signaling, IL-21 (STAT3-activating cytokine) increased expression of memory-related genes (Lef1, Sell, Tcf7) and repressed expression of effector ones (Eomes, GZMB, IL2Ra). Tcf7 is a direct target of STAT3 and is vital for memory formation [18,66]. Moreover, IL-21 reduces the expression of IL-2-driven Eomes when both cytokines are present simultaneously. Compared with IL-2, IL-21-treated T cells showed superior antitumor activity in the B16 melanoma subcutaneous model. In the acute LCMV model, IL-21 deficiency led to lower numbers of IL-2 producing CD8+ T cells [85]. IL-2 production is a feature of naïve and memory T cells and is repressed in effector cells in a BLIMP1-dependent manner [86]. Interestingly, IL-21 can also induce BLIMP1 expression through STAT3 activation [87]. The authors suggested that an IL-21/BCL6/BLIMP1 incoherent feed-forward loop can explain this discrepancy. IL-21 drives the expression of both BCL6 and BLIMP1 but BCL6 represses BLIMP1 and tunes the balance between these two transcriptional factors.
These studies, however, did not address the molecular mechanism of STAT3–STAT5 antagonism. In the Th17 differentiation setting, this mechanism was studied in greater detail. STAT5 was shown to restrict STAT3-driven RORγt expression and to compete with STAT3 DNA binding at IL-17 locus in CD4+ T cells [88,89]. STAT5 recruitment to the IL-17 locus was associated with decreased H3K4 acetylation and increased binding of NCoR2 corepressor. Although in the Th17 differentiation setting, these studies demonstrate that STAT5 can directly affect STAT3-driven gene expression, one can speculate that STAT5 may have the same direct effect on STAT3-driven genes necessary for memory formation. This idea is supported by a study indicating that IL-2 signaling suppressed BCL6 expression in Th1 cells [90]. The authors showed that STAT5 activated by IL-2 directly bound and repressed BCL6 expression. STAT3 was also shown to share almost 60% of binding sites with STAT5 in murine CD4+ cells [91].
Although the above evidence clearly argues for mutual antagonism, STAT3 and STAT5 can act cooperatively. For example, at least at low doses of IL-2, optimal CD25 (IL2Ra) expression on activated T cells requires intact STAT3 [92]. IL-21-activated STAT3 can positively regulate IL2Ra expression through super-enhancer binding [91]. This is unexpected, given the opposite roles of IL-2 and STAT3 signaling in memory formation.
4. Role of STAT3 Signaling in T-Cell Antitumor Immunity
Although the role of STAT3 in memory formation seems to be prominent, its translation into antitumor settings is less clear. There is no consensus on the role of STAT3 in antitumor immunity. Depending on the model, STAT3 activation was either beneficial or detrimental to the ability to fight the tumor. By reviewing studies on STAT3 in cancer settings, we attempt to relate the STAT3 role in memory formation to antitumor immunity. In an early study of STAT3, the Mx1–Cre–loxP system was used to knockout STAT3 in hematopoietic cells in adult mice [93]. STAT3-/- hosts rejected melanoma (B16), and bladder carcinoma (MB49), and these rejections were dependent on the T cells’ presence. STAT3-/- hosts had increased IFNγ production by CD8+ T cells, tumor infiltration, and decreased number of Tregs. This group demonstrated a similar antitumor effect using STAT3 conditional deletion in the T-cell lineage (CD4-Cre) and adoptive transfer model [94]. They also observed increased proliferation of STAT3-/- T cells. In a subsequent study, the same group elucidated the mechanism of STAT3-/- increased antitumor immunity [95]. Using the same STAT3 conditional deletion in the T-cell lineage, they demonstrated that STAT3-/- CD8+ T cells had increased IFNγ production and increased CXCR3 expression in subcutaneous melanoma (B16) and lung carcinoma (3LL) models. IFNγ promoted CXCL10 secretion by myeloid cells in the tumor and, as a result, attracted more CXCR3 expressing effector T cells. The negative effect of STAT3 signaling on CXCR3 expression was confirmed by another study [96]. The authors showed that increased IL17A in serum from colorectal cancer patients activated STAT3, leading to the downregulation of CXCR3 in CD8+ T cells and reduced tumor infiltration. High IL17A levels also correlated with worse patients’ survival. Similar to Yue et al. (2015), induction of cytokine secretion was observed in the glioma model after T-cell transfection with miR-124 [97]. MiR-124 downregulated STAT3 in T cells with a concomitant increase in IL-2, IFNγ, and TNF-α production. However, in another study, miR-124 had an opposite effect on STAT3 signaling [98]. The authors showed that the MeCP2–miR-124 axis suppresses SOCS5 expression in CD4+ T cells. SOCS5 inhibits STAT1 and STAT3 activation, thereby preventing Th1 and Th17 differentiation. The apparent contradiction in the miR-124 role can be attributed to its various effects in CD8+ and CD4+ T cells. Alternatively, STAT1 and STAT3 may have different sensitivities with respect to SOCS5.
In a recent study, the connection between tumor progression and STAT3 driven metabolism was discovered [99]. They showed that in a spontaneous breast cancer model, ablating leptin-driven STAT3 signaling in CD8+ T cells enhanced the antitumor effect by reducing fatty acid oxidation (FAO) and promoting glycolysis. Glycolysis is essential for effector T-cell expansion and synthesis of effector molecules [100,101,102]. Interestingly, they also showed that PD1 signaling, known to induce FAO and inhibit glycolysis, requires intact STAT3 [103]. However, it is not yet clear whether such a metabolic shift restricts antitumor immunity. Published studies demonstrate that it can be beneficial [104,105]. In a recent study, subdermal melanoma (CL24) and pancreatic adenocarcinoma (PanCO2) models were treated with leptin-expressing oncolytic vaccinia virus [106]. Leptin–STAT3 signaling increased mitochondrial content, IFNγ, and TNFa production by CD8+ T cells. As a result, only leptin-engineered viruses improved hosts’ survival. Although confounded by potential leptin effects on tumor cells, this study demonstrates the positive role of T-cell STAT3 signaling in antitumor immunity. As illustrated above, STAT3 signaling restricts glycolysis and promotes FAO in T cells. A similar metabolic shift is also observed during memory formation and was also shown to improve antitumor immunity [107,108].
The role of STAT3 in the antitumor activity of the CD4+ compartment is even more difficult to dissect. Besides memory formation, STAT3 signaling promotes differentiation of naïve CD4+ into Th17 and Tfh. These subsets can have both protumor and antitumor activity depending on the model [109,110,111]. For example, in one study, breast cancer patients had reduced IL6-driven STAT3 phosphorylation in CD4+ T cells. This correlated with a worse survival prognosis and was associated with impaired Th17 differentiation suggesting a decisive role of STAT3 activation [112]. In another mouse model study, IL-6/STAT3 signaling impaired CD4+ T-cell differentiation into the Th1 subset and compromised antitumor immunity [112]. Interestingly, Th17 cells were shown to exhibit prolonged longevity when compared with the Th1 subset [113,114]. Th17 cells also preserved differentiation plasticity and gave rise to Th1-like progeny that exerted antitumor activity. This suggests that Th17 cells are more stem-like than other Th subtypes. From these studies, it appears that Th17 differentiation is interconnected with memory formation potential. Another study showed that the gene expression signature of Tfh is close to the signature of CD8 T-cell memory precursors [115]. Unlike short-lived Th1 cells, Tfh cells were able to become memory cells and (similarly to CD8+ memory precursors) expressed higher levels of BCL6, TCF7, and ID3 and lower levels of IL2Ra and ID2. Thus, additional studies are required to elucidate the pleiotropic role of STAT3 in CD4+ T-cell subsets.
It is useful to look at STAT3 signaling in T cells in the context of a broader question about the role of memory T cells in antitumor immunity. Although not indisputable [116], there are reasons to believe that less differentiated T-cell subsets have superior antitumor efficacy when compared with their more differentiated counterparts [117,118,119,120,121,122,123,124]. Indeed, when preinfusion CAR T cells from 41 chronic lymphocytic leukemia patients were analyzed, CAR T cells from complete responders and partial responders were enriched for memory precursor effector cell (MPEC) genes and IL-6–STAT3 pathway [117]. CAR T cells with favorable CD27+PD1- had increased levels of gp130 expression and STAT3 phosphorylation, indicating the active role, as opposed to the mere biomarker role, of this pathway. It is noteworthy that the IL-6 signaling pathway was also enriched in CD25low effector cells in the LCMV model [82]. These cells were more likely to differentiate into memory precursors as opposed to short-lived effector cells. In a leukemia mouse model, CAR constructs incorporating both STAT5 and STAT3 binding motifs were engineered [125]. It was shown to activate both STAT5 and STAT3 and improve antitumor efficacy in leukemia (NALM6) mouse models. Although in this model, the antitumor effect of STAT3 signaling cannot be separated from STAT5, in vitro stimulated CAR T cells without STAT3 motif had decreased production of IL-2 and a lower number of less differentiated CD45RA+CD62L+CCR7+ CAR T cells. In the subcutaneous mesothelioma (M108) model, CAR T cells expanded in Th17-polarizing conditions provided superior tumor control [126]. A beneficial effect of human T-cell activation in the presence of IL-21 was shown also revealed [127]. In this study, CAR T cells targeting CD19+ were expanded in the presence of only IL-2 or in the presence of IL-2 and IL-21. In the latter case, CAR T cells showed better in vitro proliferation and a significant reduction in the leukemia NALM6 burden (NSG model). Consistent with Hinrichs et al. (2008), IL-21 treatment reduced Eomes expression in vitro. However, as opposed to Hinrichs et al. (2008), it also increased granzyme B expression, highlighting the model differences.
The benefit of less differentiated (memory) T cells for antitumor immunity lies partly in their ability to expand upon antigen encounter significantly and partly in their long-term survival advantage. Some of this survival advantage can be attributed to metabolic flexibility that can be crucial in the hostile tumor microenvironment. STAT3 both drives proper memory differentiation at early stages and modulates T-cell metabolism at the tumor site, contributing to antitumor response. Both aspects lead to better T-cell survival and persistence. On the other hand, memory T cells may have restricted cytotoxic and migration capacity, and in the short term, more differentiated T cells can have an antitumor advantage. However, it is also possible that mouse models are too short of capturing all the dynamics of antitumor response driven by memory T cells and observed in patients. In terms of metabolism, for hypoglycemic tumors, FAO induction seems to be beneficial, whereas, in an environment with sufficient nutrients, it will restrict T-cell expansion by inhibiting glycolysis. Hence, in cellular therapy, it may be reasonable to mimic physiological immune response and combine highly differentiated effector cells with less differentiated memory cells. The former will cause initial tumor reduction and prevent overstimulation of coadministered memory cells. The latter will provide antitumor effects after effector cells contract.
5. STAT3 Mutations Linked to T-Cell Memory
Rare Mendelian disorders or naturally occurring mutations in the tumor can provide much information about the physiological role of proteins harboring them. Such mutations can be correlated to their manifestation in cellular phenotype within the physiological environment.
Direct evidence of a role for STAT3 in human T-cell memory formation emerged from a study of individuals with the autosomal-dominant hyper-IgE syndrome (AD-HIES), which is caused by dominant-negative STAT3 mutations [128,129]. Individuals with AD-HIES, among other abnormalities, are susceptible to viral and bacterial infections. Importantly, in such individuals, fewer memory T cells (CD45RO+ CD27+) form, and their naïve T cells have impaired expression of BCL6 and SOCS3, which contribute to memory formation [60,130,131,132]. In line with effector–memory antagonism, increased expression of granzyme B in CD8+ TEMRA and EM subsets from AD-HIES individuals was also demonstrated [129].
Although STAT3 dysregulation often occurs in many cancer types, the role of STAT3 in large granular lymphocytic (LGL) leukemia is of particular interest [133,134]. LGL leukemia is a rare chronic lymphoproliferative disorder of mature T (CD4+, CD8+, or gamma/delta) or NK cells [135,136]. It is an indolent disease with two-thirds of symptomatic cases having splenomegaly, anemia, or neutropenia [137,138]. The origin of the disease is unknown, and the current T-cell LGL leukemia model suggests a two-step mechanism [139,140,141]. First, unknown antigen drives oligoclonal T-cell expansion. Then, dysregulation of survival pathways and, potentially persistent antigen, allow specific clones to avoid T-cell contraction and establish clonal dominance. Importantly, STAT3 seems to play a central role in this survival benefit and was shown to be activated in LGL cells, both by mutations and extrinsic factors, and its inhibition led to apoptosis of T-LGL cells in vitro [142,143,144]. STAT3 mutations appear in up to 70% of T-LGL leukemia cases. Mutations in the SH2 domain are most prevalent, with Y640F being the most common substitution. Y640F, D661V, among other mutations, make STAT3 more prone to dimerization, leading to increased transcriptional activity [145]. On the other hand, T-LGL leukemia cases without STAT3 mutations are still associated with increased STAT3 signaling. High amounts of IL-6 were observed in LGL leukemia individuals and provided antiapoptotic benefits through STAT3 activation [146]. Similarly, the downregulation of SOCS3, capable of reducing STAT3 signaling, was also observed in patients. PTPRT is a phosphatase responsible for the dephosphorylation of Tyr705 on STAT3 with subsequent STAT3 deactivation [147]. Consequently, PTPRT V995M mutation in LGL patients was also associated with increased STAT3 activity (Andersson et al. 2013). BCL11B H126R was associated with increased STAT3 signaling, although there is no known connection between BCL11B and STAT3. Interestingly, in STAT5 Y665F-mutated cases, STAT3 signaling was also activated. This STAT5 mutation is an activating one, suggesting positive crosstalk between STAT3 and STAT5, in contrast to the negative one mentioned in the context of Th17 differentiation [148]. LGL patients with STAT3 mutations are at higher risk of developing rheumatoid arthritis [149]. Interestingly, in a mouse model (adoptive transfer of bone marrow that was transduced with STAT3 mutations) activating STAT3 (Y640F or D661V) mutations by themselves could not induce LGL disease [150]. All in all, the main lesson to learn from TGL leukemia is that STAT3 signaling is associated with a significant survival advantage for T cells, even in the absence of other oncogenic mutations. Such survival is similar to cell survival in memory T cells, which can persist for a very long time after antigen encounter.
STAT3 was reported to be constitutively activated in the late-stage cutaneous T-cell lymphoma (CTCL) [151]. By transfecting tumor cells with dominant-negative STAT3, the authors showed that blocking STAT3 signaling leads to apoptosis. Interestingly, IL-21 expression is also associated with disease progression and with increased mortality in CTCL [152]. It is tempting to speculate that, in this setting, IL-21 activates STAT3 leading to survival benefit.
Mutations in STAT3 are also implicated in the development of autoimmune diseases, including type 1 diabetes [153]. Recently, a mouse model heterozygous for STAT3 K392R gain-of-function mutation was generated [154]. The authors demonstrated that STAT3 K392R mutation in CD8+ T cells counteracts exhaustion, increases cytotoxicity, and can accelerate the development of T1D.
6. STAT3 Signaling in the Context of IL-6 Blockade
Cytokine release syndrome (CRS) is a common complication after CAR T product infusion and is accompanied by an extensive release of IL-6 [155]. Now it is successfully treated with tocilizumab (anti-IL-6R antibody) [156]. There is no evidence either from CAR T clinical trials (Kymriah, Yescarta, CD22-BBz CAR T) or from other T-cell-engaging therapies (CD123/CD3 DART or CD19/CD3 BiTE antibodies) that IL-6 blockade has a negative impact on T-cell functionality and antitumor response [157,158,159,160,161,162]. The evidence that IL-6 blockade does not affect therapeutic response implies that at least shortly after infusion, IL-6-STAT3 signaling is dispensable for CAR T cells. This discrepancy is likely explained by the fact that at the end of ex vivo expansion CAR T cells differentiate and lose the expression of the gp130 receptor. Supporting this, Fraietta et al. (2018) showed that only the CD27+PD1- population (least differentiated subset) of CAR T cells expresses gp130 and activates STAT3 upon IL-6 administration [117]. Moreover, gp130 expression is largely restricted to naïve and central memory T-cell subsets from peripheral blood and is coexpressed with CCR7 (unpublished data). However, the presence of soluble gp130 and IL-6Ra receptors in patients after CAR-T infusion raises the possibility that T cells without gp130 expression can still respond to IL-6 [163]. Future studies are needed to further elucidate the role of IL-6 blockade on T cells.
7. Conclusions
Considering all the above, there is increasing evidence that STAT3 is required for T-cell memory formation. However, this is largely based on studies that used conditional knockouts in mouse T-cell precursors.
There is a need for cleaner models that interrogate STAT3 in mature T cells. They will eliminate potential effects of perturbed STAT3 that manifested at late stages of T-cell development. One option is CD8a–Cre system (Cre expression driven by the core E8I enhancer and Cd8a promoter), which deletes a gene in mature CD8+ but not CD4+ T cells [164]. GZMB–Cre system can also be used (Cui et al. 2011). Alternatively, one can consider CRISPR–Cas9-mediated gene editing in mature T cells, a system that has been validated in both human and mouse T cells [165,166,167].
STAT3 signaling in human mature T cells is still understudied and most of the evidence is still correlative. Although some parallels between human and mouse models can be drawn, no systematic effort of comparing STAT3 signaling in human and mouse T cells has been made.
In terms of antitumor immunity, STAT3 signaling in T cells seems to be highly context-dependent. There is evidence of both positive and negative roles of this signaling axis in T-cell antitumor immunity. Its negative role can be attributed to STAT3 inhibition of CXCR3-driven tumor trafficking. Hence, inhibition of STAT3 signaling in T cells may be desirable for tumors expressing CXCR3 ligands (CXCL9, CXCL10, CXCL11). Nevertheless, for tumors that do not express these chemokines or blood cancers, STAT3 activation in T cells might provide a persistence advantage with no reduction in trafficking. With STAT3 signaling promoting FAO and restricting glycolysis, STAT3 activation might be beneficial for low glycose environments, where alternative metabolic routes are required for T-cell survival and expansion. On the other hand, if glycose is abundant, STAT3 inhibition can increase T-cell expansion, at least in the short term.
Author Contributions
Writing—original draft preparation, Y.K.; writing—review and editing, J.J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT Family Member Activated by Tyrosine Phosphorylation in Response to Epidermal Growth Factor and Interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.-J.; We, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Suarez, A.A.R.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef] [Green Version]
- Babon, J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmann, U.; Gerhartz, C.; Heesel, B.; Sasse, J.; Kurapkat, G.; Grötzinger, J.; Wollmer, A.; Zhong, Z.; Darnell, J.E.; Graeve, L.; et al. Differential Activation of Acute Phase Response Factor/Stat3 and Stat1 via the Cytoplasmic Domain of the Interleukin 6 Signal Transducer gp130. J. Biol. Chem. 1996, 271, 12999–13007. [Google Scholar] [CrossRef] [Green Version]
- Kaptein, A.; Paillard, V.; Saunders, M. Dominant Negative Stat3 Mutant Inhibits Interleukin-6-induced Jak-STAT Signal Transduction. J. Biol. Chem. 1996, 271, 5961–5964. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Groner, B.; Muller, C.W. Three-dimensional structure of the Stat3 beta homodimer bound to DNA. Nature 1998, 394, 145–151. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E. Maximal activation of transcription by statl and stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.L.; Darnell, J.E. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997, 25, 2062–2067. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.-L.; Guan, Y.-J.; Chatterjee, D.; Chin, Y.E. Stat3 Dimerization Regulated by Reversible Acetylation of a Single Lysine Residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, M.; Unal, H.; Willard, B.; Yang, J.; Karnik, S.S.; Stark, G.R. Critical Role for Lysine 685 in Gene Expression Mediated by Transcription Factor Unphosphorylated STAT3. J. Biol. Chem. 2014, 289, 30763–30771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, M.; Dermawan, J.K.T.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NF kappa B. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Stark, G.R. Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Kovarik, P.; Meinke, A. GAS Elements: A Few Nucleotides with a Major Impact on Cytokine-Induced Gene Expression. J. Interf. Cytokine Res. 1997, 17, 121–134. [Google Scholar] [CrossRef]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.-W.; Kanno, Y.; Powrie, F.; et al. Diverse Targets of the Transcription Factor STAT3 Contribute to T Cell Pathogenicity and Homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Timofeeva, O.A.; Chasovskikh, S.; Lonskaya, I.; Tarasova, N.I.; Khavrutskii, L.; Tarasov, S.G.; Zhang, X.; Korostyshevskiy, V.R.; Cheema, A.; Zhang, L.; et al. Mechanisms of Unphosphorylated STAT3 Transcription actor Binding to DNA. J. Biol. Chem. 2012, 287, 14192–14200. [Google Scholar] [CrossRef] [Green Version]
- Wegrzyn, J.; Potla, R.; Chwae, Y.-J.; Sepuri, N.B.V.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of Mitochondrial Stat3 in Cellular Respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 Supports Ras-Dependent Oncogenic Transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlich, T.H.; Yagil, Z.; Kay, G.; Peretz, A.; Migalovich-Sheikhet, H.; Tshori, S.; Nechushtan, H.; Levi-Schaffer, F.; Saada, A.; Razin, E. Mitochondrial STAT3 plays a major role in IgE-antigen–mediated mast cell exocytosis. J. Allergy Clin. Immunol. 2014, 134, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; Diehl, S.A.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L.; et al. Mitochondrial Ca2+ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. eLife 2015, 4, e06376. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [Green Version]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 2018, 23, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Harhous, Z.; Booz, G.W.; Ovize, M.; Bidaux, G.; Kurdi, M. An Update on the Multifaceted Roles of STAT3 in the Heart. Front. Cardiovasc. Med. 2019, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Chou, W.-C.; Levy, D.E.; Lee, C.-K. STAT3 positively regulates an early step in B-cell development. Blood 2006, 108, 3005–3011. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Chen, X.; Dascani, P.; Hu, X.; Bolli, R.; Zhang, H.-G.; McLeish, K.R.; Yan, J. STAT3 Signaling in B Cells Is Critical for Germinal Center Maintenance and Contributes to the Pathogenesis of Murine Models of Lupus. J. Immunol. 2016, 196, 4477–4486. [Google Scholar] [CrossRef] [Green Version]
- Oladipupo, F.; Egwuagu, C.E.; Yu, C.R.; Choi, J.K. Intrinsic function of STAT3 in B cells is to promote B cell proliferation and maturation. J. Immunol. 2019, 202 (Suppl. S1), 123.10. [Google Scholar]
- Oladipupo, F.O.; Yu, C.-R.; Olumuyide, E.; Jittaysothorn, Y.; Choi, J.K.; Egwuagu, C.E. STAT3 deficiency in B cells exacerbates uveitis by promoting expansion of pathogenic lymphocytes and suppressing regulatory B cells (Bregs) and Tregs. Sci. Rep. 2020, 10, 16188. [Google Scholar] [CrossRef] [PubMed]
- Maraskovsky, E.; Brasel, K.; Teepe, M.; Roux, E.R.; Lyman, S.D.; Shortmanfi, K.; McKenna, H.J. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: Multiple dendritic cell subpopulations identified. J. Exp. Med. 1996, 184, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Clausen, B.; Kaisho, T.; Tsujimura, T.; Terada, N.; Förster, I.; Akira, S. Enhanced Th1 Activity and Development of Chronic Enterocolitis in Mice Devoid of Stat3 in Macrophages and Neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Do, N.-N.; Willenborg, S.; Eckes, B.; Jüngst, C.; Sengle, G.; Zaucke, F.; Eming, S.A. Myeloid Cell–Restricted STAT3 Signaling Controls a Cell-Autonomous Antifibrotic Repair Program. J. Immunol. 2018, 201, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, B.J.; Roberts, A.W.; Najdovska, M.; Grail, D.; Ernst, M. The threshold of gp130-dependent STAT3 signaling is critical for normal regulation of hematopoiesis. Blood 2005, 105, 3512–3520. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J.; Roberts, A.W.; Greenhill, C.J.; Najdovska, M.; Lundgren-May, T.; Robb, L.; Grail, D.; Ernst, M. Pathologic consequences of STAT3 hyperactivation by IL-6 and IL-11 during hematopoiesis and lymphopoiesis. Blood 2006, 109, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Grisouard, J.; Shimizu, T.; Duek, A.; Kubovcakova, L.; Hao-Shen, H.; Dirnhofer, S.; Skoda, R.C. Deletion of Stat3 in hematopoietic cells enhances thrombocytosis and shortens survival in a JAK2-V617F mouse model of MPN. Blood 2015, 125, 2131–2140. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Takahama, Y.; Sugawara, T.; Kosaka, H.; Itami, S.; Yoshikawa, K.; Miyazaki, J.-I.; van Ewijk, W.; Takeda, J. Stat3 in Thymic Epithelial Cells Is Essential for Postnatal Maintenance of Thymic Architecture and Thymocyte Survival. Immunity 2001, 15, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Satoh, R.; Kakugawa, K.; Yasuda, T.; Yoshìda, H.; Sibilia, M.; Katsura, Y.; Levi, B.; Abramson, J.; Koseki, Y.; Koseki, H.; et al. Requirement of Stat3 Signaling in the Postnatal Development of Thymic Medullary Epithelial Cells. PLoS Genet. 2016, 12, e1005776. [Google Scholar] [CrossRef] [Green Version]
- Lomada, D.; Jain, M.; Bolner, M.; Reeh, K.A.G.; Kang, R.; Reddy, M.; DiGiovanni, J.; Richie, E.R. Stat3 Signaling Promotes Survival And Maintenance Of Medullary Thymic Epithelial Cells. PLoS Genet. 2016, 12, e1005777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Zhang, J.; Sun, X.; Qian, X.; Zhang, Y.; Jin, R. Thymic DCs derived IL-27 regulates the final maturation of CD4+ SP thymocytes. Sci. Rep. 2016, 6, 30448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-K.; Won, C.; Yi, E.H.; Seok, S.-H.; Kim, M.-H.; Kim, S.-J.; Chung, M.-H.; Lee, H.G.; Ikuta, K.; Ye, S.-K. Signal transducer and activator of transcription 3 (Stat3) contributes to T-cell homeostasis by regulating pro-survival Bcl-2 family genes. Immunology 2013, 140, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGF beta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.; Leonard, W.J.; Littman, D.R. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Liu, X.; Lee, Y.S.; Yu, C.-R.; Egwuagu, C.E. Loss of STAT3 in CD4+ T Cells Prevents Development of Experimental Autoimmune Diseases. J. Immunol. 2008, 180, 6070–6076. [Google Scholar] [CrossRef] [Green Version]
- Nurieva, R.I.; Chung, Y.; Hwang, D.; Yang, X.O.; Kang, H.S.; Ma, L.; Wang, Y.-H.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Generation of T Follicular Helper Cells Is Mediated by Interleukin-21 but Independent of T Helper 1, 2, or 17 Cell Lineages. Immunity 2008, 29, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Vinuesa, C.; Tangye, P.S.; Moser, B.; Mackay, C. Follicular B helper T cells in antibody responses and autoimmunity. Nat. Rev. Immunol. 2005, 5, 853–865. [Google Scholar] [CrossRef]
- Liu, X.; Yan, X.; Zhong, B.; Nurieva, R.I.; Wang, A.; Wang, X.; Martin-Orozco, N.; Wang, Y.; Chang, S.H.; Esplugues, E.; et al. Bcl6 expression specifies the T follicular helper cell program in vivo. J. Exp. Med. 2012, 209, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Laurence, A.; Amarnath, S.; Mariotti, J.; Kim, Y.C.; Foley, J.; Eckhaus, M.; O’Shea, J.J.; Fowler, D.H. STAT3 Transcription Factor Promotes Instability of nTreg Cells and Limits Generation of iTreg Cells during Acute Murine Graft-versus-Host Disease. Immunity 2012, 37, 209–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, J.; Marshall, H.D.; Laidlaw, B.J.; Staron, M.M.; Kaech, S.M.; Craft, J. Transcription Factor STAT3 and Type I Interferons Are Corepressive Insulators for Differentiation of Follicular Helper and T Helper 1 Cells. Immunity 2014, 40, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Fabregas, J.; Wilmes, S.; Wang, L.; Hafer, M.; Pohler, E.; Lokau, J.; Garbers, C.; Cozzani, A.; Fyfe, P.K.; Piehler, J.; et al. Kinetics of cytokine receptor trafficking determine signaling and functional selectivity. eLife 2019, 8, e49314. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Kaisho, T.; Yoshida, N.; Takeda, J.; Kishimoto, T.; Akira, S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: Generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998, 161, 4652–4660. [Google Scholar] [CrossRef] [Green Version]
- Rochman, I.; Paul, W.E.; Ben-Sasson, S.Z. IL-6 Increases Primed Cell Expansion and Survival. J. Immunol. 2005, 174, 4761–4767. [Google Scholar] [CrossRef] [Green Version]
- Castellino, F.; Germain, R.N. Chemokine-guided CD4(+) T cell help enhances generation of IL-6R alpha(IL)-I-high-7R alpha(high) prememory CD8(+) T cells. J. Immunol. 2007, 178, 778–787. [Google Scholar] [CrossRef] [Green Version]
- Nish, S.A.; Schenten, D.; Wunderlich, F.T.; Pope, S.D.; Gao, Y.; Hoshi, N.; Yu, S.; Yan, X.; Lee, H.K.; Pasman, L.; et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. eLife 2014, 3, 47. [Google Scholar] [CrossRef]
- Betz, U.A.K.; Muller, W. Regulated expression of gp130 and IL-6 receptor alpha chain in T cell maturation and activation. Int. Immunol. 1998, 10, 1175–1184. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Liu, Y.; Weinstein, J.S.; Craft, J.; Kaech, S.M. An Interleukin-21- Interleukin-10-STAT3 Pathway Is Critical for Functional Maturation of Memory CD8+ T Cells. Immunity 2011, 35, 792–805. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Joshi, N.S.; Liu, Y.; Meng, H.; Kleinstein, S.H.; Kaech, S.M. TLR4 Ligands Lipopolysaccharide and Monophosphoryl Lipid A Differentially Regulate Effector and Memory CD8+ T Cell Differentiation. J. Immunol. 2014, 192, 4221–4232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.Y.; Best, J.A.; Knell, J.; Yang, E.; Sheridan, A.; Jesionek, A.K.; Li, H.S.; Rivera, R.R.; Lind, K.C.; D’Cruz, L.M.; et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat. Immunol. 2011, 12, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Foulds, K.E.; Rotte, M.; Seder, R.A. IL-10 Is Required for Optimal CD8 T Cell Memory followingListeria monocytogenesInfection. J. Immunol. 2006, 177, 2565–2574. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.X.; Amezquita, R.A.; Guan, T.; Marshall, H.D.; Joshi, N.; Kleinstein, S.H.; Kaech, S.M. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med. 2015, 212, 2041–2056. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 2019, 51, 840–855.e5. [Google Scholar] [CrossRef] [PubMed]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional Repressor Blimp-1 Promotes CD8+ T Cell Terminal Differentiation and Represses the Acquisition of Central Memory T Cell Properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Xin, A.; Masson, F.; Liao, Y.; Preston, S.; Guan, T.; Gloury, R.; Olshansky, M.; Lin, J.-X.; Li, P.; Speed, T.P.; et al. A molecular threshold for effector CD8+ T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat. Immunol. 2016, 17, 422–432. [Google Scholar] [CrossRef]
- Backer, R.A.; Hombrink, P.; Helbig, C.; Amsen, D. The Fate Choice between Effector and Memory T Cell Lineages: Asymmetry, Signal Integration, and Feedback to Create Bistability. In Advances in Immunology; Alt, F., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2018; Volume 137, pp. 43–82. [Google Scholar]
- Dong, H.; Franklin, N.A.; Roberts, E.J.; Yagita, H.; Glennie, M.J.; Bullock, T.N.J. CD27 Stimulation Promotes the Frequency of IL-7 Receptor-Expressing Memory Precursors and Prevents IL-12–Mediated Loss of CD8+ T Cell Memory in the Absence of CD4+ T Cell Help. J. Immunol. 2012, 188, 3829–3838. [Google Scholar] [CrossRef] [Green Version]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.; Phan, A.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef]
- Urban, S.L.; Welsh, R.M. Out-of-Sequence Signal 3 as a Mechanism for Virus-Induced Immune Suppression of CD8 T Cell Responses. PLoS Pathog. 2014, 10, e1004357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjabi, S.; Mosaheb, M.M.; Flavell, R.A. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 2009, 31, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.-H.; Lee, P.-H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363, eaau0135. [Google Scholar] [CrossRef] [PubMed]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e4. [Google Scholar] [CrossRef]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Ciucci, T.; Vacchio, M.S.; Bosselut, R. A STAT3-dependent transcriptional circuitry inhibits cytotoxic gene expression in T cells. Proc. Natl. Acad. Sci. USA 2017, 114, 13236–13241. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.-M.; Yu, C.-R.; Golestaneh, N.; Amadi-Obi, A.; Lee, Y.S.; Eseonu, A.; Mahdi, R.M.; Egwuagu, C.E. STAT3 Protein Promotes T-cell Survival and Inhibits Interleukin-2 Production through Up-regulation of Class O Forkhead Transcription Factors. J. Biol. Chem. 2011, 286, 30888–30897. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.R.; Li, Q.; Bupp, M.R.G.; Shrikant, P.A. Transcription Factor Foxo1 Represses T-bet-Mediated Effector Functions and Promotes Memory CD8+ T Cell Differentiation. Immunity 2012, 36, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Obar, J.; Molloy, M.J.; Jellison, E.R.; Stoklasek, T.A.; Zhang, W.; Usherwood, E.J.; Lefrançois, L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc. Natl. Acad. Sci. USA 2009, 107, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Arsenio, J.; Kakaradov, B.; Metz, P.J.; Kim, S.H.; Yeo, G.W.; Chang, J.T. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses. Nat. Immunol. 2014, 15, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Kalia, V.; Sarkar, S.; Subramaniam, S.; Haining, W.N.; Smith, K.A.; Ahmed, R. Prolonged Interleukin-2R alpha Expression on Virus-Specific CD8(+) T Cells Favors Terminal-Effector Differentiation In Vivo. Immunity 2010, 32, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matek, T.R. The biology of interleukin-2. In Annual Review of Immunology; Annual Reviews: Palo Alto, CA, USA, 2008; pp. 453–479. [Google Scholar]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.S.; Ingram, J.T.; Zajac, A.J. IL-21 Deficiency Influences CD8 T Cell Quality and Recall Responses following an Acute Viral Infection. J. Immunol. 2010, 185, 4835–4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, G.A.; Cimmino, L.; Liao, J.; Magnusdottir, E.; Calame, K. Blimp-1 directly repressesIl2and theIl2activatorFos, attenuating T cell proliferation and survival. J. Exp. Med. 2008, 205, 1959–1965. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Thierry-Mieg, D.; Thierry-Mieg, J.; Kim, H.-P.; Oh, J.; Tunyaplin, C.; Carotta, S.; Donovan, C.E.; Goldman, M.L.; Tailor, P.; et al. Analysis of Interleukin-21-Induced Prdm1 Gene Regulation Reveals Functional Cooperation of STAT3 and IRF4 Transcription Factors. Immunity 2009, 31, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurence, A.; Tato, C.M.; Davidson, T.S.; Kanno, Y.; Chen, Z.; Yao, Z.; Blank, R.B.; Meylan, F.; Siegel, R.; Hennighausen, L.; et al. Interleukin-2 Signaling via STAT5 Constrains T Helper 17 Cell Generation. Immunity 2007, 26, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.-W.; Wei, L.; Vahedi, G.; et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Oestreich, K.J.; Mohn, S.E.; Weinmann, A.S. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat. Immunol. 2012, 13, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Mitra, S.; Spolski, R.; Oh, J.; Liao, W.; Tang, Z.; Mo, F.; Li, X.; West, E.E.; Gromer, D.; et al. STAT5-mediated chromatin interactions in superenhancers activate IL-2 highly inducible genes: Functional dissection of the Il2ra gene locus. Proc. Natl. Acad. Sci. USA 2017, 114, 12111–12119. [Google Scholar] [CrossRef] [Green Version]
- Akaishi, H.; Takeda, K.; Kaisho, T.; Shineha, R.; Satomi, S.; Takeda, J.; Akira, S. Defective IL-2-mediated IL-2 receptor alpha chain expression in Stat3-deficient T lymphocytes. Int. Immunol. 1998, 10, 1747–1751. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, M.; Zhang, C.; Herrmann, A.; Reckamp, K.; Scuto, A.; Jensen, M.; Deng, J.; Forman, S.; Figlin, R.; Yu, H. Targeting STAT3 in Adoptively Transferred T Cells Promotes Their In Vivo Expansion and Antitumor Effects. Cancer Res. 2010, 70, 9599–9610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, C.; Shen, S.; Deng, J.; Priceman, S.J.; Li, W.; Huang, A.; Yu, H. STAT3 in CD8(+) T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis. Cancer Immunol. Res. 2015, 3, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yu, W.; Lian, J.; Wu, Q.; Liu, S.; Yang, L.; Li, F.; Huang, L.; Chen, X.; Zhang, Z.; et al. Th17 cells inhibit CD8+ T cell migration by systematically downregulating CXCR3 expression via IL-17A/STAT3 in advanced-stage colorectal cancer patients. J. Hematol. Oncol. 2020, 13, 68. [Google Scholar] [CrossRef]
- Wei, J.; Wang, F.; Kong, L.-Y.; Xu, S.; Doucette, T.; Ferguson, S.D.; Yang, Y.; McEnery, K.; Jethwa, K.; Gjyshi, O.; et al. miR-124 Inhibits STAT3 Signaling to Enhance T Cell–Mediated Immune Clearance of Glioma. Cancer Res. 2013, 73, 3913–3926. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Li, C.; McRae, G.; Lykken, E.; Sevilla, J.; Liu, S.-Q.; Wan, Y.; Li, Q.-J. MeCP2 Reinforces STAT3 Signaling and the Generation of Effector CD4 + T Cells by Promoting miR-124–Mediated Suppression of SOCS5. Sci. Signal. 2014, 7, ra25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2019, 31, 148–161.e5. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.M.; Opipari, A.W.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8+ T Cells and Facilitates Anti–PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivadeneira, D.B.; DePeaux, K.; Wang, Y.; Kulkarni, A.; Tabib, T.; Menk, A.V.; Sampath, P.; Lafyatis, R.; Ferris, R.L.; Sarkar, S.; et al. Oncolytic Viruses Engineered to Enforce Leptin Expression Reprogram Tumor-Infiltrating T Cell Metabolism and Promote Tumor Clearance. Immunity 2019, 51, 548–560.e4. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. In Annual Review of Immunology; Littman, D.R., Yokoyama, W.M., Eds.; Annual Reviews: Palo Alto, CA, USA, 2013; Volume 31, pp. 259–283. [Google Scholar]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Senju, S.; Matsumura, K.; Swain, S.L.; Nishimura, Y. IL-6-mediated environmental conditioning of defective Th1 differentiation dampens antitumour immune responses in old age. Nat. Commun. 2015, 6, 6702. [Google Scholar] [CrossRef] [Green Version]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of Th17 cell functions in tumor immunity. Cell. Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Wang, L.; Miyahira, A.K.; Simons, D.L.; Lu, X.; Chang, A.Y.; Wang, C.; Suni, M.A.; Maino, V.C.; Dirbas, F.M.; Yim, J.; et al. IL6 Signaling in Peripheral Blood T Cells Predicts Clinical Outcome in Breast Cancer. Cancer Res. 2016, 77, 1119–1126. [Google Scholar] [CrossRef] [Green Version]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Muranski, P.; Borman, Z.A.; Kerkar, S.P.; Klebanoff, C.; Ji, Y.; Sanchez-Perez, L.; Sukumar, M.; Reger, R.N.; Yu, Z.; Kern, S.J.; et al. Th17 Cells Are Long Lived and Retain a Stem Cell-like Molecular Signature. Immunity 2011, 35, 972–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Yang, J.A.; Yusuf, I.; Johnston, R.J.; Greenbaum, J.; Peters, B.; Crotty, S. Bcl6 Expressing Follicular Helper CD4 T Cells Are Fate Committed Early and Have the Capacity To Form Memory. J. Immunol. 2013, 190, 4014–4026. [Google Scholar] [CrossRef] [PubMed]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific Lymphocyte Subsets Predict Response to Adoptive Cell Therapy Using Expanded Autologous Tumor-Infiltrating Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J.; Rosenberg, S.A. Cutting Edge: Persistence of Transferred Lymphocyte Clonotypes Correlates with Cancer Regression in Patients Receiving Cell Transfer Therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.W.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef]
- Gattinoni, L.; Zhong, X.-S.; Palmer, D.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pós, Z.; Paulos, C.M.; Quigley, M.F.; de Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell–like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Pruteanu, I.; Cohen, A.D.; Garfall, A.L.; Tian, L.; Lacey, S.F.; Fraietta, J.A.; Brogdon, J.; Davis, M.; Gonzalez, V.E.; et al. Response to Anti-Bcma CAR T Cell Therapy Correlates with T Cell Exhaustion and Activation Status in T Cells at Baseline in Myeloma. Blood 2019, 134, 1909. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Carpenito, C.; Plesa, G.; Suhoski, M.M.; Varela-Rohena, A.; Golovina, T.N.; Carroll, R.G.; Riley, J.L.; June, C.H. The Inducible Costimulator (ICOS) Is Critical for the Development of Human TH17 Cells. Sci. Transl. Med. 2010, 2, 55ra78. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Figliola, M.J.; Dawson, M.J.; Huls, H.; Olivares, S.; Switzer, K.; Mi, T.; Maiti, S.; Kebriaei, P.; Lee, D.; et al. Reprogramming CD19-Specific T Cells with IL-21 Signaling Can Improve Adoptive Immunotherapy of B-Lineage Malignancies. Cancer Res. 2011, 71, 3516–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, A.M.; Heimall, J.; Freeman, A.F.; Hsu, A.P.; Brittain, E.; Brenchley, J.M.; Douek, D.C.; Fahle, G.H.; Cohen, J.I.; Holland, S.M.; et al. A Critical Role for STAT3 Transcription Factor Signaling in the Development and Maintenance of Human T Cell Memory. Immunity 2011, 35, 806–818. [Google Scholar] [CrossRef] [Green Version]
- Ives, M.L.; Ma, C.; Palendira, U.; Chan, A.; Bustamante, J.; Boisson-Dupuis, S.; Arkwright, P.; Engelhard, D.; Averbuch, D.; Magdorf, K.; et al. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper-IgE syndrome impair human CD8+ T-cell memory formation and function. J. Allergy Clin. Immunol. 2013, 132, 400–411.e9. [Google Scholar] [CrossRef] [Green Version]
- Ichii, H.; Sakamoto, A.; Kuroda, Y.; Tokuhisa, T. Bc16 acts as an amplifier for the generation and proliferative capacity of central memory CD8(+) T cells. J. Immunol. 2004, 173, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Ichii, H.; Sakamoto, A.; Arima, M.; Hatano, M.; Kuroda, Y.; Tokuhisa, T. Bcl6 is essential for the generation of long-term memory CD4+ T cells. Int. Immunol. 2007, 19, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S.; Johnston, R.J.; Schoenberger, S.P. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol. 2010, 11, 114–120. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. In Annual Review of Immunology; Littman, D.R., Yokoyama, W.M., Eds.; Annual Reviews: Palo Alto, CA, USA, 2017; Volume 35, pp. 533–550. [Google Scholar]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Loughran, T.P.; Kadin, M.E.; Starkebaum, G.; Abkowitz, J.L.; Clark, E.A.; Disteche, C.; Lum, L.G.; Slichter, S.J. Leukemia of large granular lymphocytes: Association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann. Intern. Med. 1985, 102, 169–175. [Google Scholar] [CrossRef]
- Loughran, T.P. Clonal diseases of large granular lymphocytes. Blood 1993, 82, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanikommu, S.R.; Clemente, M.J.; Chomczynski, P.; Ii, M.G.A.; Jerez, A.; Thota, S.; Patel, B.; Hirsch, C.; Nazha, A.; Desamito, J.; et al. Clinical features and treatment outcomes in large granular lymphocytic leukemia (LGLL). Leuk. Lymphoma 2017, 59, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.R.; Maciejewski, J.P. Diagnosis and therapy of neutropenia in large granular lymphocyte leukemia. Curr. Opin. Hematol. 2009, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Lamy, T.; Moignet, A.; Loughran, T.P. LGL leukemia: From pathogenesis to treatment. Blood Am. Soc. Hematol. 2017, 129, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, Y.; Kallemeijn, M.J.; Dik, W.A.; Tielemans, D.; Wolvers-Tettero, I.L.M.; van Gastel-Mol, E.J.; Szczepanski, T.; Pol, Y.; Darzentas, N.; van Dongen, J.J.M.; et al. Lack of common TCRA and TCRB clonotypes in CD8(+)/TCR alpha beta(+) T-cell large granular lymphocyte leukemia: A review on the role of antigenic selection in the immunopathogenesis of CD8(+) T-LGL. Blood Cancer J. 2014, 4, e172. [Google Scholar] [CrossRef]
- Clemente, M.J.; Przychodzen, B.; Jerez, A.; Dienes, B.E.; Afable, M.G.; Husseinzadeh, H.; Rajala, H.L.M.; Wlodarski, M.; Mustjoki, S.; Maciejewski, J.P. Deep sequencing of the T-cell receptor repertoire in CD8+ T-large granular lymphocyte leukemia identifies signature landscapes. Blood 2013, 122, 4077–4085. [Google Scholar] [CrossRef]
- Shi, M.; He, R.; Feldman, A.L.; Viswanatha, D.S.; Jevremovic, D.; Chen, D.; Morice, W.G. STAT3 mutation and its clinical and histopathologic correlation in T-cell large granular lymphocytic leukemia. Hum. Pathol. 2018, 73, 74–81. [Google Scholar] [CrossRef]
- Fasan, A.; Kern, W.; Grossmann, V.; Haferlach, C.; Schnittger, S.; Haferlach, T. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia 2012, 27, 1598–1600. [Google Scholar] [CrossRef]
- Andersson, E.; Rajala, H.L.M.; Eldfors, S.; Ellonen, P.; Olson, T.; Jerez, A.; Clemente, M.J.; Kallioniemi, O.; Porkka, K.; Heckman, C.; et al. Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation. Blood Cancer J. 2013, 3, e168. [Google Scholar] [CrossRef] [Green Version]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; Van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. SomaticSTAT3Mutations in Large Granular Lymphocytic Leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teramo, A.; Gattazzo, C.; Passeri, F.; Lico, A.; Tasca, G.; Cabrelle, A.; Martini, V.; Frezzato, F.; Trimarco, V.; Ave, E.; et al. Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia. Blood 2013, 121, 3843–3854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, A.; Yu, J.; Possemato, A.; Chen, Y.; Zheng, W.; Polakiewicz, R.D.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.; et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc. Natl. Acad. Sci. USA 2007, 104, 4060–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajala, H.L.M.; Eldfors, S.; Kuusanmäki, H.; van Adrichem, A.J.; Olson, T.; Lagström, S.; Andersson, E.I.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajala, H.L.M.; Olson, T.; Clemente, M.J.; Lagström, S.; Ellonen, P.; Lundan, T.; Hamm, D.E.; Zaman, S.A.U.; Marti, J.M.L.; Andersson, E.I.; et al. The analysis of clonal diversity and therapy responses using STAT3 mutations as a molecular marker in large granular lymphocytic leukemia. Haematologica 2014, 100, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Yan, D.; Hutchison, R.E.; Mohi, G. STAT3mutations are not sufficient to induce large granular lymphocytic leukaemia in mice. Br. J. Haematol. 2016, 180, 911–915. [Google Scholar] [CrossRef]
- Sommer, V.H.; Clemmensen, O.J.; Nielsen, O.; Wasik, M.; Lovato, P.; Brender, C.; Eriksen, K.W.; Woetmann, A.; Kaestel, C.G.; Nissen, M.H.; et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia 2004, 18, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, S.; Litvinov, I.; Mongan, N.P.; Schiele, S.; Willerslev-Olsen, A.; Petersen, D.L.; Krejsgaard, T.; Sibbesen, N.; Nastasi, C.; Bonefeld, C.M.; et al. The Expression of IL-21 Is Promoted by MEKK4 in Malignant T Cells and Associated with Increased Progression Risk in Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2016, 136, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Fung, E.Y.M.G.; Smyth, D.J.; Howson, J.M.; Cooper, J.D.; Walker, N.M.; Stevens, H.; Wicker, L.S.; Todd, J.A. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 2009, 10, 188–191. [Google Scholar] [CrossRef]
- Warshauer, J.T.; Belk, J.A.; Chan, A.Y.; Wang, J.; Gupta, A.R.; Shi, Q.; Skartsis, N.; Peng, Y.; Phipps, J.D.; Acenas, D.; et al. A human mutation in STAT3 promotes type 1 diabetes through a defect in CD8+ T cell tolerance. J. Exp. Med. 2021, 218, e20210759. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.A.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.C.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2014, 16, 57–66. [Google Scholar] [CrossRef]
- Si, S.; Teachey, D.T. Spotlight on Tocilizumab in the Treatment of CAR-T-Cell-Induced Cytokine Release Syndrome: Clinical Evidence to Date. Ther. Clin. Risk Manag. 2020, 16, 705–714. [Google Scholar]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Jeffrey, F.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, Y.; Minato, Y.; Ishifune, C.; Kurihara, T.; Kitamura, A.; Kojima, H.; Yagita, H.; Sakata-Yanagimoto, M.; Saito, T.; Taniuchi, I.; et al. Notch2 integrates signaling by the transcription factors RBP-J and CREB1 to promote T cell cytotoxicity. Nat. Immunol. 2008, 9, 1140–1147. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.A.; Seki, A.; Rutz, S. Ribonucleoprotein Transfection for CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. Curr. Protoc. Immunol. 2018, 124, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Johansen, K.H.; Schwartzberg, P.L. Efficient CRISPR/Cas9-Mediated Mutagenesis in Primary Murine T Lymphocytes. Curr. Protoc. Immunol. 2018, 124, e62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematics of cytokine-mediated STAT3 activation. Created with BioRender.com (accessed on 7 September 2021).
Figure 1.
Schematics of cytokine-mediated STAT3 activation. Created with BioRender.com (accessed on 7 September 2021).

Figure 2.
Four groups of genes modulated by STAT3 signaling. Created with BioRender.com (accessed on 7 September 2021).
Figure 2.
Four groups of genes modulated by STAT3 signaling. Created with BioRender.com (accessed on 7 September 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kaminskiy, Y.; Melenhorst, J.J. STAT3 Role in T-Cell Memory Formation. Int. J. Mol. Sci. 2022, 23, 2878. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052878
AMA Style
Kaminskiy Y, Melenhorst JJ. STAT3 Role in T-Cell Memory Formation. International Journal of Molecular Sciences. 2022; 23(5):2878. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052878
Chicago/Turabian StyleKaminskiy, Yaroslav, and Jan Joseph Melenhorst. 2022. "STAT3 Role in T-Cell Memory Formation" International Journal of Molecular Sciences 23, no. 5: 2878. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052878
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.