
Hyperglycemia Promotes Endothelial Cell Senescence through AQR/PLAU Signaling Axis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
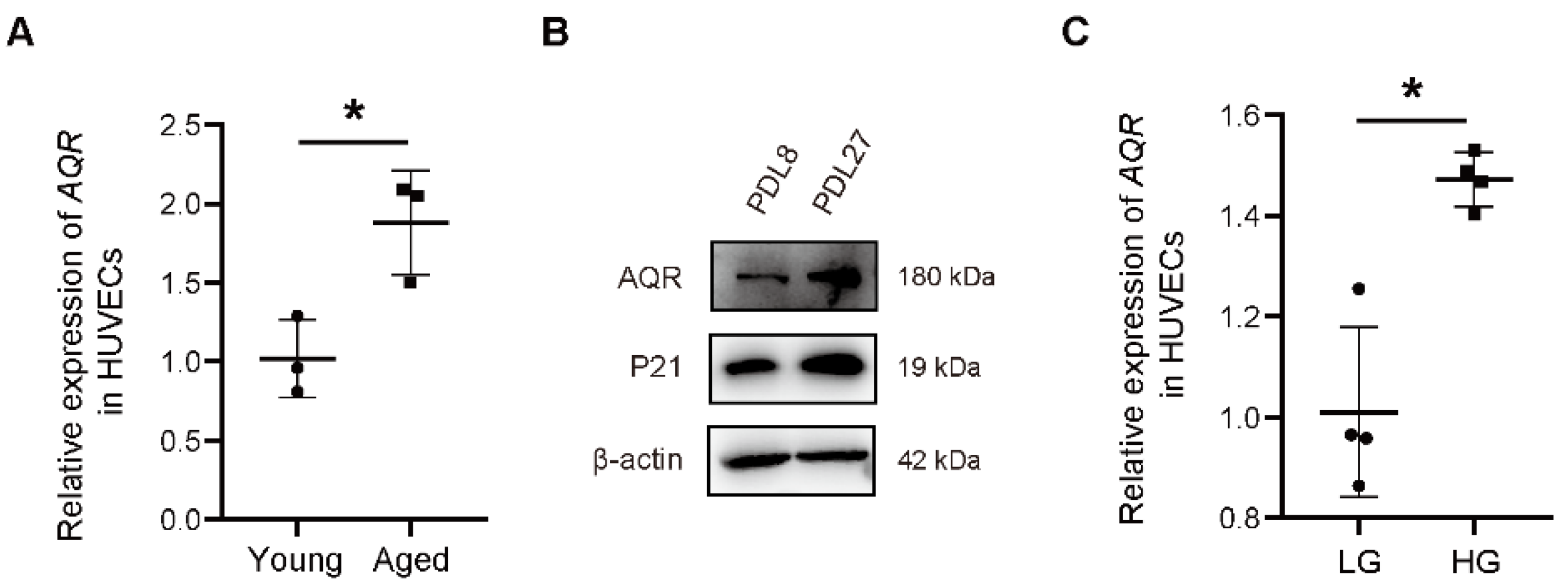
2.1. The Expression of AQR Is Increased with Aging
2.2. AQR Arrests Cell Cycle at G2/M and Promotes Cellular Senescence in HUVECs
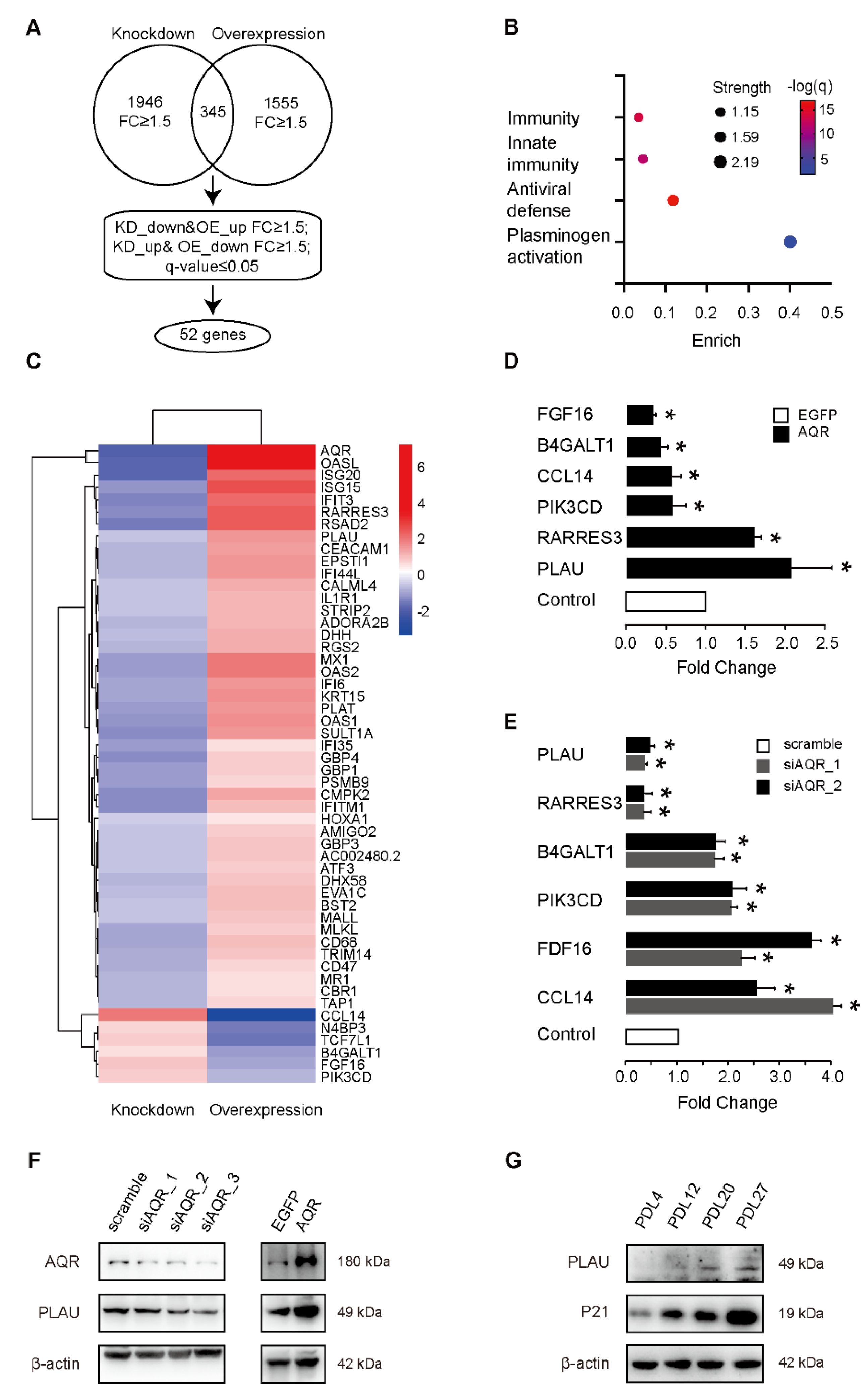
2.3. PLAU Is Co-Expressed with AQR during HUVEC Senescence
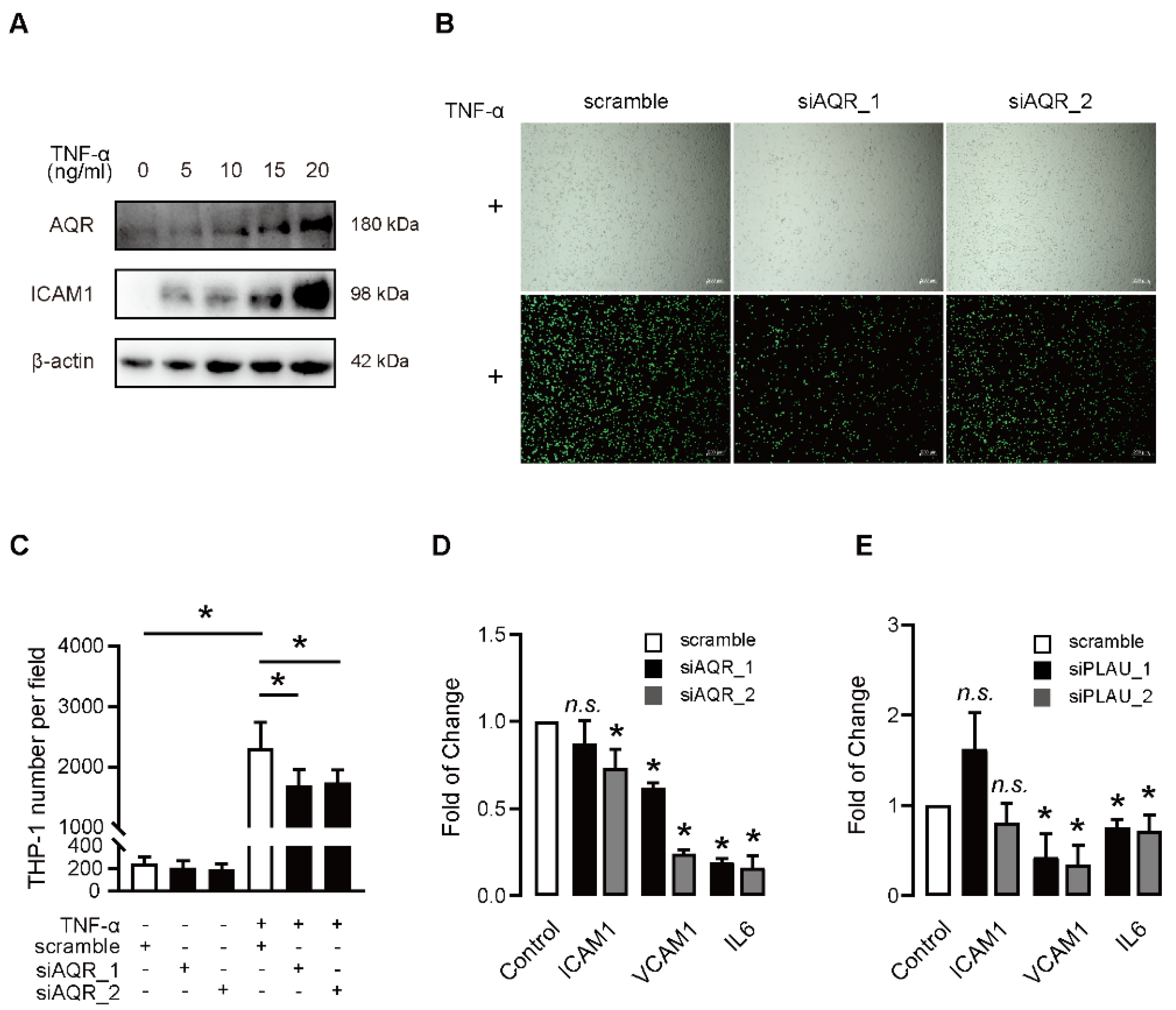
2.4. PLAU Knockdown Rescues the AQR-Induced Endothelial Cell Dysfunction
2.5. Functional Involvement AQR and PLAU Is Validated in Senescent Model Induced by TNF-α
3. Discussion
4. Material and Methods
4.1. Cell Lines and Culture
4.2. siRNAs Transfection and Adenovirus Infection
4.3. Confocal Laser Scanning Microscopy Analysis
4.4. Quantitative RT-PCR
4.5. Western Blot Analysis
4.6. Cell Cycle Analysis
4.7. Monocyte-HUVEC Adhesion Assays
4.8. Colony Formation Assay
4.9. RNA Sequencing Analysis
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R. Epigenetic Mechanisms in Diabetic Vascular Complications and Metabolic Memory: The 2020 Edwin Bierman Award Lecture. Diabetes 2021, 70, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.D.F.; Cooper, M.E. 50 years forward: Mechanisms of hyperglycaemia-driven diabetic complications. Diabetologia 2015, 58, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Papatheodorou, K.; Banach, M.; Bekiari, E.; Rizzo, M.; Edmonds, M. Complications of Diabetes 2017. J. Diabetes Res. 2018, 2018, 3086167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Miao, L.-F.; Qian, L.-L.; Wang, N.; Qi, M.-M.; Zhang, Y.-M.; Dang, S.-P.; Wu, Y.; Wang, R.-X. Molecular Mechanisms of Glucose Fluctuations on Diabetic Complications. Front. Endocrinol. 2019, 10, 640. [Google Scholar] [CrossRef]
- Qian, L.-L.; Liu, X.-Y.; Chai, Q.; Wang, R.-X. Hydrogen Sulfide in Diabetic Complications: Focus on Molecular Mechanisms. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 470–476. [Google Scholar] [CrossRef]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.E.; El-Osta, A. Epigenetics: Mechanisms and implications for diabetic complications. Circ. Res. 2010, 107, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.D.; Nadler, J.L. Inflammatory mechanisms of diabetic complications. Curr. Diabetes Rep. 2007, 7, 242–248. [Google Scholar] [CrossRef]
- Senthil, K.K.J.; Gokila, V.M.; Wang, S.Y. Activation of Nrf2-mediated anti-oxidant genes by antrodin C prevents hyperglycemia-induced senescence and apoptosis in human endothelial cells. Oncotarget 2017, 8, 96568–96587. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhan, J.-K.; Wang, Y.-J.; Lin, X.; Zhong, J.-Y.; Wang, Y.; Tan, P.; He, J.-Y.; Cui, X.-J.; Chen, Y.-Y.; et al. Exosomes from hyperglycemia-stimulated vascular endothelial cells contain versican that regulate calcification/senescence in vascular smooth muscle cells. Cell Biosci. 2019, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuki, S.; Imanishi, T.; Kobayashi, K.; Matsuo, Y.; Obana, M.; Akasaka, T. Hyperglycemia Accelerated Endothelial Progenitor Cell Senescence via the Activation of p38 Mitogen-Activated Protein Kinase. Circ. J. 2006, 70, 1076–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, M.; Zhang, Y.; Yu, H.; Li, X. Role of Hyperglycemia in the Senescence of Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2021, 9, 665412. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Pratt, G.A.; Yee, B.A.; Wheeler, E.C.; Blue, S.M.; Mueller, J.; Park, S.S.; Garcia, K.E.; Gelboin-Burkhart, C.; Nguyen, T.B.; et al. Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol. 2020, 21, 90. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Roy, R. Increased levels of hydrogen peroxide induce a HIF-1-dependent modification of lipid metabolism in AMPK compromised C. elegans dauer larvae. Cell Metab. 2012, 16, 322–335. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Yan, H.; Wang, H.; Zhang, Y.; Cao, H.; Wan, Y.; Kong, L.; Chen, S.; Xu, H.; Pan, B.; et al. AQR is a novel type 2 diabetes-associated gene that regulates signaling pathways critical for glucose metabolism. J. Genet. Genom. 2018, 45, 111–120. [Google Scholar] [CrossRef]
- Qian, H.; Kowalski, M.H.; Kramer, H.J.; Tao, R.; Lash, J.P.; Stilp, A.M.; Cai, J.; Li, Y.; Franceschini, N. Genome-Wide Association of Kidney Traits in Hispanics/Latinos Using Dense Imputed Whole-Genome Sequencing Data: The Hispanic Community Health Study/Study of Latinos. Circ. Genom. Precis. Med. 2020, 13, e002891. [Google Scholar] [CrossRef]
- Fowler, M.J. Microvascular and Macrovascular Complications of Diabetes. Clin. Diabetes 2008, 26, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Chawla, R.; Chawla, A.; Jaggi, S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J. Endocrinol. Metab. 2016, 20, 546–551. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, H.-X.; Yan, H.; Zhu, S.-L.; Guo, S.-W.; Peng, W.-J.; Luo, D. Rutaecarpine prevents high glucose-induced endothelial cell senescence through TRPV1/ SIRT1 pathway. J. Cardiovasc. Pharmacol. 2021, 79, e129–e137. [Google Scholar] [CrossRef]
- Wang, G.; Han, B.; Zhang, R.; Liu, Q.; Wang, X.; Huang, X.; Liu, D.; Qiao, W.; Yang, M.; Luo, X.; et al. C1q/TNF-Related Protein 9 Attenuates Atherosclerosis by Inhibiting Hyperglycemia-Induced Endothelial Cell Senescence Through the AMPKalpha/KLF4 Signaling Pathway. Front Pharmacol. 2021, 12, 758792. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, H.; Wang, J.; Zhong, L.; Chen, X.; Zhang, R.; Wang, H. Glucose restriction delays senescence and promotes proliferation of HUVECs via the AMPK/SIRT1-FOXA3-Beclin1 pathway. Exp. Gerontol. 2020, 139, 111053. [Google Scholar] [CrossRef] [PubMed]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.-H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2020, 24, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C., IV; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arter. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef] [PubMed]
- Lyu, G.; Guan, Y.; Zhang, C.; Zong, L.; Sun, L.; Huang, X.; Huang, L.; Zhang, L.; Tian, X.-L.; Zhou, Z.; et al. TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat. Commun. 2018, 9, 2560. [Google Scholar] [CrossRef]
- Min, X.; Cai, M.; Shao, T.; Xu, Z.; Liao, Z.; Liu, D.; Zhou, M.; Wu, W.; Zhou, Y.; Mo, M.; et al. A circular intronic RNA ciPVT1 delays endothelial cell senescence by regulating the miR-24-3p/CDK4/pRb axis. Aging Cell 2022, 21, e13529. [Google Scholar] [CrossRef] [PubMed]
- Sam, M.; Wurst, W.; Klüppel, M.; Jin, O.; Heng, H.; Bernstein, A. Aquarius, a novel gene isolated by gene trapping with an RNA-dependent RNA polymerase motif. Dev. Dyn. 1998, 212, 304–317. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Yang, J. Genome-wide association studies of stress score in a Korean Cohort. Stress 2021, 24, 1016–1022. [Google Scholar] [CrossRef]
- White, T.; Deshpande, N.; Kumar, V.; Gauthier, A.G.; Jurkunas, U.V. Cell cycle re-entry and arrest in G2/M phase induces senescence and fibrosis in Fuchs Endothelial Corneal Dystrophy. Free Radic. Biol. Med. 2021, 164, 34–43. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gire, V.; Dulić, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyyriäinen, J.; Tapiala, J.; Lipponen, A.; Ndode-Ekane, X.E.; Pitkänen, A. Plau/Plaur double-deficiency did not worsen lesion severity or vascular integrity after traumatic brain injury. Neurosci. Lett. 2020, 729, 134935. [Google Scholar] [CrossRef] [PubMed]
- Ai, C.; Zhang, J.; Lian, S.; Ma, J.; Győrffy, B.; Qian, Z.; Han, Y.; Feng, Q. FOXM1 functions collaboratively with PLAU to promote gastric cancer progression. J. Cancer 2020, 11, 788–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyyriäinen, J.; Bolkvadze, T.; Koivisto, H.; Lipponen, A.; Pérez, L.O.; Ndode-Ekane, X.E.; Tanila, H.; Pitkänen, A. Deficiency of urokinase-type plasminogen activator and its receptor affects social behavior and increases seizure susceptibility. Epilepsy Res. 2019, 151, 67–74. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Fernandes, A.; Aguilar-Pimentel, J.A.; De Angelis, M.H.; Guedes, J.R.; Brito, M.A.; Ortolano, S.; Pani, G.; Athanasopoulou, S.; Gonos, E.S.; et al. Towards frailty biomarkers: Candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res. Rev. 2018, 47, 214–277. [Google Scholar] [CrossRef]
- Jiang, H.; Liang, L.; Qin, J.; Lu, Y.; Li, B.; Wang, Y.; Lin, C.; Zhou, Q.; Feng, S.; Yip, S.; et al. Functional networks of aging markers in the glomeruli of IgA nephropathy: A new therapeutic opportunity. Oncotarget 2016, 7, 33616–33626. [Google Scholar] [CrossRef] [Green Version]
- Lehallier, B.; Shokhirev, M.N.; Wyss-Coray, T.; Johnson, A.A. Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell 2020, 19, e13256. [Google Scholar] [CrossRef]
- Corley, S.M.; Mendoza-Reinoso, V.; Giles, N.; Singer, E.S.; Common, J.E.; Wilkins, M.R.; Beverdam, A. Plau and Tgfbr3 are YAP-regulated genes that promote keratinocyte proliferation. Cell Death Dis. 2018, 9, 1106. [Google Scholar] [CrossRef]
- Pan, X.; Wu, B.; Fan, X.; Xu, G.; Ou, C.; Chen, M. YAP accelerates vascular senescence via blocking autophagic flux and activating mTOR. J. Cell. Mol. Med. 2021, 25, 170–183. [Google Scholar] [CrossRef]
- Xu, X.; Shen, X.; Feng, W.; Yang, D.; Jin, L.; Wang, J.; Wang, M.; Ting, Z.; Xue, F.; Zhang, J.; et al. D-galactose induces senescence of glioblastoma cells through YAP-CDK6 pathway. Aging 2020, 12, 18501–18521. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shen, X.; Wang, J.; Feng, W.; Wang, M.; Miao, X.; Wu, Q.; Wu, L.; Wang, X.; Ma, Y.; et al. YAP prevents premature senescence of astrocytes and cognitive decline of Alzheimer’s disease through regulating CDK6 signaling. Aging Cell 2021, 20, e13465. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhang, Z.; Gao, M.; Yu, H.; Sheng, H.; Huang, J. MicroRNA-193a-3p suppresses the colorectal cancer cell proliferation and progression through downregulating the PLAU expression. Cancer Manag. Res. 2019, 11, 5353–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, L.; Deng, H.; Li, Y.; Zhang, C.; Liu, X.; Liu, Q.; Zhang, D.; Wang, L.; Pu, Y.; Zhang, H.; et al. The DNA methylation-regulated miR-193a-3p dictates the multi-chemoresistance of bladder cancer via repression of SRSF2/PLAU/HIC2 expression. Cell Death Dis. 2014, 5, e1402. [Google Scholar] [CrossRef]
- Chen, G.; Sun, J.; Xie, M.; Yu, S.; Tang, Q.; Chen, L. PLAU Promotes Cell Proliferation and Epithelial-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Front. Genet. 2021, 12, 651882. [Google Scholar] [CrossRef]
- Cunningham, O.; Campion, S.; Perry, V.H.; Murray, C.; Sidenius, N.; Docagne, F.; Cunningham, C. Microglia and the urokinase plasminogen activator receptor/uPA system in innate brain inflammation. Glia 2009, 57, 1802–1814. [Google Scholar] [CrossRef] [Green Version]
- Connolly, B.M.; Choi, E.Y.; Gardsvoll, H.; Bey, A.L.; Currie, B.M.; Chavakis, T.; Liu, S.; Molinolo, A.; Ploug, M.; Leppla, S.H.; et al. Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood 2010, 116, 1593–1603. [Google Scholar] [CrossRef] [Green Version]
- Theilade, S.; Lyngbaek, S.; Hansen, T.; Eugen-Olsen, J.; Fenger, M.; Rossing, P.; Jeppesen, J.L. Soluble urokinase plasminogen activator receptor levels are elevated and associated with complications in patients with type 1 diabetes. J. Intern. Med. 2015, 277, 362–371. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Y.; Liu, Z.; Wu, A.; Khan, A.H.; Zhu, Y.; Ding, S.; Li, X.; Zhao, Y.; Dai, X.; Zhou, J.; et al. Hyperglycemia Promotes Endothelial Cell Senescence through AQR/PLAU Signaling Axis. Int. J. Mol. Sci. 2022, 23, 2879. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052879
Wan Y, Liu Z, Wu A, Khan AH, Zhu Y, Ding S, Li X, Zhao Y, Dai X, Zhou J, et al. Hyperglycemia Promotes Endothelial Cell Senescence through AQR/PLAU Signaling Axis. International Journal of Molecular Sciences. 2022; 23(5):2879. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052879
Chicago/Turabian StyleWan, Yiqi, Zhirui Liu, Andong Wu, Abdul Haseeb Khan, Ying Zhu, Shuangjin Ding, Xueer Li, Ya Zhao, Ximo Dai, Jin Zhou, and et al. 2022. "Hyperglycemia Promotes Endothelial Cell Senescence through AQR/PLAU Signaling Axis" International Journal of Molecular Sciences 23, no. 5: 2879. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052879