
Improved Binding Affinity of Omicron’s Spike Protein for the Human Angiotensin-Converting Enzyme 2 Receptor Is the Key behind Its Increased Virulence


Abstract
:1. Introduction
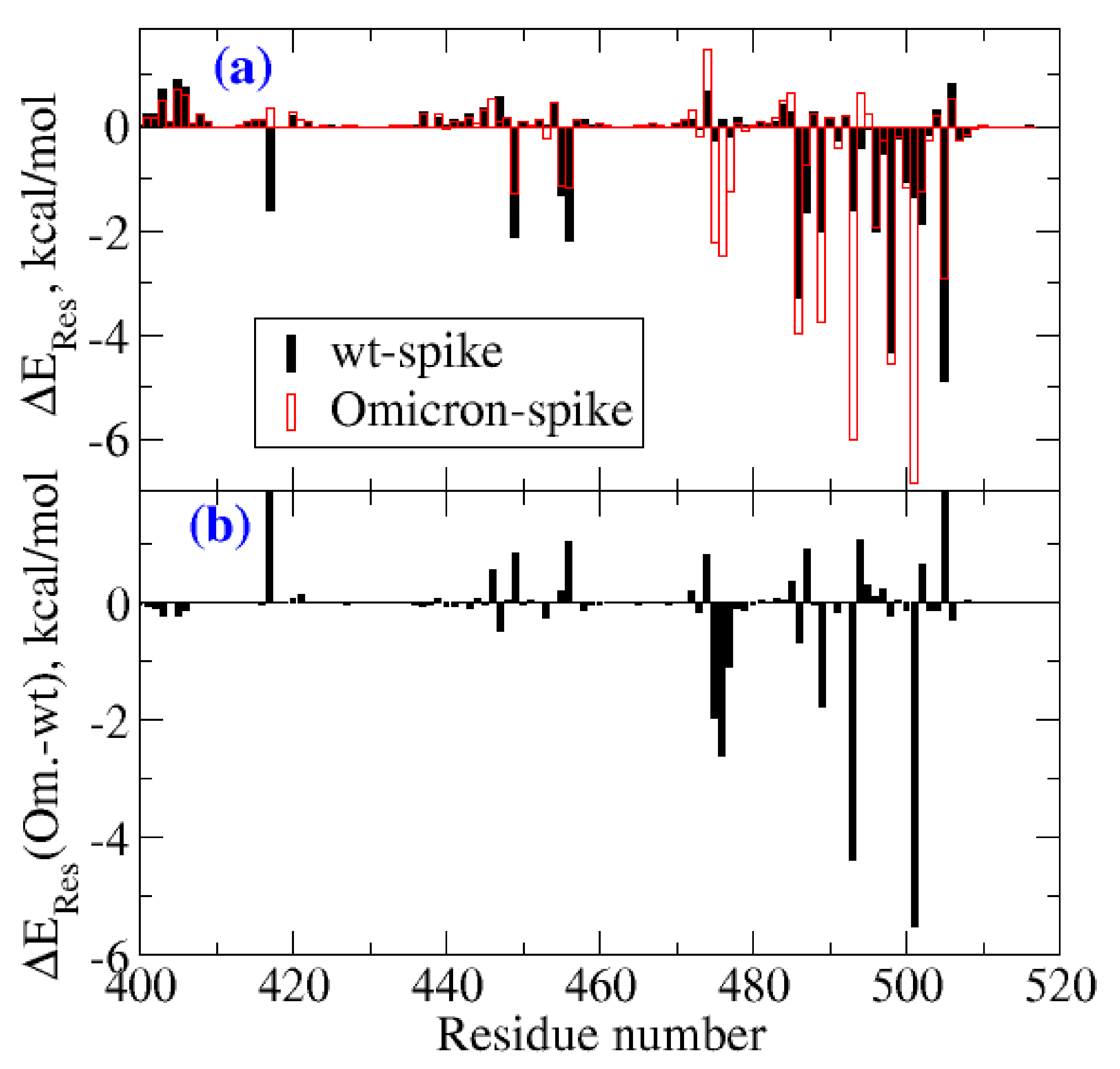
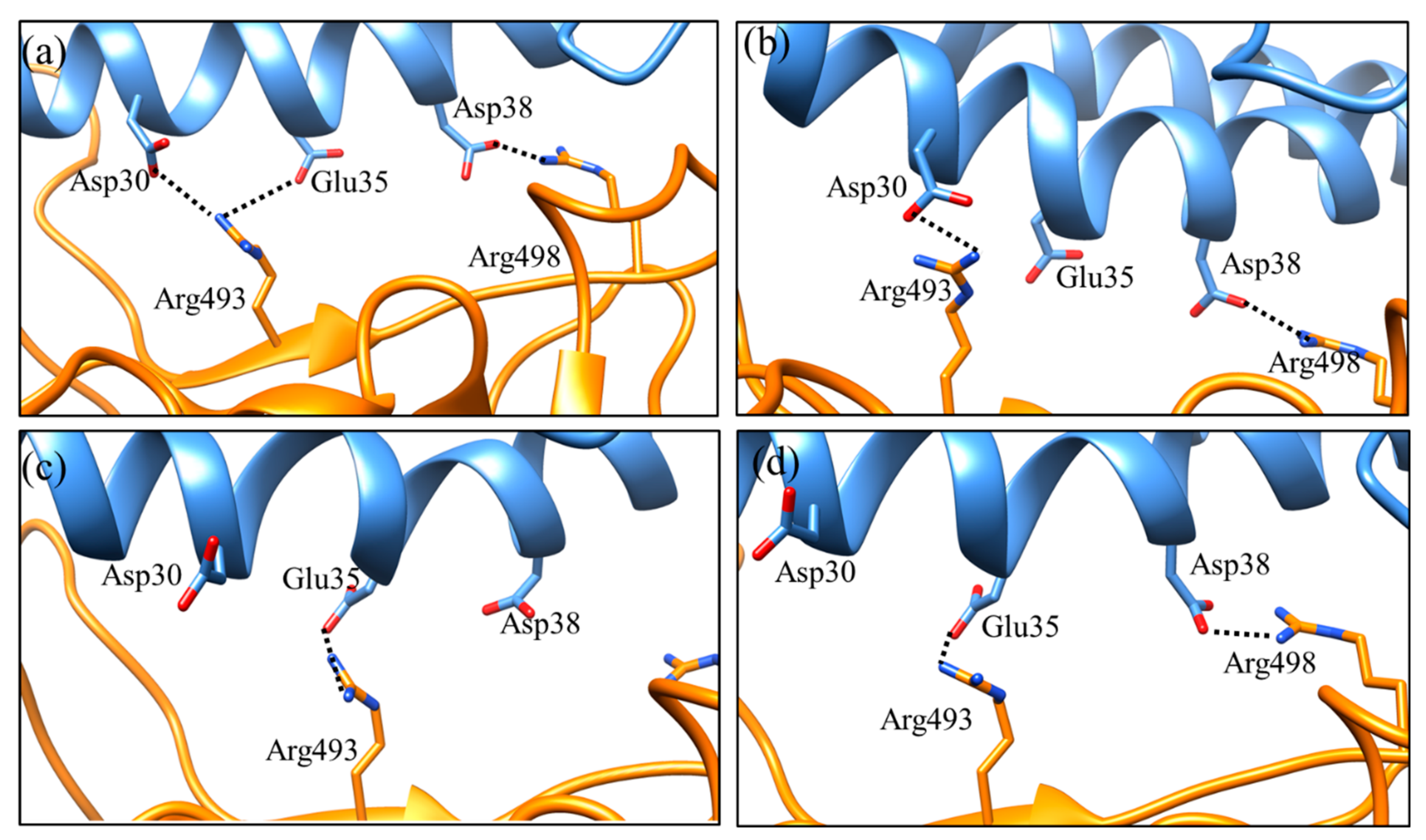
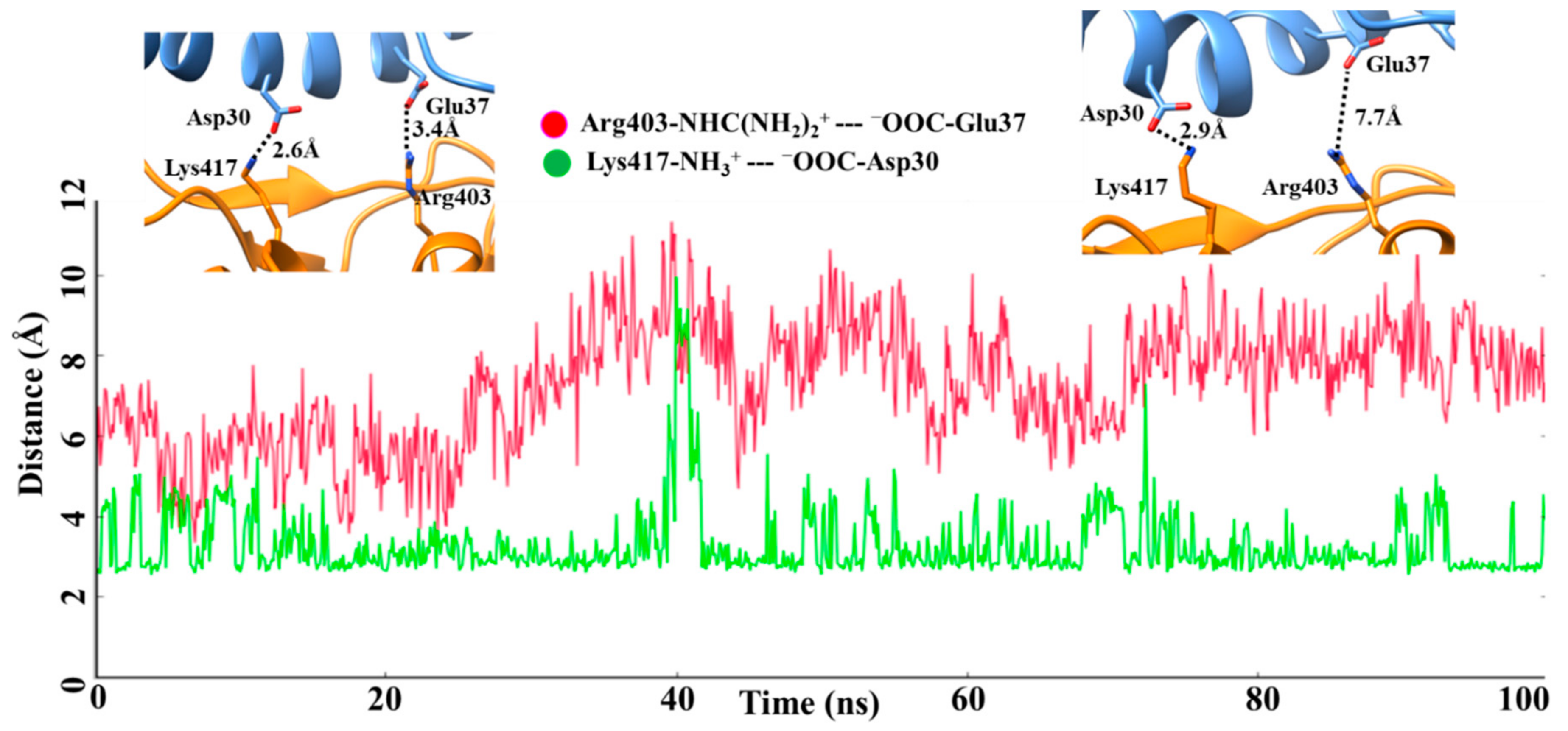
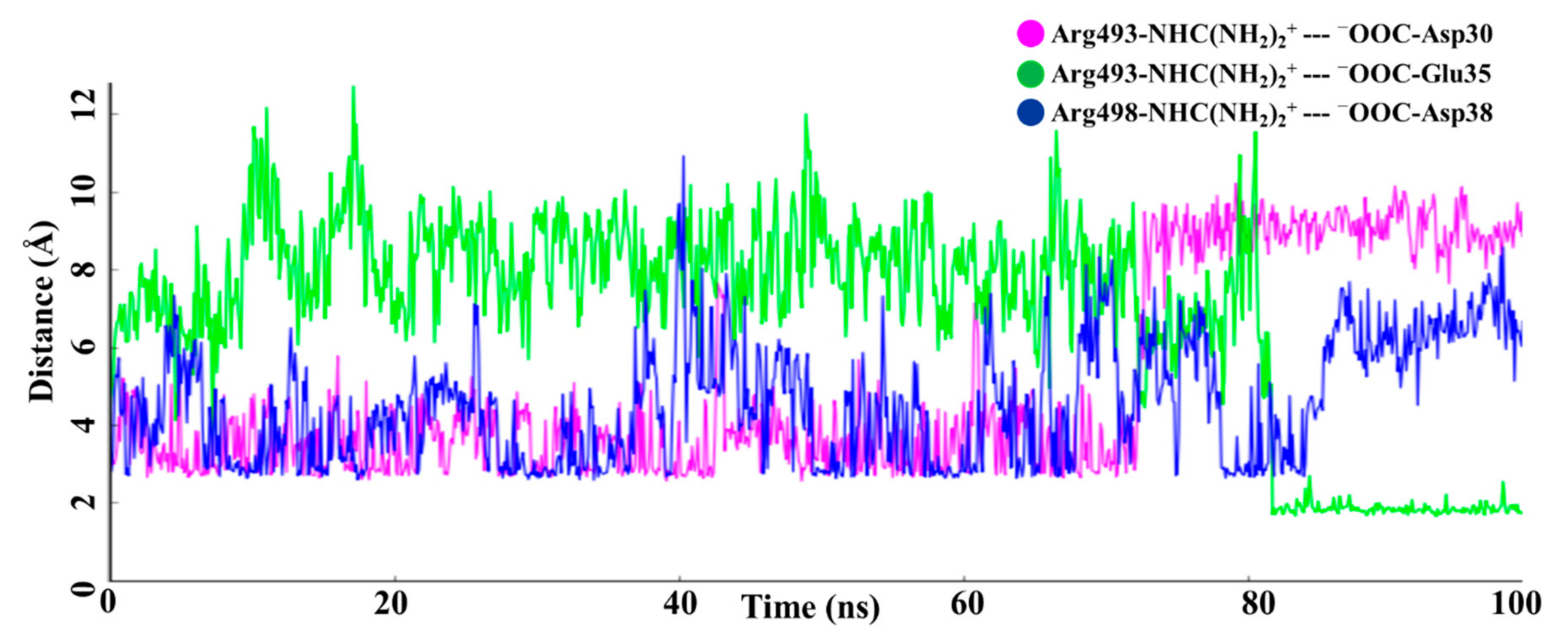
2. Result and Discussion
3. Materials and Methods
3.1. Sequence Analysis and Structure Modelling
3.2. Molecular Dynamics Simulation and Molecular Mechanics-Generalized Born Surface Area Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satarker, S.; Nampoothiri, M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Centre for Disease Prevention and Control. SARS-CoV-2 Variants of Concern as of 13 January 2022. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 10 December 2021).
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef]
- CDC. Science Brief: Omicron (B.1.1.529) Variant; US Department of Health and Human Services, CDC: Atlanta, GA, USA, 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-omicron-variant.html (accessed on 2 December 2021).
- HKUMed Finds Omicron SARS-CoV-2 Can Infect Faster and Better than Delta in Human Bronchus but with Less Severe Infection in Lung. Available online: https://www.med.hku.hk/en/news/press/20211215-omicron-sars-cov-2-infection?utm_medium=social&utm_source=twitter&utm_campaign=press_release (accessed on 15 January 2022).
- Grabowski, F.; Kochańczyk, M.; Lipniacki, T. Omicron strain spreads with the doubling time of 3.2–3.6 days in South Africa province of Gauteng that achieved herd immunity to Delta variant. Viruses 2022, 14, 294. [Google Scholar] [CrossRef]
- World Health Organization. Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern 2021. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 15 December 2021).
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV-a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Ye, F.; Lin, X.; Chen, Z.; Yang, F.; Lin, S.; Yang, J.; Chen, H.; Sun, H.; Wang, L.; Wen, A.; et al. S19W, T27W, and N330Y mutations in ACE2 enhance SARS-CoV-2 S-RBD binding toward both wild-type and antibody-resistant viruses and its molecular basis. Sig. Transduct. Target Ther. 2021, 6, 343. [Google Scholar] [CrossRef]
- Niu, S.; Wang, J.; Bai, B.; Wu, L.; Zheng, A.; Chen, Q.; Du, P.; Han, P.; Zhang, Y.; Jia, Y.; et al. Molecular basis of cross-species ACE2 interactions with SARS-CoV-2-like viruses of pangolin origin. EMBO J. 2021, 40, e107786. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Harari, D.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Li, C.; Marton, I.; et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021, 6, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Chelli, R.; Gervasio, F.L.; Procacci, P.; Schettino, V. Stacking and T-shape competition in aromatic-aromatic amino acid interactions. J. Am. Chem. Soc. 2002, 124, 6133–6143. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S. E484K and N501Y SARS-CoV-2 spike mutants Increase ACE2 recognition but reduce affinity for neutralizing antibody. Int. Immunopharmacol. 2022, 102, 108424. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2021, 602, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Lee, T.S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-accelerated molecular dynamics and free energy methods in Amber18: Performance enhancements and new features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | ΔEvdw | ΔEelec | ΔEpsol | ΔEnpsol | ΔG, kcal/mol |
|---|---|---|---|---|---|
| Wild Type | −85.28 | −856.42 | 922.17 | −12.91 | −32.43 |
| B.1.1.529 | −100.84 | −1714.73 | 1787.97 | −13.39 | −41.00 |
| No. | Wild Type-RBM: hACE2 | H-Bond Occupancy (%) | Omicron-RBM: hACE2 | H-Bond Occupancy (%) | Salt Bridges (Wild Type-RBM--hACE2) | Salt Bridges (Omicron-RBM--hACE2) |
|---|---|---|---|---|---|---|
| 1 | Tyr449-Asp38 | 70.50 | Tyr449-Asp38 | 17.10 | Lys417- -Asp30 | Arg493- -Asp30 |
| 2 | Thr500-Asp355 | 38.10 | Thr500-Asp355 | 54.20 | Arg493- -Glu35 | |
| 3 | Thr500-Tyr41 | 18.40 | - | - | Arg498- -Asp38 | |
| 4 | Gln493-Glu35 | 38.20 | Arg493-Asp30 | 39.20 | ||
| 5 | - | - | Arg493-Glu35 | 11.10 | ||
| 6 | Gln498-Asp38 | 26.90 | - | - | ||
| 7 | Gln498-Lys353 | 20.80 | Arg498-Asp38 | 28.80 | ||
| 8 | Gly502-Lys353 | 48.60 | Gly502-Lys353 | 48.60 | ||
| 9 | Tyr505-Glu37 | 22.70 | His505-Ala386 | 16.50 | ||
| 10 | Gly496-Lys353 | 14.00 | Ser496-Asp38 | 16.10 | ||
| 11 | Ser494-His34 | 14.90 | Ser494-His34 | 12.20 | ||
| 12 | Asn487-Tyr83 | 28.30 | Asn487-Tyr83 | 9.10 | ||
| 13 | Ala475-Gln24 | 11.10 | ||||
| 14 | Lys417-Asp30 | 33.50 | - | - | ||
| 15 | - | Asn487-Gln24 | 13.90 | |||
| 16 | Gly446-Gln42 | 9.50 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Murugan, N.A.; Srivastava, V. Improved Binding Affinity of Omicron’s Spike Protein for the Human Angiotensin-Converting Enzyme 2 Receptor Is the Key behind Its Increased Virulence. Int. J. Mol. Sci. 2022, 23, 3409. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063409
Kumar R, Murugan NA, Srivastava V. Improved Binding Affinity of Omicron’s Spike Protein for the Human Angiotensin-Converting Enzyme 2 Receptor Is the Key behind Its Increased Virulence. International Journal of Molecular Sciences. 2022; 23(6):3409. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063409
Chicago/Turabian StyleKumar, Rajender, Natarajan Arul Murugan, and Vaibhav Srivastava. 2022. "Improved Binding Affinity of Omicron’s Spike Protein for the Human Angiotensin-Converting Enzyme 2 Receptor Is the Key behind Its Increased Virulence" International Journal of Molecular Sciences 23, no. 6: 3409. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063409