
Rotamers in Crystal Structures of Xylitol, D-Arabitol and L-Arabitol

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
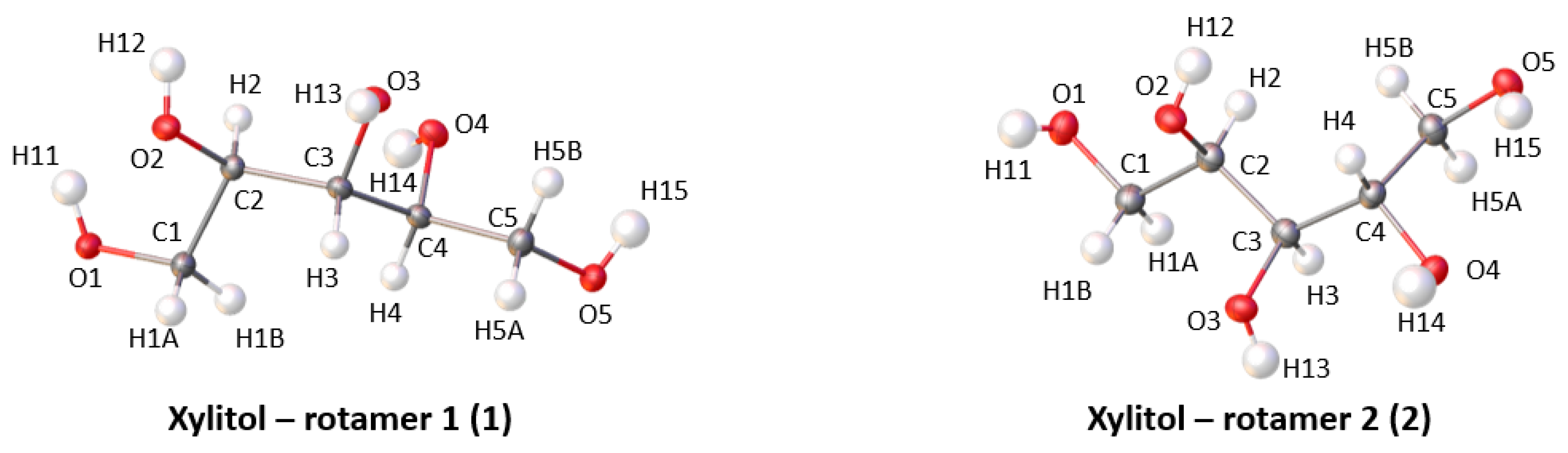
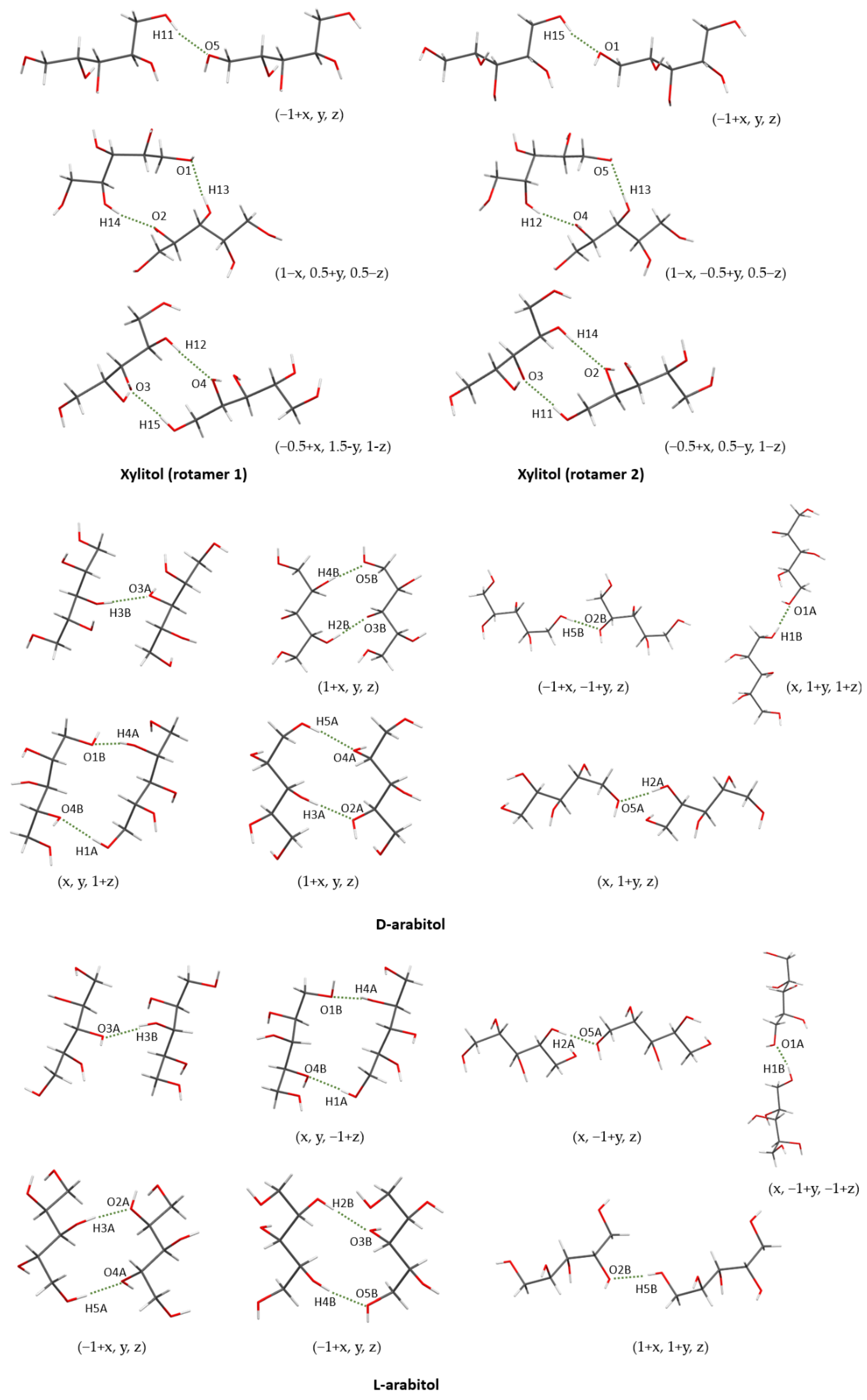
2.1. Crystal Structures of Xylitol
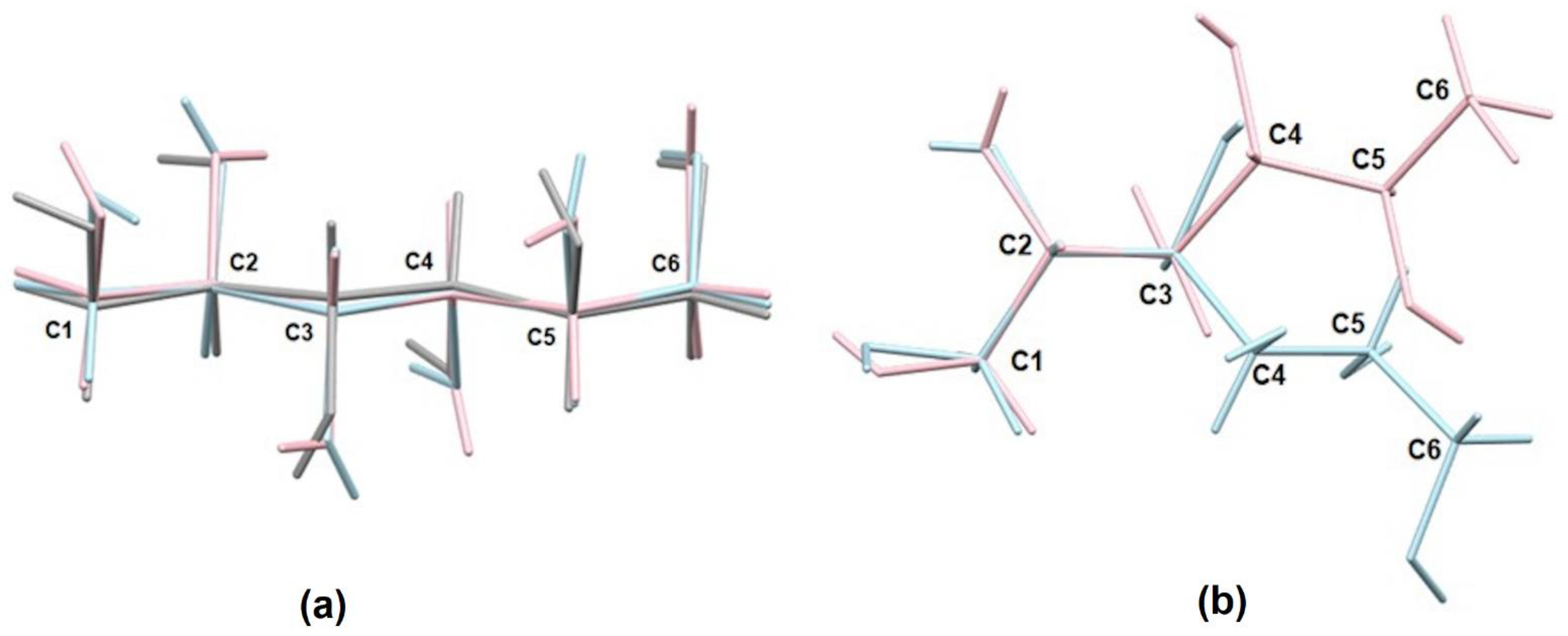
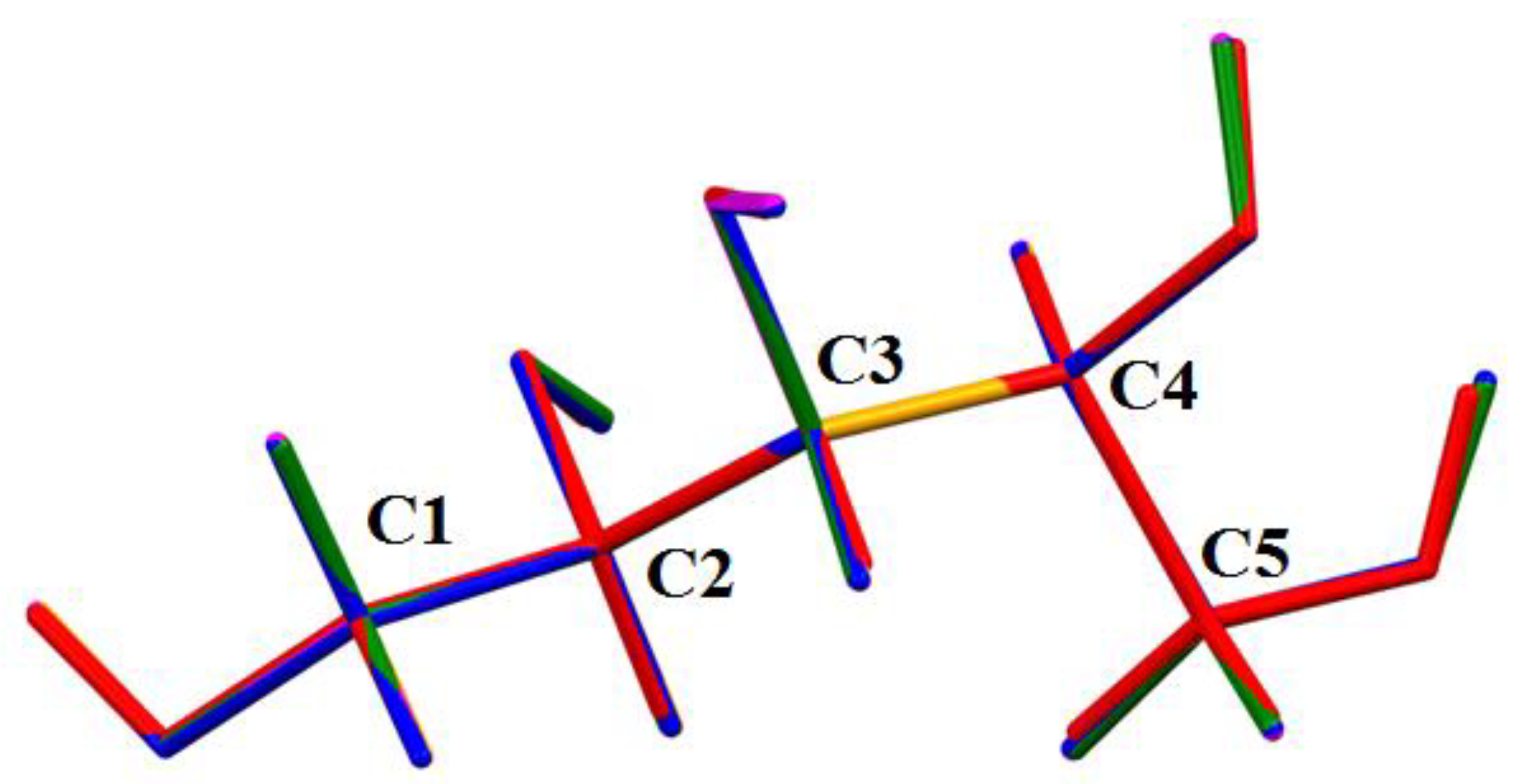
2.2. Geometry
2.3. Crystal Structures of L-Arabitol and D-Arabitol
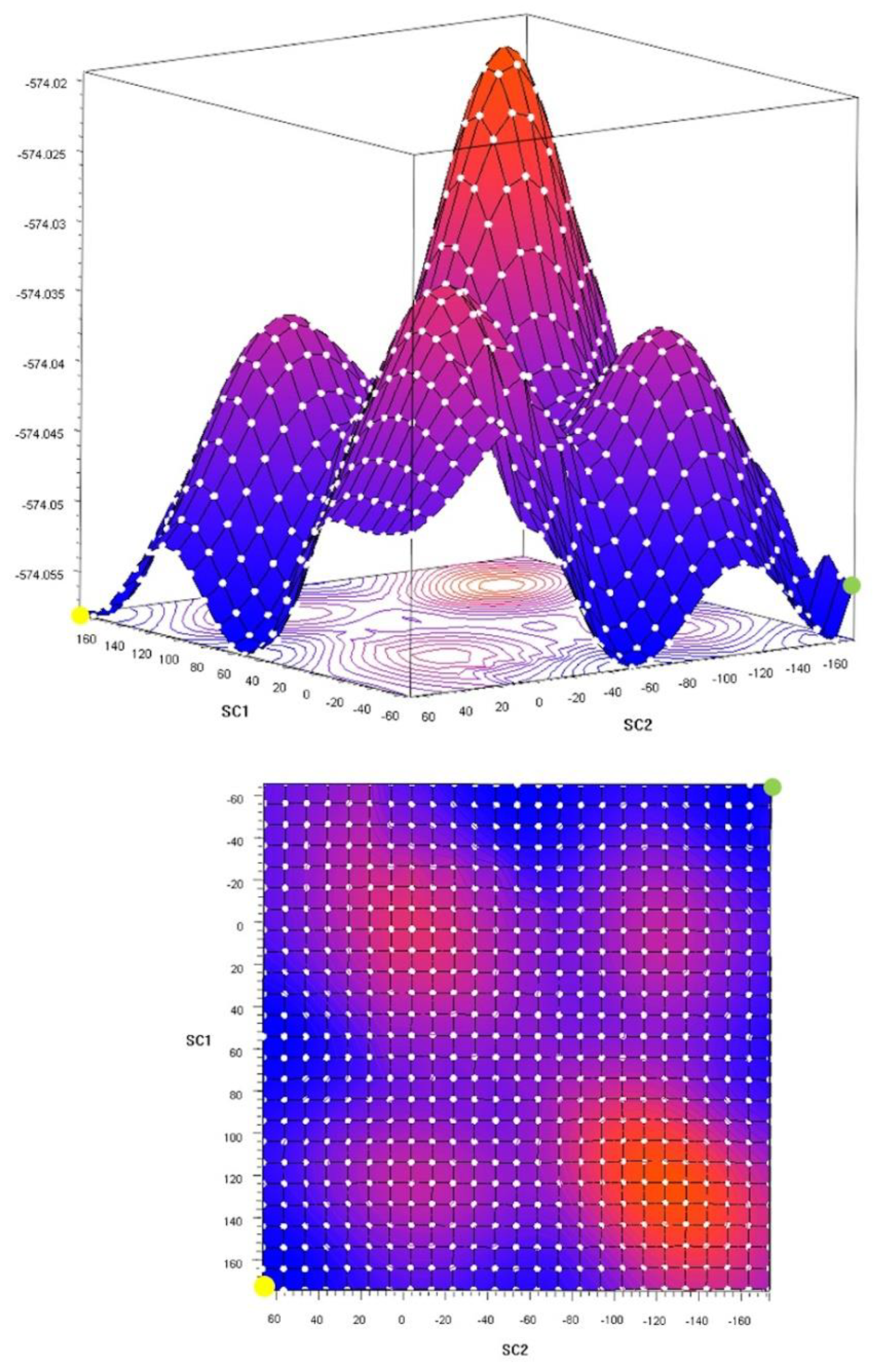
2.4. Theoretical Calculations
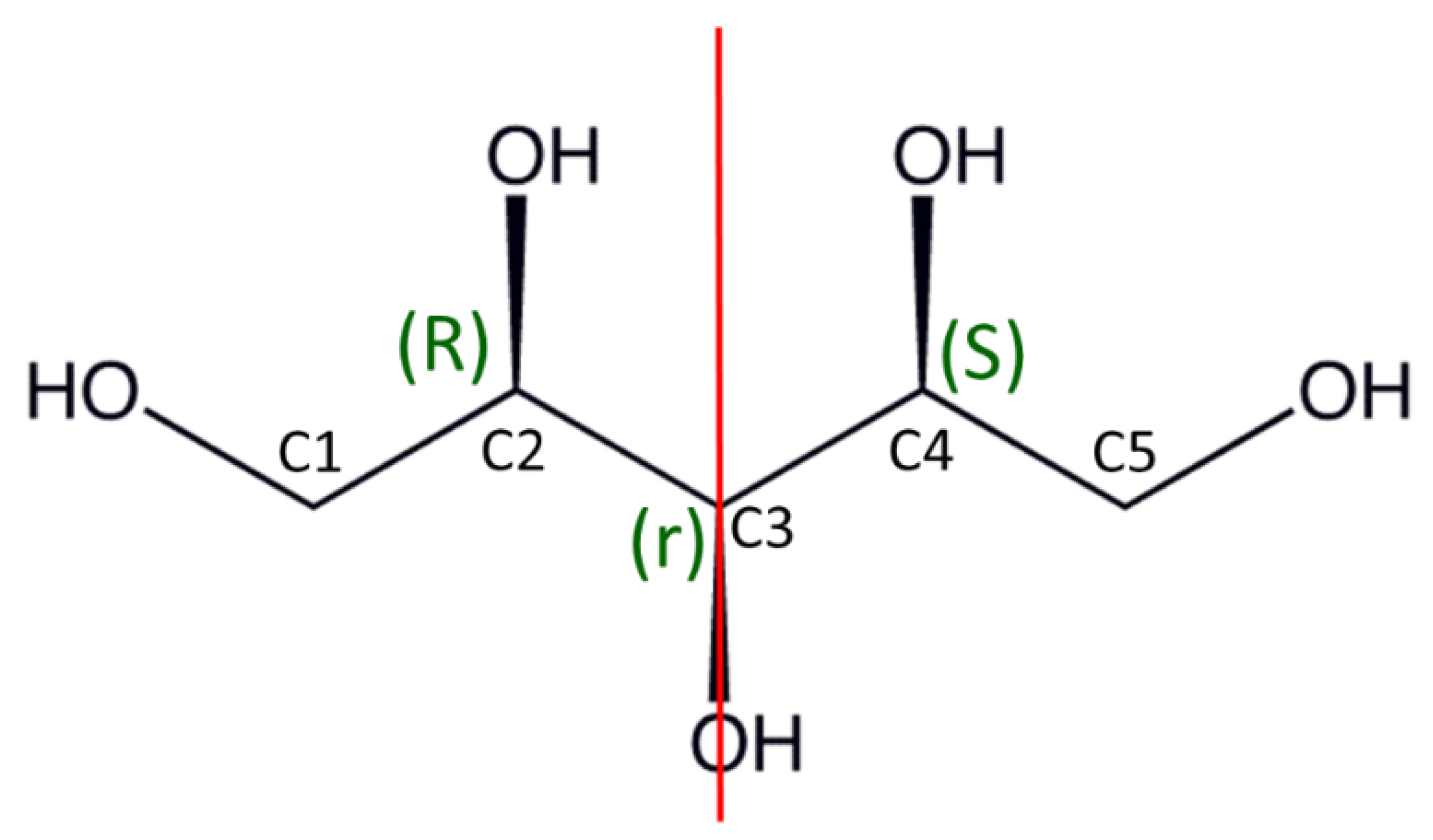
2.5. Absolute Configuration of Xylitol
2.6. Numbering System in Deposited Compounds
3. Materials and Methods
3.1. Preparation of Crystals
- Recrystallization of xylitol (2);
- Co-crystallization of xylitol (1) and xylitol (2);
- Co-crystallization of xylitol (1) with other sugars (i.e., D-arabitol, L-arabitol, and ribitol).
3.2. Single Crystal X-ray Studies
3.3. CSD Search
3.4. Computational Studies
3.5. Powder X-ray Diffraction Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen Atoms Can Be Located Accurately and Precisely by X-Ray Crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanat, M.; Malinska, M.; Gutmann, M.J.; Cooper, R.I.; Wozniak, K. HAR, TAAM and BODD Refinements of Model Crystal Structures Using Cu Kα and Mo Kα X-Ray Diffraction Data. Acta Cryst. B 2021, 77, 41–53. [Google Scholar] [CrossRef]
- Wanat, M.; Malinska, M.; Hoser, A.A.; Woźniak, K. Further Validation of Quantum Crystallography Approaches. Molecules 2021, 26, 3730. [Google Scholar] [CrossRef] [PubMed]
- Hoser, A.A.; Madsen, A.Ø. Dynamic Quantum Crystallography: Lattice-Dynamical Models Refined against Diffraction Data. II. Applications to l-Alanine, Naphthalene and Xylitol. Acta Cryst. A 2017, 73, 102–114. [Google Scholar] [CrossRef]
- Madsen, A.Ø.; Sørensen, H.O.; Flensburg, C.; Stewart, R.F.; Larsen, S. Modeling of the Nuclear Parameters for H Atoms in X-Ray Charge-Density Studies. Acta Cryst. A 2004, 60, 550–561. [Google Scholar] [CrossRef]
- Madsen, A.Ø.; Mattson, R.; Larsen, S. Understanding Thermodynamic Properties at the Molecular Level: Multiple Temperature Charge Density Study of Ribitol and Xylitol. J. Phys. Chem. A 2011, 115, 7794–7804. [Google Scholar] [CrossRef]
- Madsen, A.Ø.; Mason, S.; Larsen, S. A Neutron Diffraction Study of Xylitol: Derivation of Mean Square Internal Vibrations for H Atoms from a Rigid-Body Description. Acta Cryst. B 2003, 59, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Guex, W.; Klaeui, H.; Pauling, H.; Voirol, F. Reusable Heat Devices Containing Xylitol as Heat Storage Material. U.S. Patent 4,296,801, 27 October 1981. [Google Scholar]
- Palomo Del Barrio, E.; Cadoret, R.; Daranlot, J.; Achchaq, F. New Sugar Alcohols Mixtures for Long-Term Thermal Energy Storage Applications at Temperatures between 70 °C and 100 °C. Solar Energy Mater. Solar Cells 2016, 155, 454–468. [Google Scholar] [CrossRef]
- Paleta, O.; Dlouhá, I.; Kaplánek, R.; Kefurt, K.; Kodíček, M. Novel Amphiphilic Fluoroalkylated Derivatives of Xylitol, d-Glucose and d-Galactose for Medical Applications: Hemocompatibility and Co-Emulsifying Properties. Carbohydr. Res. 2002, 337, 2411–2418. [Google Scholar] [CrossRef]
- Zarif, L.; Greiner, J.; Pace, S.; Riess, J.G. Synthesis of Perfluoroalkylated Xylitol Ethers and Esters: New Surfactants for Biomedical Uses. J. Med. Chem. 1990, 33, 1262–1269. [Google Scholar] [CrossRef]
- Fronczek, F.R.; Kamel, H.N.; Slattery, M. Three Polymorphs (α, β, and δ) of d-Mannitol at 100 K. Acta Cryst. C 2003, 59, o567–o570. [Google Scholar] [CrossRef] [PubMed]
- Schouten, A.; Kanters, J.A.; Kroon, J.; Comini, S.; Looten, P.; Mathlouthi, M. Conformational Polymorphism of D-Sorbitol (d-Glucitol): The Crystal and Molecular Structures of d-Glucitol 2/3-Hydrate and Epsilond-Glucitol. Carbohydr. Res. 1998, 312, 131–137. [Google Scholar] [CrossRef]
- Park, Y.J.; Jeffrey, G.A.; Hamilton, W.C. Determination of the Crystal Structure of the A Form of D-Glucitol by Neutron and X-ray Diffraction. Acta Cryst. B 1971, 27, 2393–2401. [Google Scholar] [CrossRef]
- Dierks, T.M.; Korter, T.M. Comparison of Intermolecular Forces in Anhydrous Sorbitol and Solvent Cocrystals. J. Phys. Chem. A 2017, 121, 5720–5727. [Google Scholar] [CrossRef] [PubMed]
- Rukiah, M.; Lefebvre, J.; Hernandez, O.; van Beek, W.; Serpelloni, M. Ab Initio Structure Determination of the Γ Form of D-Sorbitol (d-Glucitol) by Powder Synchrotron X-Ray Diffraction. J. Appl. Cryst. 2004, 37, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Jeffrey, G.A. The Crystal Structure of Xylitol. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1969, 25, 2607–2613. [Google Scholar] [CrossRef]
- Wolfrom, M.L.; Kohn, E.J. Crystalline Xylitol. J. Am. Chem. Soc. 1942, 64, 1739. [Google Scholar] [CrossRef]
- Franks, F.; Kay, R.L.; Dadok, J. A Nuclear Magnetic Resonance Study of Isomeric Pentitols in Aqueous and Non-Aqueous Solutions. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1988, 84, 2595–2601. [Google Scholar] [CrossRef]
- Roberts, J.K.; Jardetzky, O. Monitoring of Cellular Metabolism by NMR. Biochim. Biophys. Acta (BBA)-Rev. Bioenerg. 1981, 639, 53–76. [Google Scholar] [CrossRef]
- Fenn, T.D.; Ringe, D.; Petsko, G.A. Xylose Isomerase in Substrate and Inhibitor Michaelis States: Atomic Resolution Studies of a Metal-Mediated Hydride Shift. Biochemistry 2004, 43, 6464–6474. [Google Scholar] [CrossRef]
- Botzki, A.; Rigden, D.J.; Braun, S.; Nukui, M.; Salmen, S.; Hoechstetter, J.; Bernhardt, G.; Dove, S.; Jedrzejas, M.J.; Buschauer, A. L-Ascorbic Acid 6-Hexadecanoate, a Potent Hyaluronidase Inhibitor: X-ray structure and molecular modeling of enzyme-inhibitor complexes. J. Biol. Chem. 2004, 279, 45990–45997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safari, F.; Katrusiak, A. Structure–Property Relationships of Molecular Shape and Orientation with Com pression and Expansion of Xylitol. Acta Cryst. B 2021, 77, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Woinska, M.; Wanat, M.; Taciak, P.; Pawinski, T.; Minor, W.; Wozniak, K. Energetics of Interactions in the Solid State of 2-Hydroxy-8-X-Quinoline Derivatives (X = Cl, Br, I, S-Ph): Comparison of Hirshfeld Atom, X-Ray Wavefunction and Multipole Refinements. IUCrJ 2019, 6, 868–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derollez, P.; Guinet, Y.; Affouard, F.; Danède, F.; Carpentier, L.; Hédoux, A. Structure Determination of L-Arabinitol by Powder X-Ray Diffraction. Acta Cryst. B 2012, 68, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, L.; Filali Rharrassi, K.; Derollez, P.; Guinet, Y. Crystallization and Polymorphism of L-Arabitol. Thermochim. Acta 2013, 556, 63–67. [Google Scholar] [CrossRef]
- Kopf, J.; Morf, M.; Zimmer, B.; Köll, P. Kristall- und molekülstruktur von d-arabinitol. Carbohydr. Res. 1991, 218, 9–13. [Google Scholar] [CrossRef]
- Hoser, A.A.; Madsen, A.Ø. Dynamic Quantum Crystallography: Lattice-Dynamical Models Refined against Diffraction Data. I. Theory. Acta Cryst. A 2016, 72, 206–214. [Google Scholar] [CrossRef]
- Prince, E.; Spiegelman, C.H. International Tables for Crystallography; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992; Volume C, pp. 622–624. [Google Scholar]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of Intensity Quotients and Differences in Absolute Structure Refinement. Acta Cryst. B 2013, 69, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Capelli, S.C.; Bürgi, H.-B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld Atom Refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. GAUSSIAN09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J. CRYSTAL17. In CRYSTAL17 User’s Manual Torino; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond Lengths in Organic and Metal-Organic Compounds Revisited: X—H Bond Lengths from Neutron Diffraction Data. Acta Cryst. B 2010, 66, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Jarzembska, K.N.; Dominiak, P.M. New version of the theoretical databank of transferable aspherical pseudoatoms, UBDB2011—Towards nucleic acid modelling. Acta Cryst. A 2012, A68, 139–147. [Google Scholar] [CrossRef]
- Volkov, A.; Li, X.; Koritsanszky, T.; Coppens, P. Ab initio quality electrostatic atomic and molecular properties including intermolecular energies from a transferable theoretical pseudoatom databank. J. Phys. Chem. A 2004, 108, 4283–4300. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP Augmented with an Empirical Dispersion Term (B3LYP-D*) as Applied to Molecular Crystals. CrystEngComm 2008, 10, 405–410. [Google Scholar] [CrossRef]
- DiLabio, G.A.; Otero-de-la-Roza, A. Noncovalent Interactions in Density Functional Theory. In Reviews in Computational Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016; pp. 1–97. ISBN 978-1-119-14873-9. [Google Scholar]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How Does Basis Set Superposition Error Change the Potential Surfaces for Hydrogen-Bonded Dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Streek, J.V.D. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Dimers | Gaussian Calculations (kJ/mol) |
|---|---|---|
| Rotamer 1 (1) | H12…O4(−0.5+x, 1.5−y, 1−z) O3…H15(−0.5+x, 1.5−y, 1−z) | −48.37 |
| H14…O2(1−x, 0.5+y, 0.5−z) O1…H13(1−x, 0.5+y, 0.5−z) | −50.56 | |
| H11…O5(−1+x, y, z) | −13.28 | |
| Rotamer 2 (D) | H14…O2(−0.5+x, 0.5−y, 1−z) O3…H11(−0.5+x, 0.5−y, 1−z) | −48.47 |
| H12…O4(1−x, −0.5+y, 0.5−z) O5…H13(1−x, −0.5+y, 0.5−z) | −50.56 | |
| H15…O1(−1+x, y, z) | −13.26 |
| Structure | Dimers | Gaussian Calculations [kJ/mol] |
|---|---|---|
| D-arabitol (3D) | O1A…H1B(x, 1+y, 1+z) | −15.78 |
| H1A…O4B(x, y, 1+z), H4A…O1B(x, y, 1+z) | −61.74 | |
| O3A…H3B | −31.56 | |
| H3A…O2A(1+x, y, z), H5A…O4A(1+x, y, z) | −48.45 | |
| H2A…O5A(x, 1+y, z) | −18.23 | |
| O2B…H5B(−1+x, −1+y, z) | −20.48 | |
| O3B…H2B(1+x, y, z), O5B…H4B(1+x, y, z) | −51.96 | |
| L-arabitol (3L) | O1A…H1B(x, −1+y, −1+z) | −15.90 |
| H1A…O4B(x, y, −1+z), H4A…O1B(x, y, −1+z) | −61.78 | |
| O3A…H3B | −31.46 | |
| H3A…O2A(−1+x, y, z), H5A…O4A(−1+x, y, z) | −48.36 | |
| H2A…O5A(x, −1+y, z) | −18.34 | |
| O2B…H5B(1+x, 1+y, z) | −20.67 | |
| H2B…O3B(−1+x, y, z), H4B…O5B(−1+x, y, z) | −52.03 |
| Crystal Data | 1 | 2 | 3L | 3D |
|---|---|---|---|---|
| Chemical formula | C5H12O5 | |||
| Mr | 152.15 | |||
| Crystal system, space group | Orthorhombic, P212121 | Triclinic, P1 | ||
| Temperature (K) | 122 | 123 | 100 | 100 |
| a, b, c (Å) | 8.2664(1), 8.8978(1), 8.9132(2) | 8.2707(2), 8.9022(2), 8.9217(2) | 4.8000(2), 7.6568(3), 9.6384(4), 95.910(3), 96.094(4), 106.833(4) | 4.8055(2), 7.6526(3), 9.6387(4), 95.908(3), 96.052(4), 106.862(4) |
| V (Å3) | 655.59(2) | 656.88(3) | 333.80(2) | 333.99(3) |
| Z | 4 | 2 | 2 | |
| Radiation type | Cu Kα | |||
| µ (mm−1) | 1.21 | 1.20 | 1.19 | 1.19 |
| Crystal size (mm) | 0.23 × 0.17 × 0.09 | 0.18 × 0.22 × 0.35 | 0.28 × 0.09 × 0.05 | 0.17 × 0.15 × 0.12 |
| Data Collection | ||||
| Diffractometer | SuperNova, Dual, CuKα, and Atlas detector | |||
| Tmin, Tmax | 0.837, 1.000 | 0.383, 1.000 | 0.825, 1.000 | 0.901, 1.000 |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 23,329, 1325, and 1316 | 14,853, 1383, and 1370 | 11,435, 2336, and 2230 | 8367, 2449, and 2385 |
| Rint | 0.026 | 0.046 | 0.027 | 0.027 |
| (sin θ/λ)max (Å−1) | 0.624 | 0.631 | 0.625 | 0.624 |
| Refinement | ||||
| R[F2 > 2σ(F2)], wR(F2), S | 0.022, 0.057, and 1.07 | 0.028, 0.077, and 1.09 | 0.032, 0.084, and 1.05 | 0.028, 0.074, and 1.04 |
| No. of reflections | 1325 | 1383 | 2336 | 2449 |
| No. of parameters | 102 | 99 | 277 | 191 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | |||
| Δρmax, Δρmin (e Å−3) | 0.27, −0.15 | 0.32, −0.21 | 0.33, −0.22 | 0.30, −0.18 |
| Absolute structure | Flack × determined using 525 quotients [(I+) − (I−)]/[(I+) + (I−)] [30] | Flack × determined using 550 quotients [(I+) − (I−)]/[(I+) + (I−)] [30] | Flack × determined using 995 quotients [(I+) − (I−)]/[(I+) + (I−)] [30] | Flack × determined using 1055 quotients [(I+) − (I−)]/[(I+) + (I−)] [30] |
| Absolute structure parameter | 0.01(4) | 0.03(7) | −0.02(13) | 0.07(10) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanat, M.; Malinska, M.; Kucia, M.; Sicinski, R.R.; Woźniak, K. Rotamers in Crystal Structures of Xylitol, D-Arabitol and L-Arabitol. Int. J. Mol. Sci. 2022, 23, 3875. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073875
Wanat M, Malinska M, Kucia M, Sicinski RR, Woźniak K. Rotamers in Crystal Structures of Xylitol, D-Arabitol and L-Arabitol. International Journal of Molecular Sciences. 2022; 23(7):3875. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073875
Chicago/Turabian StyleWanat, Monika, Maura Malinska, Malgorzata Kucia, Rafal R. Sicinski, and Krzysztof Woźniak. 2022. "Rotamers in Crystal Structures of Xylitol, D-Arabitol and L-Arabitol" International Journal of Molecular Sciences 23, no. 7: 3875. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073875