Time Dependent Coupled Cluster Approach to Resonance Raman Excitation Profiles from General Anharmonic Surfaces

School of Chemistry, University of Hyderabad, Hyderabad - 500 046, India
Int. J. Mol. Sci. 2002, 3(5), 447-458; https://0-doi-org.brum.beds.ac.uk/10.3390/i3050447
Submission received: 15 November 2001
/
Accepted: 3 January 2002
/
Published: 31 May 2002
(This article belongs to the Special Issue Recent Advances in Coupled Cluster Theory)
{kind=link}
{kind=link}
{kind=link}
Abstract
:A time dependent coupled cluster approach to the calculation of Resonance Raman excitation profiles on general anharmonic surfaces is presented. The vibrational wave functions on the ground electronic surface are obtained by the coupled cluster method (CCM). It is shown that the propagation of the vibrational ground state on the upper surface is equivalent to propagation of the vacuum state by an effective hamiltonian generated by the similarity transformation of the vibrational hamiltonian of that surface by the CCM wave operator of the lower surface up to a normalization constant. This time propagation is carried out by the time-dependent coupled cluster method in a time dependent frame. Numerical studies are presented to asses the validity of the approach.
Introduction
In this paper, we develop a coupled cluster method (CCM) based approach to the calculation of resonance Raman excitation profiles for polyatomic molecues with general anharmonic potential surfaces. Calculations on molecules with several degrees of freedom, with arbitrary displacements, Duischinsky rotation and higher order terms in the potential become possible by this method.
The standard method for the calculation of Raman excitation profiles invokes the Kramers-Heisenberg-Dirac sum over states expression for the polarizability tensor [1]. Such summation-over-states form becomes rapidly prohibitive with increasing size of the system because the basis set required to converge the eigenfunctions increases exponentially with the number of degrees of freedom in the system. Consequently the vibrational eigenstates are extremely difficult to calculate when anharmonicities are present. Even in the harmonic limit, the Franck-Condon overlaps have to be calculated, a formidable task in itself when large displacements and Duchinsky rotations are present in the excited surface. These difficulties have prompted several authors to develop alternative formulations to the calculation of the polarizability tensor [2,3,4,5,6,7,8]. All these approaches are based on the time-dependent picture of Raman scattering, and evaluate the polarizability tensor as the half-Fourier transformation of the Raman kernel. For harmonic surfaces (even in the presence of coordinate rotation in the excited surface), these approaches lead to closed set of equations. Tanner and Heller [3] have presented a detailed study of this situation, and discussed the advantages of the time dependent approach over the summation of states method. The doorway state in the time-dependent approach (being the vibrational ground surface) is a gaussian in this case. The advantages of the time-dependent approach stem from the fact, that the time evolved wave packet remains a gaussian for all times when propagated over harmonic surfaces when the initial state is a gaussian, and thus can be parameterized and propagated efficiently by the Gaussian wave packet propagation method [9]. The situation becomes complicated in the presence of anharomonicities on either surface. Neither is the doorway state strictly a gaussian in such a situation, nor does it follow the classical trajectory during its evolution. It becomes necessary then to either expand the wave packet in a suitable basis [8] or use some other algorithm to evolve the wave packet [6]. The computational effort once again scales exponentially with the number of degrees of freedom. The coupled cluster method [10,11,12,13,14] has come into prominence in recent years as a method of choice for describing many-body dynamics both in terms of the accuracy it can provide, and in the economy of effort. In addition, it has the desirable property of being size consistent. While most of the applications to date have been to the electronic structure problem, a few attempts have been made to apply it to the molecular vibrational problems as well [15,16]. The time dependent version of the coupled cluster method has been applied to the calculation of Franck-Condon spectra on harmonic [17] and coupled surfaces [18] with considerable success. In the present work we develop the CCM approach to the calculation of resonance Raman excitation profiles from general anharmonic surfaces. Section II presents the basic steps in the approach. Representative numerical studies are presented in section III along with a discussion on the advantages of the present approach.
Theory
Preliminary remarks:
The expression for the resonance Raman excitation profile in the time dependent picture is given by the half-Fourier integral of the Raman kernel [2]
Here, ωL is the incident radiation frequency, φf, φi are the final and initial vibrational states of the molecule, and He is the vibrational hamiltonian on the upper surface. The scattered light frequency is determined by the conservation of energy. Condon approximation is invoked in deriving Eq.1, and the counter resonant term for the polarizability tensor has been dropped. Thus the calculation of the resonance Raman excitation profiles by the time dependent approach consists of three parts: (a) Calculation of the vibrational eigenstates φf and φi ; (b) Propagation of φi on the upper surface and evaluation of its overlap with φf ; (c) Evaluation of the half-Fourier integral and the intensity. We now address these issues.
Ifi (ωL) = | ∫o∞ <φf | exp (-iHet) | φi >. exp (itωL) dt | 2
Vibrational states of the ground surface:
Ignoring the Coriolis interaction, the vibrational hamiltonian of the ground surface for a molecule of N vibrational modes can be written as
Here, qi are the mass weighted normal coordinates, and pi are the associated momenta. We assume that the potential energy Vg can be approximated by a quartic polynomial in the normal coordinates.
Hg = Σ pi2/2 + Vg
Vg (q) = Σ fgii qi2 + Σfgijk qiqjqk + Σ fgijkl qiqjqkql
The coupled cluster approach to molecular vibrational energy levels consists of three steps [15]. In the first step a reference gaussian function is constructed variationally for the ground state:
The harmonic oscillator ladder operators ai and ai+ are now defined with respect to this optimized vacuum state.
and the hamiltonian is expanded in terms of these operators. By definition, the vacuum state of eq.(4) satisfies
| 0 > = Ng.exp [Σ ωi (qi - qio)2 ].
ai = √ωi/2 (qi - qio + d/dωiqi),
ai+ = √ωi/2 (qi - qio - d/dωiqi),
ai | 0 > = 0 .
In the second step, the correlated vibrational ground state is posited as
The cluster operators Sg and Dg are expanded as
Note that in the traditional coupled cluster approach, the deexcitation operators Dg are not included. However we prefer to include them for providing stability to the dynamical calculations in the second step. From the Schrödinger equation for the ground state
one obtains in the usual fashion
where,
Eq.(9b) is an infinite set of coupled non-linear equations which on solution yield the cluster matrix elements {si}and {di} of eq (8). The exact vibrational ground state energy Eo is obtained directly from eq (9a) once si are known.
| Ψo > = Ug | 0 > ,
Ug = exp (Sg).exp (Dg)
Sg = Σsi ai+ + Σ sij ai+aj+ + Σ sijk ai+aj+ak + + Σ sijkl ai+aj+ak+al+ + ......
Dg = Σdi ai + Σ dij aiaj + Σ dijk aiajak + Σ dijkl aiajakal + ......
H |Ψo> = Eo |Ψo> ,
0 > = Eo ,
< n1n2....| Heff | 0 > = 0
< 0| Heff | n1n2.... > = 0
Heff = Ug-1.Hg.Ug
In the final step, the excited states and their energies are obtained by using what is variously termed as the equations of motion coupled cluster theory or the coupled cluster linear response theory [21]. In this approach, one formally writes the excited states as the result of the action of an excitation operator on the exact ground state. The excitation operator is expanded in a complete set of operators. The coefficients in the linear expansion and the transition energies are then obtained by invoking the equation of motion for the excitation operator. In practice, this involves diagonalizing Heff in the nb (= Σni) ≠ 0 boson state manifold [15]. Since Heff is just a similarity transform of the original hamiltonian (via eq.(10)), its eigenvalues are identical to those of the original hamiltonian. Note that the general response function (or the excitation operator) consists of both excitation and deexcitation operators. However the equations of motion for the two sets are decoupled by virtue of eq.9, yielding positive and negative excitation energies respectively.
We next turn to the calculation of matrix element of the time evolution operator exp(-itHe) between the two states φf and φi. While the eigenvalues of Hg and Heff are identical, their eigenvectors are not. However, they are related through the same similarity transformation used to construct the effective hamiltonian Heff. [19] Since Heff is not manifestly hermitian, its right and left eigenvectors
are not identical. They are related to the eigenvectors of the original hamiltonian via the relations
up to some normalization constants Nr and Nl. Consequently, the matrix element of any arbitrary operator X is given by [19]
Thus the Raman kernel of eq.(1) is given by
upto an (irrelevant) scaling constant N = Nri .Nlf. Here,
is the similarity transformed upper surface hamiltonian. Thus the time propagation of the eigenstate of the lower surface on the surface reduces to the propagation of the right eigenstate of the transformed hamiltonian Heff on the upper surface but with the modified effective hamiltonian Heffe.
Heff | Ri > = Ei | Ri >
< Li | Heff = Ei < Li |
| φi > = Ug | Ri >. Nri ,
<φi | = Nli .< Li | Ug-1 ,
< φi | X | φj > = < Li | Xeff | Rj >. Nrj .Nli ,
Xeff = Ug-1 . X. Ug .
<φf | exp (-iHet) | φi > = <Lf | exp (-i.Heffe.t) | Ri >.N,
Heffe = Ug-1.He.Ug ,
Time propagation :
We now turn to the propagation of the doorway state φi. As a consequence of the coupled cluster equations (9), the right eigenvector corresponding to the vibrational ground state is the vacuum state itself.
Thus we need to propagate only the vacuum state, though by the effective hamiltonian (15). This is the major gain by using the effective hamiltonian route. The time-dependent coupled cluster approach (TDCCM) [20] provides a particularly convenient ansatz for the propagation of such states in the form of
where the time dependent cluster operator Se contains only excitation operators.
Here s0 is a time-dependent scalar that contains the normalization and the overall phase information of the wave packet. Substitution of ansatz (17) into the time dependent Schrödinger equation
provides the working equations for the cluster amplitudes. It has been noted in earlier studies [18] that the working equations by the TDCCM approach often become singular for long propagation times. One reason for this is due to the loss of the hermiticity of the hamiltonian implicit in the approximation calculations. Moreover, in the present context, the similarity transformed hamiltonian Heffe itself is manifestly non-hermitian. When it is approximated, it may develop complex eigenvalues [22]. Such complex eigenvalues could also lead to norm violations. It is to reduce the effect of such hermiticity violating terms in the effective hamiltonian, we use the double exponential ansatz 7. Second, the overlap between the exact wave packet and reference state becomes progressively smaller as the propagation time increases. As a consequence, the cluster amplitudes (which are linearly related to the ratioes of the coefficients of the appropriate states to the coefficient of the reference state) increase, and in some pathological cases even become infinite. This renders the working equations to be stiff and nonintegrable. As a consequence, the simple ansatz (17) is unlikely to provide reliable results. To avoid this potential pitfall, we define the TDCCM evolution operator with respect to a time-dependent vacuum state | 0t > such that | 0t > has significantly large overlap with the exact wave packet throughout the period of time evolution that we are interested in. The two vacua are related through the auxiliary operator U0
The ladder operators at associated with | 0t > are related to the original set via the transformation
We now posit the TDCCM ansatz with respect to the time-dependent vacuum | 0t > .
as before. The cluster operator Set is defined as in eq(18), the only difference being the time-dependent operators at+ would appear in the expansion. Combining eq (20-22), it can be shown the final form of the time evolved wave packet in this time dependent reference frame is
where U0 and U have the meaning as defined in eq (20) and (17) respectively. Since the primary motion of the wave packet in the initial stages of evolution is to slide down the potential surface, we chose the following ansatz for U0:
Note that we have chosen U0 to be unitary. Thus this transformation would preserve the norm of the wave packet. Second, it involves the annihilation operators as well which provide a degree of balance to the working equations. Third, since the one boson operators are included in the definition of U0, these are not included in the definition of U in eq (18). The working equations for the amplitudes ti and si are obtained by substituting the ansatz (23) into the Schrödinger equation (19), premultiplying by (U0U)-1 , and projecting on to the various states of the Fock space [20]. We present the some numerical studies in the next section based on this formalism.
|Ri> = |0> .
| t > = U |0>,
U = exp (Se) ,
Se = s0e + Σsie ai+ + Σ sije ai+aj+ + Σ sijke ai+aj+ak + + Σ sijkle ai+aj+ak+al+ + ......
i ∂ | t >/∂t = Heffe | t >
| 0t > = U0(t) | 0 > .
at = U0(t).a. U0(t)-1 .
| t > = Ut | 0t >,
Ut = exp (Set),
| t > = U0.U | 0 >,
U0 = exp (T),
T = Σ (ti ai+ - ti* ai ) .
Results and discussion
We have applied the methodology developed in the previous section to the calculation of the resonance Raman excitation profiles on a model 1-d system. The potential surfaces characterising the system are given by
Vg = 0.5q2 - 0.021q3 + 0.00096q4
Ve = 2.3q + 0.66q2 - 0.026q3 + 0.0008q4
Based on our earlier studies [15,16] we have restricted the cluster operator Sg, Dg and Se to contain at most four creation operators through out these calculations. The non-linear equations of eq.(9) were solved by quasi-linearization. The expansion for the excited states was also limited such that states containing more than four bosons were not included in it. In addition, all terms in Heff that contain more than four ladder operators were neglected. The dynamical equations were integrated by fourth order Runge-Kutta method.
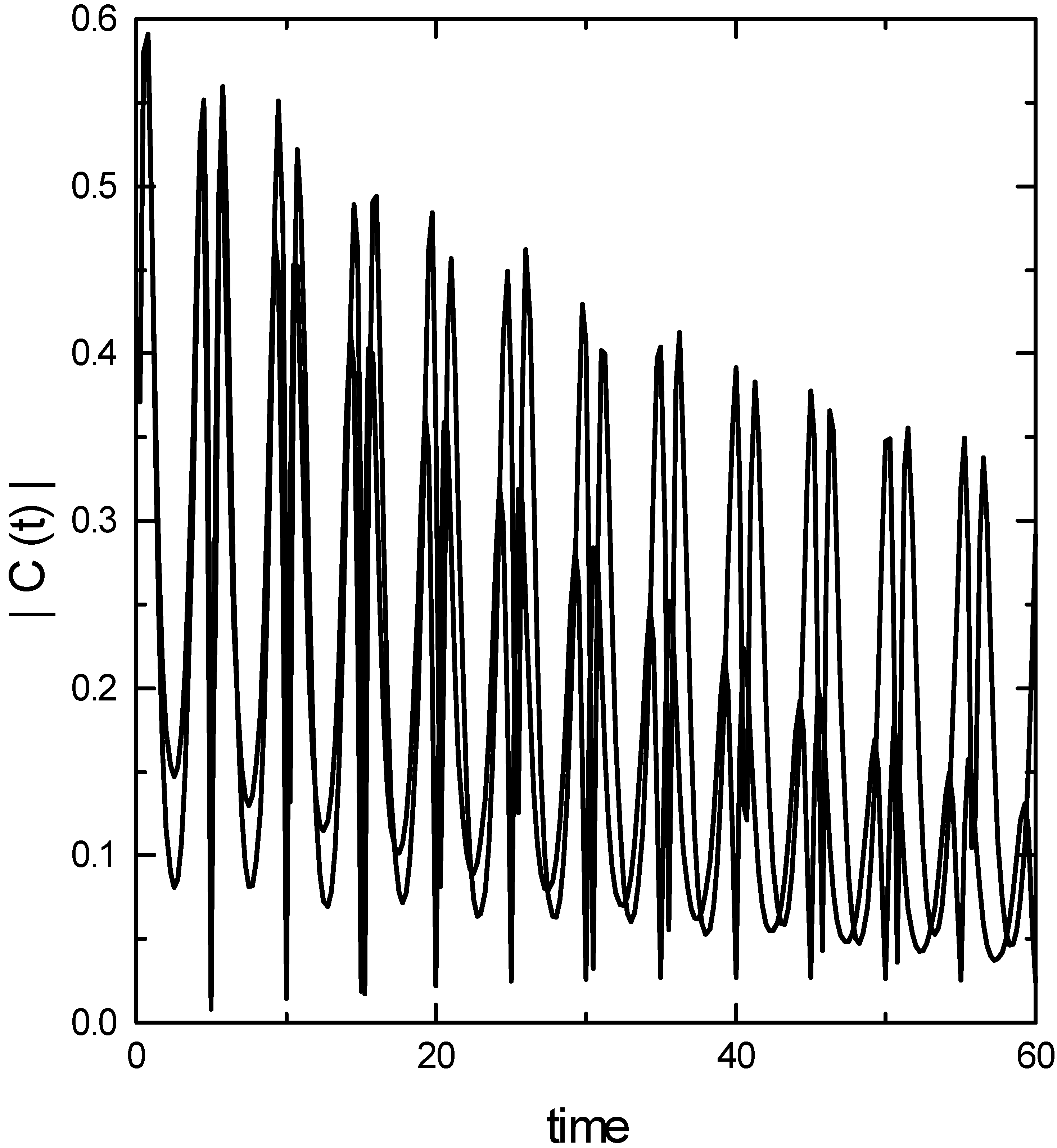
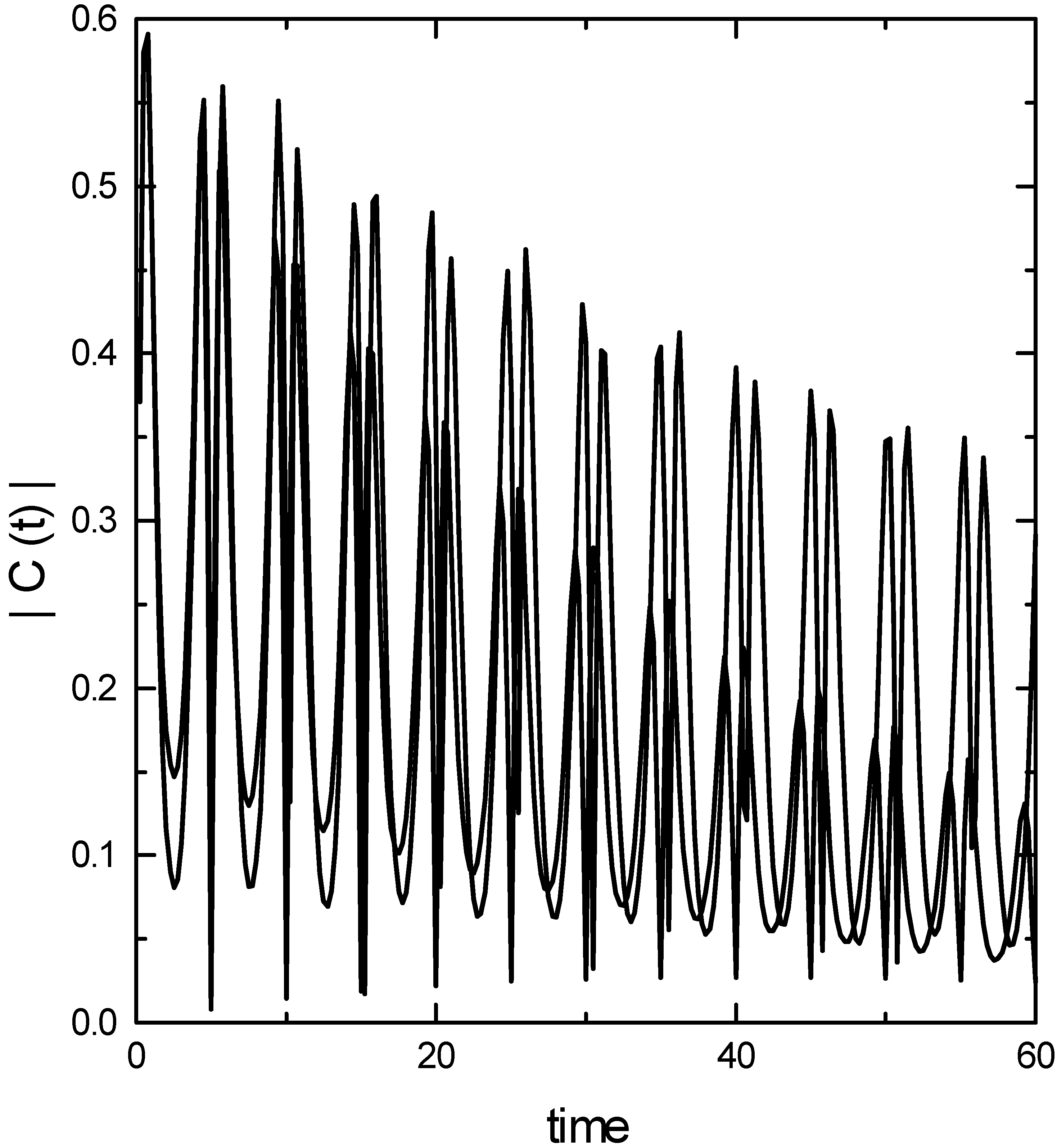
The modulus of Raman kernel for the fundamental transition is presented in Fig. 1. Two features of the correlation function are noteworthy. First, it is oscillatory representing the return of the wave packet to the Franck-Condon zone. Second, on a slower time scale, the correlation function decays. This is due to the dephasing of the wave packet by the anharmonicity of the potential surface. The
![Ijms 03 00447 g001]() coupled cluster method is able to reproduce the oscillatory nature of the correlation function correctly. However, it underestimates the dephasing part, particularly at longer times. This would imply that the spectral line widths would be underestimated by the TDCCM approach.
coupled cluster method is able to reproduce the oscillatory nature of the correlation function correctly. However, it underestimates the dephasing part, particularly at longer times. This would imply that the spectral line widths would be underestimated by the TDCCM approach.
Fig.1.
Modulus of the Raman kernel for the fundamental transition. (Continous line – converged basis set calculation; dash-dotted line - coupled cluster calculation.)
Fig.1.
Modulus of the Raman kernel for the fundamental transition. (Continous line – converged basis set calculation; dash-dotted line - coupled cluster calculation.)
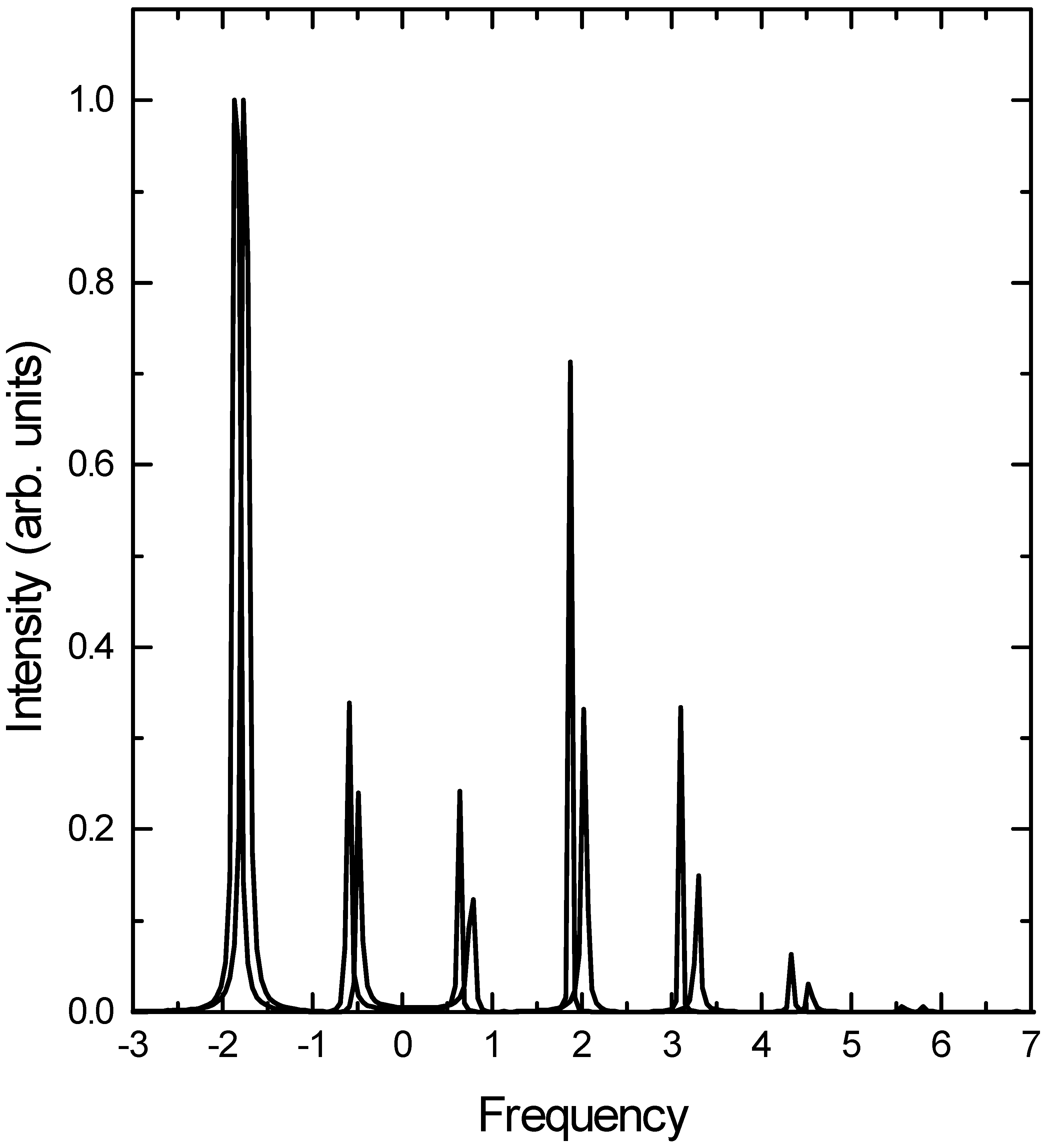
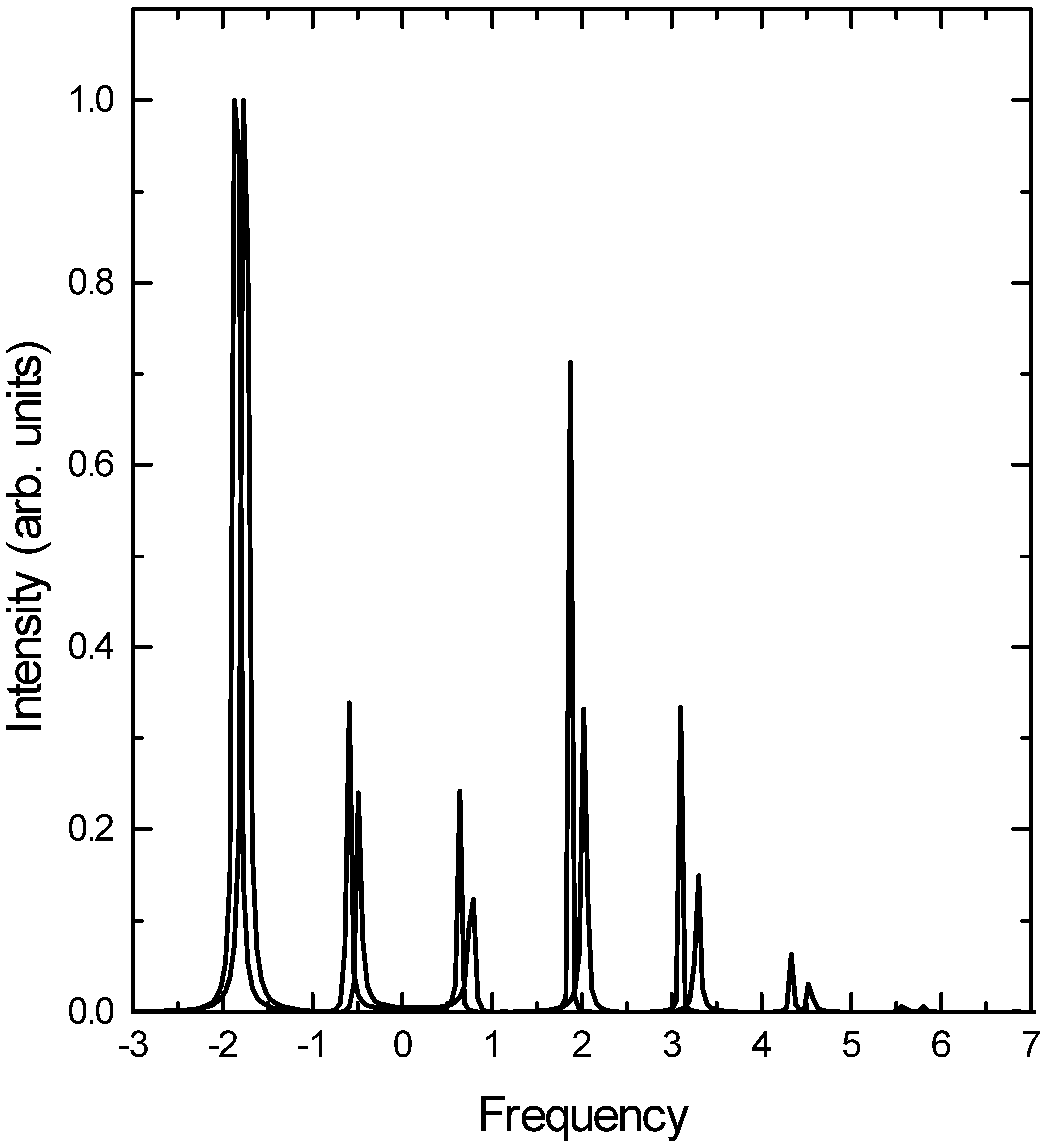
The Raman excitation profiles for the fundamental and the first overtone are presented in Fig. 2 and Fig. 3 respectively. The overall agreement between the coupled cluster calculation and the converged basis set calculation is good. In particular, the bimodal distribution of intensity is reproduced quite well by the TDCCM approach in both cases. Among the discrepancies, two features are worth noting. First, the TDCCM approach overestimates the intensity of the higher overtones. This is particularly noticeable in Fig. 3, where the intensities of the v = 0 and v = 1 transitions are underestimated, while the intensities of the rest of the transitions are overestimated. It appears that the TDCCM approach overestimates the role of the higher excited states during the propagation at the cost of the lower energy
![Ijms 03 00447 g002]() states. Second, the TDCCM calculation underestimates the energies of the higher overtones. This is understandable in view of the approximations that we have made. Since we restricted the Se operator to contain no more than four boson operators, the implicit effective hamiltonian does not have terms beyond n2 in terms of the quantum number n of the states concerned. Since the overall hamiltonian is quartic, the energy level spacing increases beyond a threshold, and the present approximation is unable to describe it correctly.
states. Second, the TDCCM calculation underestimates the energies of the higher overtones. This is understandable in view of the approximations that we have made. Since we restricted the Se operator to contain no more than four boson operators, the implicit effective hamiltonian does not have terms beyond n2 in terms of the quantum number n of the states concerned. Since the overall hamiltonian is quartic, the energy level spacing increases beyond a threshold, and the present approximation is unable to describe it correctly.
Fig.2.
Raman excitation profile for the fundamental transition. (Figure conventions are the same as above.)
Fig.2.
Raman excitation profile for the fundamental transition. (Figure conventions are the same as above.)
To summarize, we have presented a method to calculate the resonance Raman excitation profiles from anharmonic potential surfaces by the time dependent coupled cluster method. In this approach, the vibrational states of the lower surface are obtained by the stationary state coupled cluster method. It is shown that the propagation of the vibrational ground state on the upper surface can be replaced with propagation of the vacuum state by an effective hamiltonian. This can be done efficiently by the closed shell version of the TDCCM approach. The advantages of the TDCCM approach appear at two
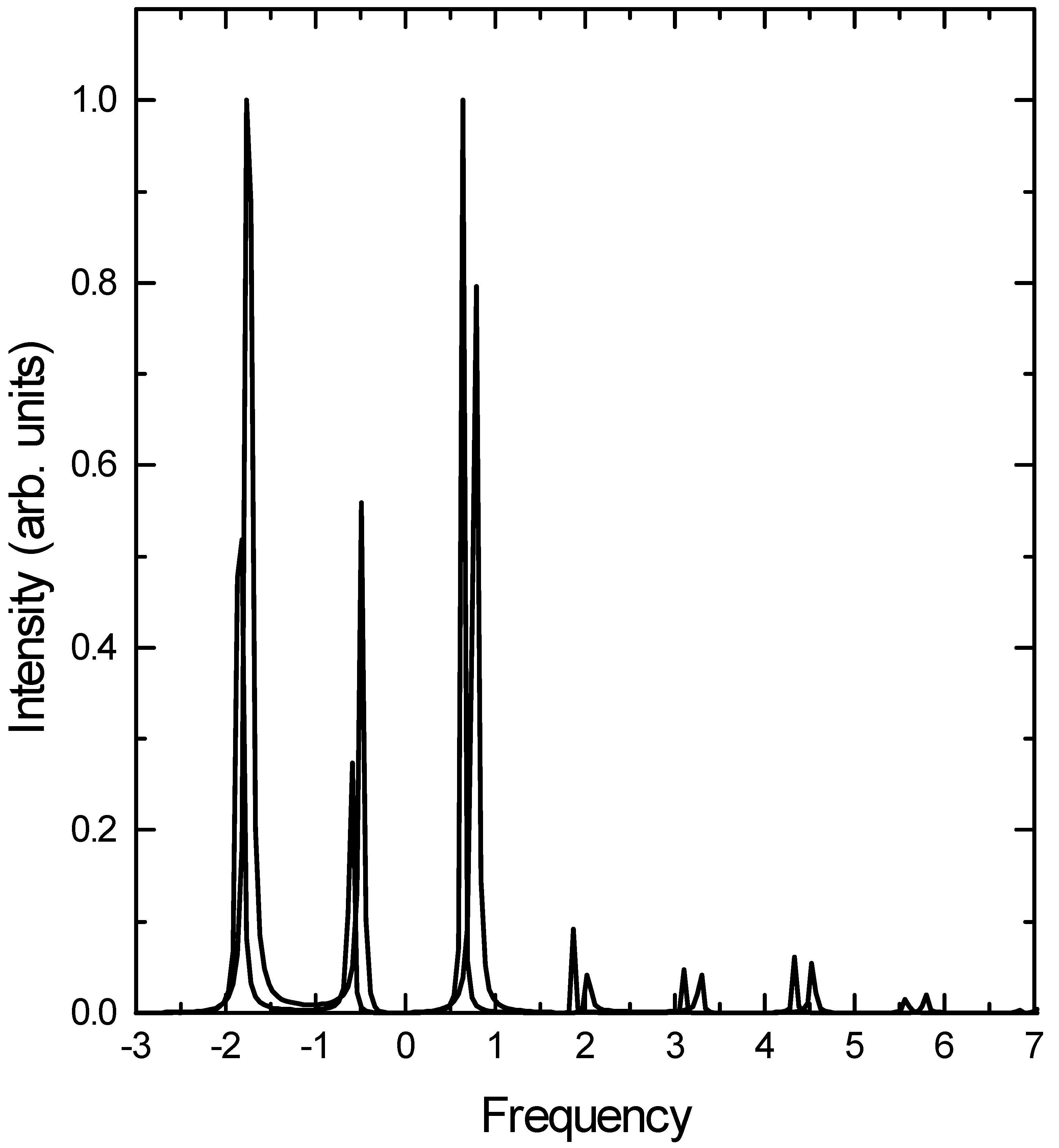
![Ijms 03 00447 g003]() levels. At the formal level, the TDCCM approach is size consistent. Size consistency in the present context implies not only the additive separability of energy, but also the multiplicative separability of the intensities of overtone and combination bands in the non-interacting limit. In addition, in the context of molecular vibrations, the basic motif of energy levels is the harmonic oscillator. Hence the energy levels are proportional to the number of quanta present in the system to the leading order even for a single mode. Finite basis expansions do not satisfy this requirement even for a simple displaced harmonic oscillator. The CCM ansatz avoids this problem due to its exponential structure.
levels. At the formal level, the TDCCM approach is size consistent. Size consistency in the present context implies not only the additive separability of energy, but also the multiplicative separability of the intensities of overtone and combination bands in the non-interacting limit. In addition, in the context of molecular vibrations, the basic motif of energy levels is the harmonic oscillator. Hence the energy levels are proportional to the number of quanta present in the system to the leading order even for a single mode. Finite basis expansions do not satisfy this requirement even for a simple displaced harmonic oscillator. The CCM ansatz avoids this problem due to its exponential structure.
Fig.3.
Raman excitation profile for the first overtone transition. (Figure conventions are the same as above.)
Fig.3.
Raman excitation profile for the first overtone transition. (Figure conventions are the same as above.)

The second advantage of the TDCCM approach is from the computational perspective. Note that the harmonic ladder operators of eq.5 are defined for each mode. Consequently, the number of cluster matrix elements scales in terms of the number of modes N as against the number of basis functions bN in a basis set calculation. Obviously, this is a great saving from the computational point of view, particularly for large systems.
Acknowledgements
Financial support from DST is gratefully acknowledged.
References
- Kramers, H.A.; Heisenberg, W. Z. Phys. 1925, 31, 681.Dirac, P. Proc. R. Soc., London 1927, 114, 710. [CrossRef]
- Lee, S.-Y.; Heller, E.J. J. Chem. Phys. 1979, 71, 4777.
- Tanner, D. J.; Heller, E.J. J. Chem. Phys. 1985, 82, 1982.
- Heller, E.J. Acc. Chem. Res. 1981, 14, 368. [CrossRef]
- Friesner, R.; Pettitt, M.; Jean, J.M. J. Chem. Phys. 1985, 82, 2918. [CrossRef]
- Coalson, R.D.; Kinsey, J.L. J. Chem. Phys. 1986, 85, 4322.
- Page, B.; Tonks, D.L. J. Chem. Phys. 1988, 88, 738.
- Gerdts, T.; Manthe, U. J. Chem. Phys. 1997, 107, 6584.
- Heller, E.J. J. Chem. Phys. 1978, 68, 2066.
- Cizek, J. Adv. Chem. Phys. 1969, 14, 35.
- Bartlett, R.J. Ann. Rev. Phys. Chem 1981, 32, 359.
- Mukherjee, D.; Pal, S. Adv. Quantum Chem. 1988, 20, 291.
- Lindgren, I.; Mukherjee, D. Phys. Rept. 1987, 151, 93. [CrossRef]
- Bartlett, R.J. J. Phys. Chem., 93 1989, 93, 1697. [CrossRef]
- Nagalakshmi, V.; Lakshminarayana, V.; Sumitra, G.; Durga Prasad, M. Chem. Phys. Lett. 1994, 217, 279.
- Durga Prasad, M. Ind. J. Chem 2000, 39A, 196.
- Patwari, G.N.; Wagegaonkar, S.; Durga Prasad, M. Chem. Phys. Lett. 2001, 344, 229.
- Sreelatha, G.; Durga Prasad, M. J. Chem. Phys. 1996, 105, 2972.
- Durga Prasad, M. Theor. Chim. Acta 1994, 88, 383. [CrossRef]
- Durga Prasad, M. J. Chem. Phys. 1988, 88, 7005.
- Monkhorst, H. Int. J. Quantum Chem. 1977, S11, 421.Mukherjee, D.; Mukherjee, P.K. Chem. Phys. 1979, 37, 325.Emrich, K. Nucl. Phys. A. 1981, 351, 379.Koch, H.; Jorgenson, P. J. Chem. Phys 1990, 93, 3333. [CrossRef]Stanton, J.F.; Bartlett, R.J. J. Chem. Phys. 1993, 98, 7029.
- Sree Lath, G.; Durga Prasad, M. Theor. Chim. Acta. 1993, 86, 511. [CrossRef]
© 2002 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Prasad, M.D. Time Dependent Coupled Cluster Approach to Resonance Raman Excitation Profiles from General Anharmonic Surfaces. Int. J. Mol. Sci. 2002, 3, 447-458. https://0-doi-org.brum.beds.ac.uk/10.3390/i3050447
AMA Style
Prasad MD. Time Dependent Coupled Cluster Approach to Resonance Raman Excitation Profiles from General Anharmonic Surfaces. International Journal of Molecular Sciences. 2002; 3(5):447-458. https://0-doi-org.brum.beds.ac.uk/10.3390/i3050447
Chicago/Turabian StylePrasad, M. Durga. 2002. "Time Dependent Coupled Cluster Approach to Resonance Raman Excitation Profiles from General Anharmonic Surfaces" International Journal of Molecular Sciences 3, no. 5: 447-458. https://0-doi-org.brum.beds.ac.uk/10.3390/i3050447