Quantum Chemical and FTIR Spectroscopic Studies on the Linkage Isomerism of Carbon Monoxide in Alkali-Metal-Exchanged Zeolites: A Review of Current Research

Abstract
:1. Introduction
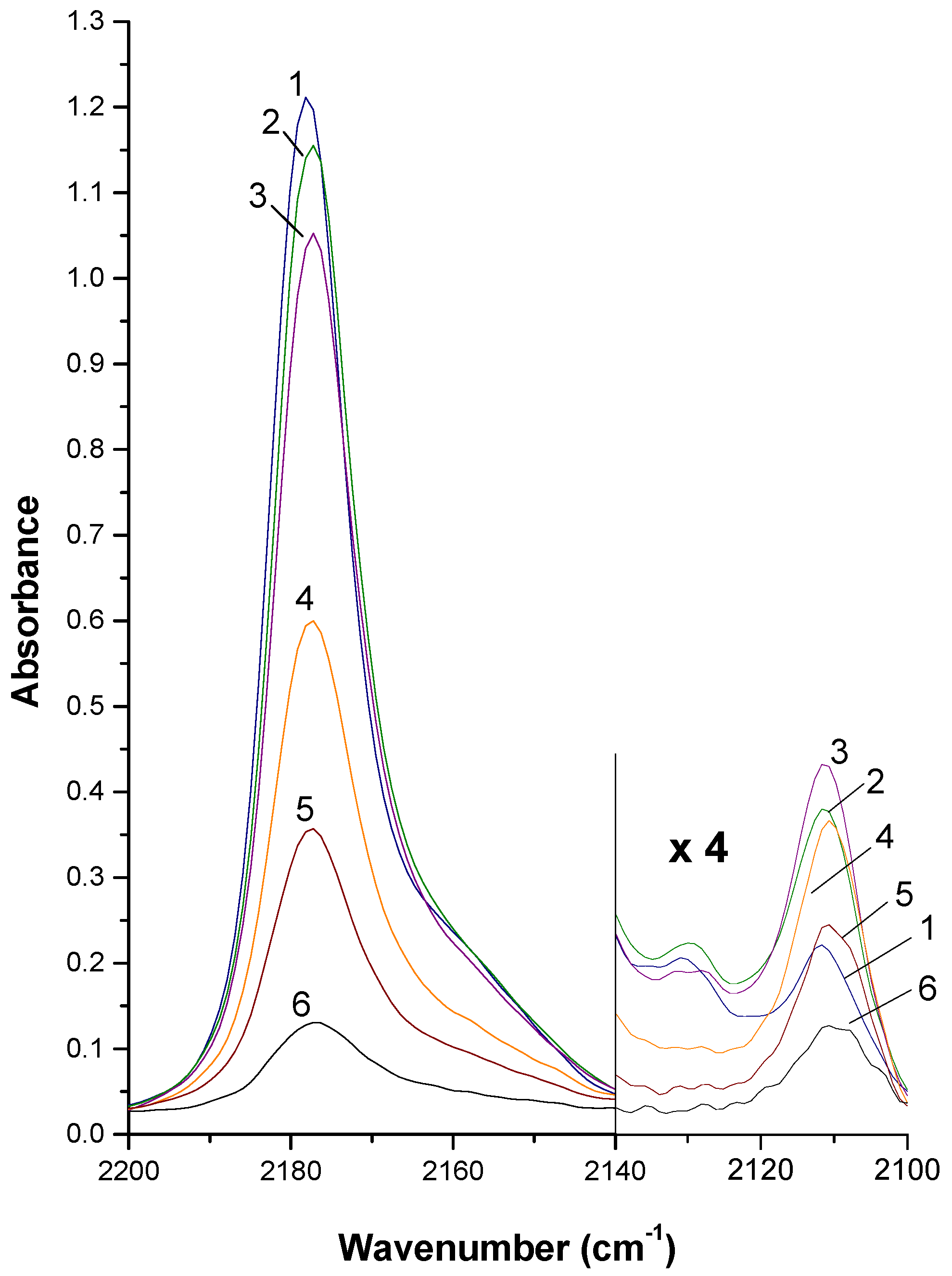
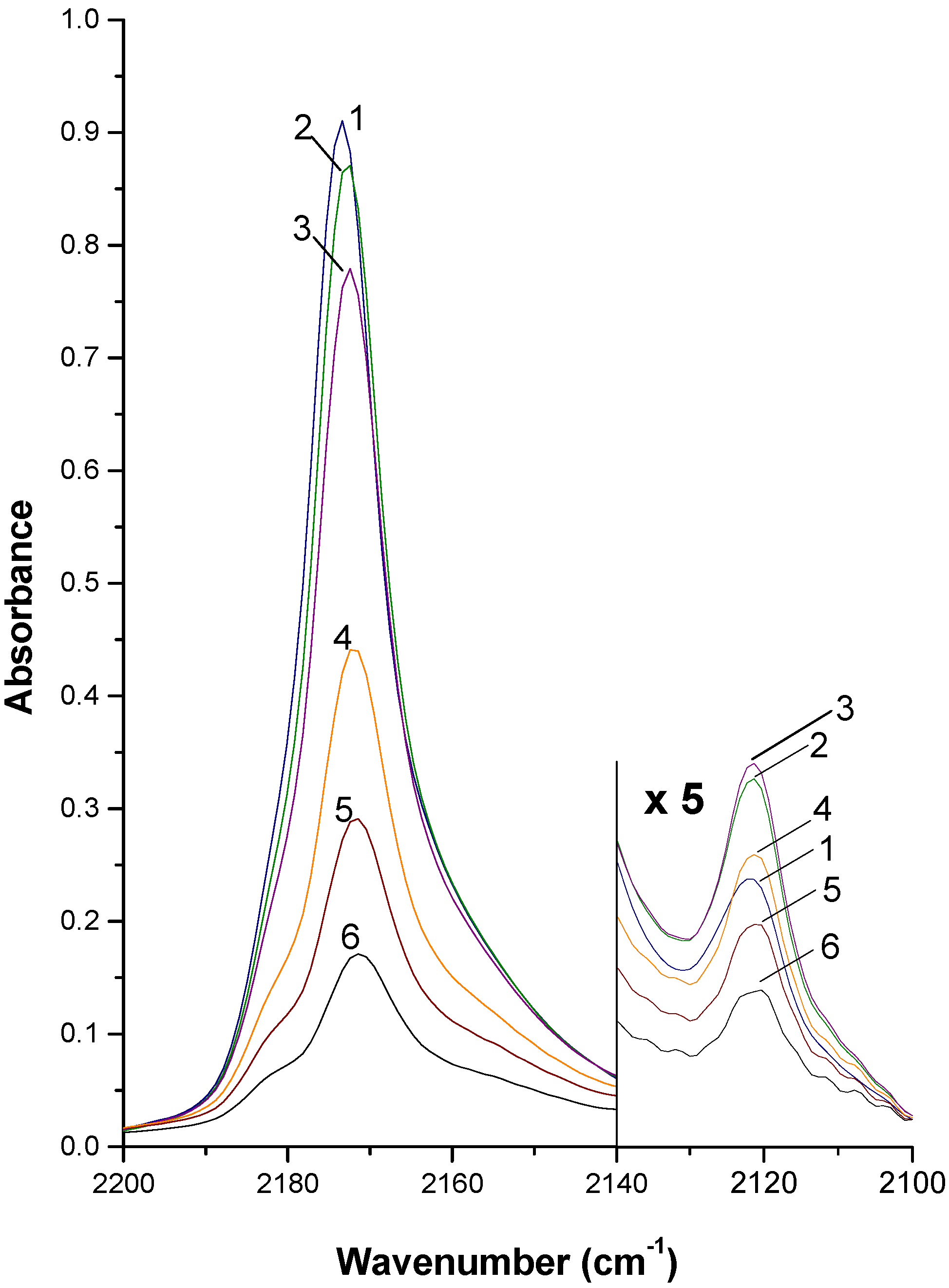
2. Summary of experimental procedures and results

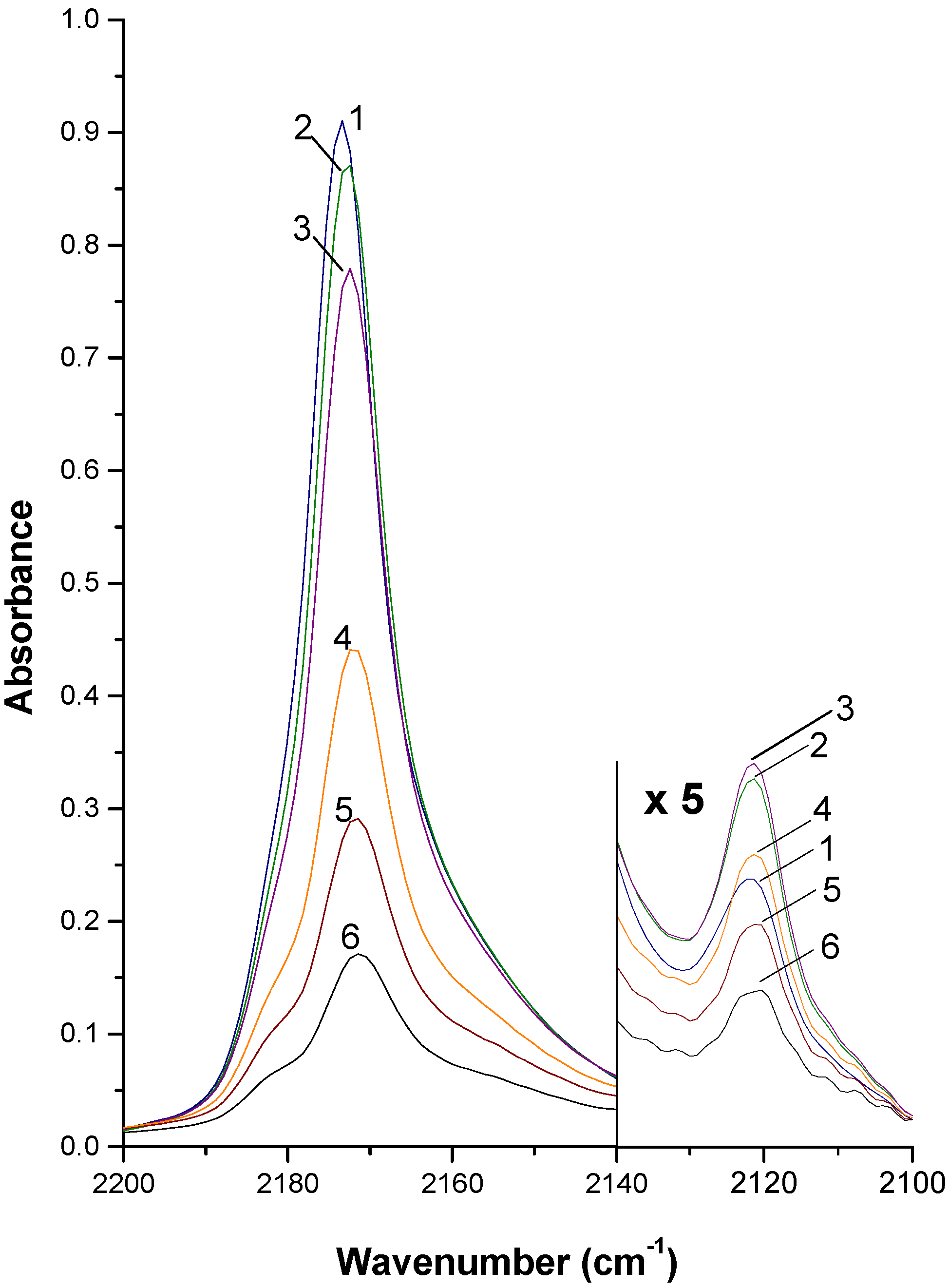
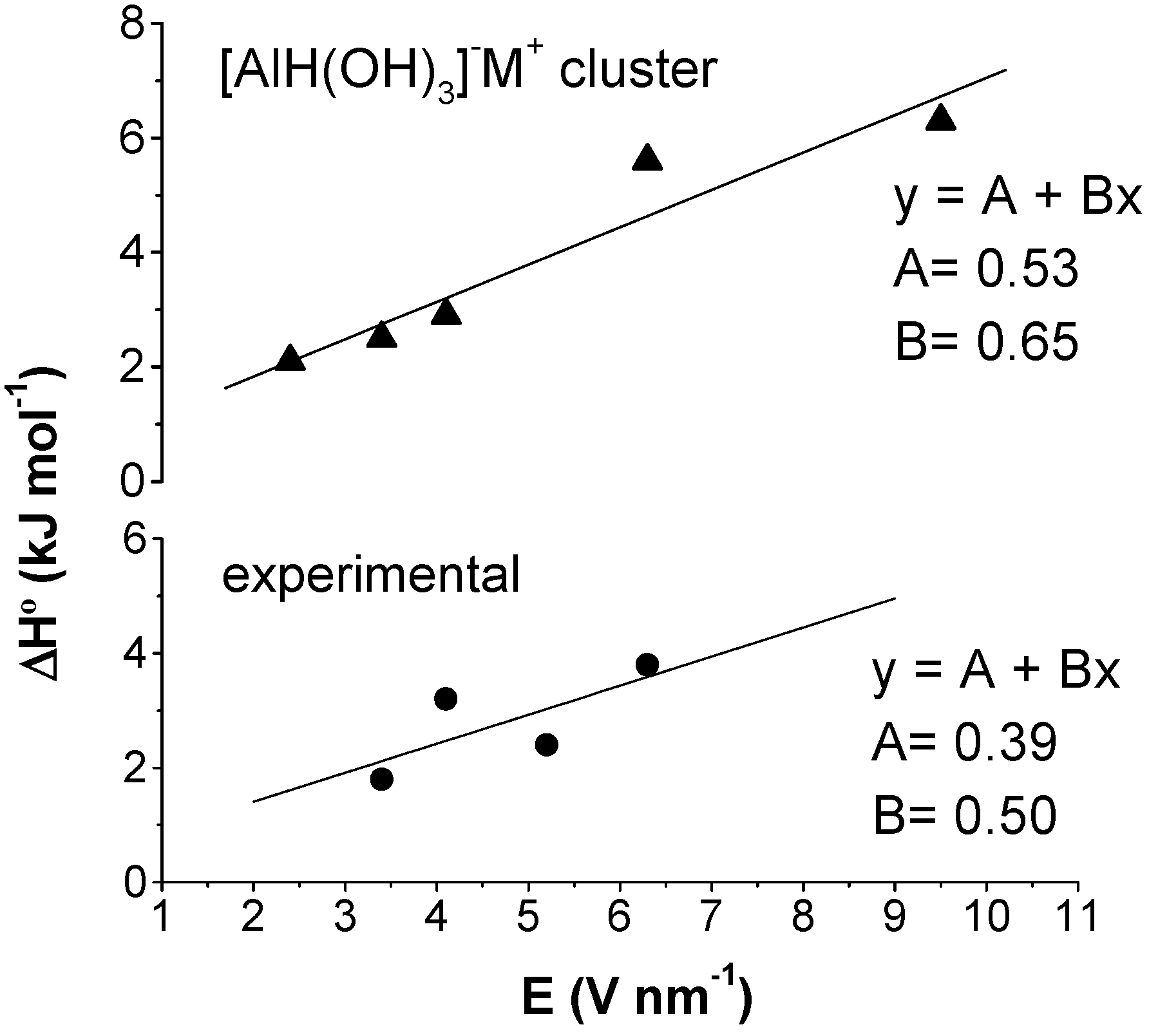
3. Outline of Quantum Chemical Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | ν(HF), cm-1 | ν(LF), cm-1 | ΔH°, kJ mol-1 |
| CO/Na-ZSM-5 | 2178 | 2112 | 3.8 |
| CO/K-ZSM-5 | 2166 | 2117 | 3.2 |
| CO/Rb-ZSM-5 | 2161 | 2120 | 1.8 |
| CO/Na-Y | 2171 | 2122 | 2.4 |
| M+ | ΔH°/bare cation | ΔH°/[AlH(OH)3]-M+ | ΔH°/experimental |
|---|---|---|---|
| Li+ | 16.3 | 6.3 | - |
| Na+ | 12.2 | 5.6 | 3.8 |
| K+ | 6.3 | 2.9 | 3.2 |
| Rb+ | 5.4 | 2.5 | 1.8 |
| Cs+ | 4.6 | 2.1 | - |
4. Discussion
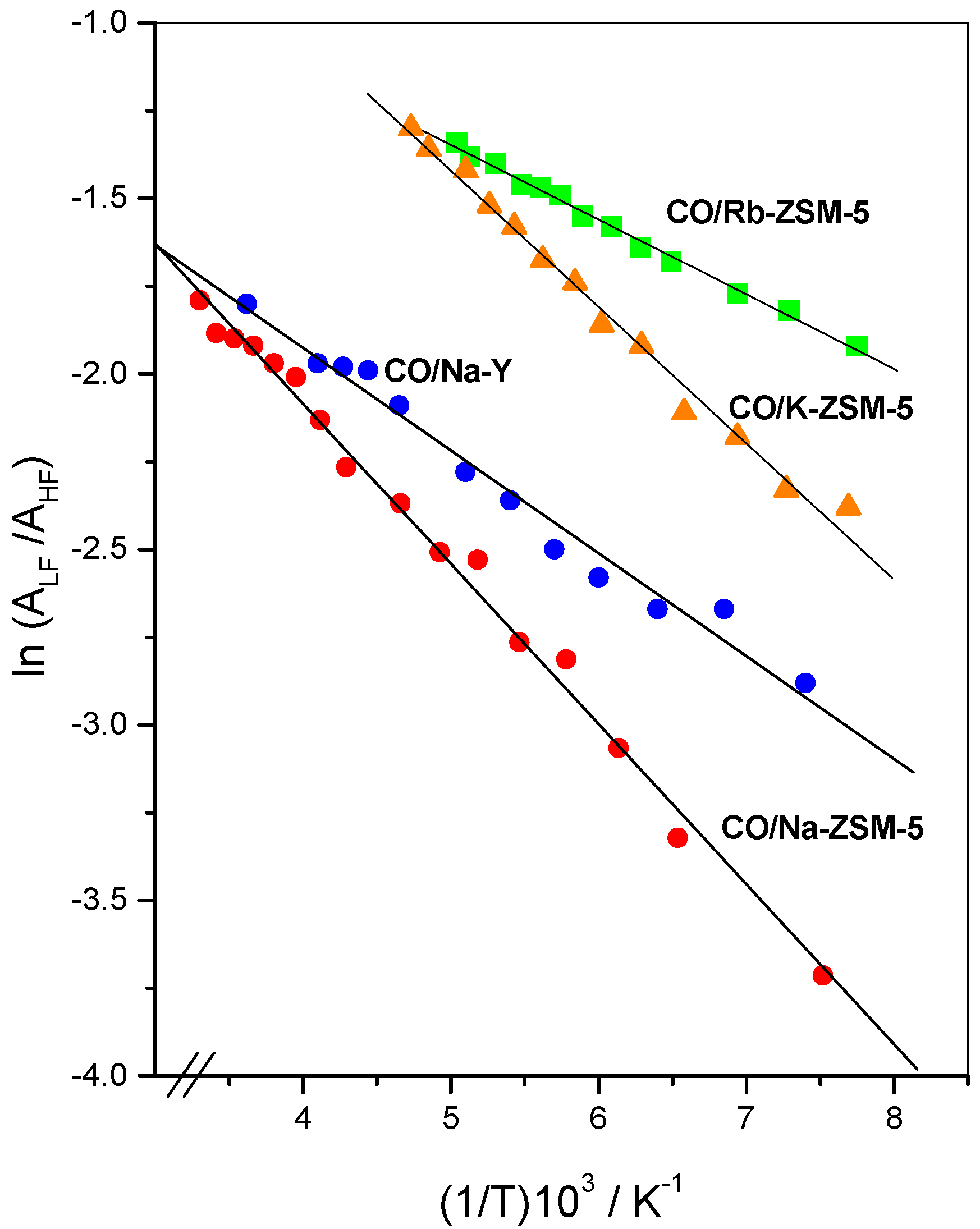
4.1 Thermal isomerization equilibrium
4.2 Isomerization enthalpy as a function of electric field

5. Conclusions and outlook
References
- Cohen de Lara, E.; Nguyen Tan, T. Potential energy of a molecule adsorbed in synthetic zeolites. Application to the analysis of infrared spectra. 1. Electrostatic field in the zeolites NaA and CaA. J. Phys. Chem. 1976, 80, 1917–1921. [Google Scholar] [CrossRef]
- Otero Areán, C. Zeolites and intrazeolite chemistry: insights from infrared spectroscopy. Comments Inorg. Chem. 2000, 22, 241–273. [Google Scholar] [CrossRef]
- Makarova, M. A.; Al-Ghefaili, K. M.; Dwyer, J. Brønsted acid strength in US-Y: FTIR study of CO adsorption. J. Chem. Soc. Faraday Trans. 1994, 90, 383–386. [Google Scholar] [CrossRef]
- Gruver, V.; Fripiat, J. J. Lewis acid sites and surface aluminum in aluminas and mordenites: an infrared study of CO chemisorption. J. Phys. Chem. 1994, 98, 8549–8554. [Google Scholar] [CrossRef]
- Bordiga, S.; Garrone, E.; Lamberti, C.; Zecchina, A.; Otero Areán, C.; Kazansky, V. B.; Kustov, L. M. Comparative IR spectroscopic study of low temperature H2 and CO adsorption on Na zeolites. J. Chem. Soc. Faraday Trans. 1994, 90, 3367–3372. [Google Scholar] [CrossRef]
- Wakabayashi, F.; Kondo, J. N.; Domen, K.; Hirose, C. Direct comparison of N2 and CO as IR spectrocopic probes of acid sites in H-ZSM-5 zeolite. J. Phys. Chem. 1995, 99, 10573–10580. [Google Scholar] [CrossRef]
- Borovkov, V. Yu.; Karge, H. G. Use of combination modes and overtones of metal carbonyls for the IR study of cation states in zeolites. J. Chem. Soc. Faraday Trans. 1995, 91, 2035–2039. [Google Scholar] [CrossRef]
- Lercher, J. A.; Gründling, C.; Eder-Mirth, G. Infrared studies of the surface acidity of oxides and zeolites using adsorbed probe molecules. Catal. Today 1996, 27, 353–376. [Google Scholar] [CrossRef]
- Zecchina, A.; Otero Areán, C. Diatomic molecular probes for Mid-IR studies of zeolites. Chem. Soc. Rev. 1996, 25, 187–197. [Google Scholar] [CrossRef]
- Lavalley, J. C. Infrared spectrometric studies of the surface basicity of metal oxides and zeolites using adsorbed probe molecules. Catal. Today 1996, 27, 377–401. [Google Scholar] [CrossRef]
- Otero Areán, C.; Turnes Palomino, G.; Escalona Platero, E.; Peñarroya Mentruit, M. Zeolite-supported metal carbonyls: sensitive probes for infrared spectroscopic characterization of the zeolite surface. J. Chem. Soc. Dalton Trans 1997, 873–879. [Google Scholar]
- Onida, B.; Gabelica, Z.; Lourenço, J. P.; Ribeiro, M. F.; Garrone, E. Spectroscopic characterization of the hydroxyl groups in SAPO-40. 2. Interaction with CO and N2. J. Phys. Chem. B 1997, 101, 9244–9249. [Google Scholar] [CrossRef]
- Knözinger, H.; Huber, S. IR spectroscopy of small and weakly interacting molecular probes for acidic and basic zeolites. J. Chem. Soc. Faraday Trans. 1998, 94, 2047–2059. [Google Scholar] [CrossRef]
- Katoh, M.; Yamazaki, T.; Ozawa, S. IR spectroscopic study of adsorption of binary gases over ion-exchanged ZSM-5 zeolites. J. Colloid Interf. Sci. 1998, 203, 447–455. [Google Scholar] [CrossRef]
- Li, P.; Xiang, Y.; Grassian, V. H.; Larsen, S. C. CO adsorption as a probe of acid sites and the electric field in alkaline earth exchanged zeolite beta using FTIR and ab initio quantum calculations. J. Phys. Chem. B 1999, 103, 5058–5062. [Google Scholar] [CrossRef]
- Otero Areán, C.; Turnes Palomino, G.; Zecchina, A.; Spoto, G.; Bordiga, S.; Roy, P. Cation-carbon stretching vibration of adducts formed upon CO adsorption on alkaline zeolites. Phys. Chem. Chem. Phys 1999, 1, 4139–4140. [Google Scholar]
- Otero Areán, C.; Turnes Palomino, G.; Zecchina, A.; Bordiga, S.; Llabrés i Xamena, F. X.; Pazè, C. Vibrational spectroscopy of carbon monoxide and dinitrogen adsorbed on magnesium-exchanged ETS-10 molecular sieve. Catal. Lett. 2000, 66, 231–235. [Google Scholar]
- Hadjiivanov, K.; Knözinger, H.; Ivanova, E.; Dimitrov, L. FTIR study of low-temperature CO and 15N2 adsorption on a CaNaY zeolite: formation of site-specific Ca2+(CO)3 and Ca2+(15N2)3 complexes. Phys. Chem. Chem. Phys. 2001, 3, 2531–2536. [Google Scholar] [CrossRef]
- Otero Areán, C.; Manoilova, O. V.; Rodríguez Delgado, M.; Tsyganenko, A. A.; Garrone, E. Formation of several types of coordination complexes upon CO adsorption on the zeolite Li-ZSM-5. Phys. Chem. Chem. Phys 2001, 3, 4187–4188. [Google Scholar]
- Garrone, E.; Rodríguez Delgado, M.; Otero Areán, C. Trends in infrared spectroscopy of zeolites. Trends Inorg. Chem. in press.
- Zecchina, A.; Bordiga, S.; Lamberti, C.; Spoto, G.; Carnelli, L.; Otero Areán, C. Low-temperature Fourier transform infrared study of the interaction of CO with cations in alkali-metal-exchanged ZSM-5 zeolites. J. Phys. Chem. 1994, 98, 9577–9582. [Google Scholar] [CrossRef]
- Pacchioni, G.; Cogliandro, G.; Bagus, P. S. Molecular orbital cluster model study of bonding and vibrations of CO adsorbed on MgO surfaces. Int. J. Quantum Chem. 1992, 42, 1115–1139. [Google Scholar] [CrossRef]
- Ferrari, A. M.; Ugliengo, P.; Garrone, E. Ab initio study of the adducts of carbon monoxide with alkaline cations. J. Chem. Phys. 1996, 105, 4129–4139. [Google Scholar] [CrossRef]
- Böse, H.; Förster, H. Sorption states of isoelectronic linear molecules in zeolites. J. Mol. Struct. 1990, 218, 393–398. [Google Scholar] [CrossRef]
- Katoh, M.; Yamazaki, T.; Ozawa, S. IR spectra for adsorbed CO on various alkali metal ion-exchanged ZSM-5 zeolites. Bull. Chem. Soc. Jpn. 1994, 67, 1246–1253. [Google Scholar] [CrossRef]
- Turnes Palomino, G.; Otero Areán, C.; Geobaldo, F.; Ricchiardi, G.; Bordiga, S.; Zecchina, A. FTIR study of CO adsorbed at low temperature on zeolite L. Evidence for an ordered distribution of aluminium atoms. J. Chem. Soc. Faraday Trans 1997, 93, 189–191. [Google Scholar]
- Garrone, E.; Fubini, B.; Bonelli, B.; Onida, B.; Otero Areán, C. Thermodynamics of CO adsorption on the zeolite Na-ZSM-5. A combined microcalorimetric and FTIR spectroscopic study. Phys. Chem. Chem. Phys. 1999, 1, 513–518. [Google Scholar] [CrossRef]
- Ugliengo, P.; Garrone, E.; Ferrari, A. M.; Zecchina, A.; Otero Areán, C. Quantum chemical calculations and experimental evidence for O-bonding of carbon monoxide to alkali metal cations in zeolites. J. Phys. Chem. B 1999, 103, 4839–4846. [Google Scholar] [CrossRef]
- Ferrari, A. M.; Neyman, K. M.; Rösch, N. CO interaction with alkali metal cations in zeolites: A density functional model cluster study. J. Phys. Chem. B 1997, 101, 9292–9298. [Google Scholar] [CrossRef]
- Hush, N. S.; Williams, M. L. Carbon monoxide bond length, force constant and infrared intensity variations in strong electric fields: Valence-shell calculations with applications to properties of adsorbed and complexed CO. J. Mol. Spectrosc. 1974, 50, 349–368. [Google Scholar] [CrossRef]
- Larsson, R.; Lykvist, R.; Rebenstorf, B. On the IR frequency shift of carbon monoxide adsorbed on positive surface ions. Z. Phys. Chem. Leipzig 1982, 263, 1089–1104. [Google Scholar]
- Lupinetti, A. J.; Strauss, S. H.; Frenking, G. Nonclassical metal carbonyls. Progr. Inorg. Chem. 2001, 49, 1–112. [Google Scholar]
- Otero Areán, C.; Tsyganenko, A. A.; Escalona Platero, E.; Garrone, E.; Zecchina, A. Two coordination modes of CO in zeolites: A temperature-dependent equilibrium. Angew. Chem. Int. Ed. 1998, 37, 3161–3163. [Google Scholar]
- Manoilova, O. V.; Peñarroya Mentruit, M.; Turnes Palomino, G.; Tsyganenko, A. A.; Otero Areán, C. Variable-temperature infrared spectrometry of carbon monoxide adsorbed on the zeolite K-ZSM-5. Vib. Spectrosc. 2001, 26, 107–111. [Google Scholar] [CrossRef]
- Otero Areán, C.; Peñarroya Mentruit, M.; Rodríguez Delgado, M.; Turnes Palomino, G.; Manoilova, O. V.; Tsyganenko, A. A.; Garrone, E. Variable temperature FTIR spectroscopy of carbon monoxide adsorbed on protonic and rubidium-exchanged ZSM-5 zeolites. Stud. Surf. Sci. Catal. in press.
- Tsyganenko, A. A.; Escalona Platero, E.; Otero Areán, C.; Garrone, E.; Zecchina, A. Variable-temperature IR spectroscopic studies of CO adsorbed on Na-ZSM-5 and Na-Y zeolites. Catal. Lett. 1999, 61, 187–192. [Google Scholar] [CrossRef]
- Szostak, R. Molecular Sieves; Van Nostrand Reinhold: New York, 1989. [Google Scholar]
- Otero Areán, C.; Manoilova, O. V.; Tsyganenko, A. A.; Turnes Palomino, G.; Peñarroya Mentruit, M.; Geobaldo, F.; Garrone, E. Thermodynamics of hydrogen bonding between CO and the supercage Brønsted acid sites of the H-Y zeolite: Studies from variable temperature IR spectroscopy. Eur. J. Inorg. Chem 2001, 1739–1743. [Google Scholar]
- Muenter, J. S. Electric dipole moment of carbon monoxide. J. Mol. Spectrosc. 1975, 55, 490–491. [Google Scholar] [CrossRef]
- Zecchina, A.; Otero Areán, C.; Turnes Palomino, G.; Geobaldo, G.; Lamberti, C.; Spoto, G.; Bordiga, S. The vibrational spectroscopy of H2, N2, CO and NO adsorbed on the titanosilicate molecular sieve ETS-10. Phys. Chem. Chem. Phys. 1999, 1, 1649–1657. [Google Scholar] [CrossRef]
© 2002 by MDPI (http://www.mdpi.org).
Share and Cite
Areán, C.O.; Palomino, G.T.; Tsyganenko, A.A.; Garrone, E. Quantum Chemical and FTIR Spectroscopic Studies on the Linkage Isomerism of Carbon Monoxide in Alkali-Metal-Exchanged Zeolites: A Review of Current Research. Int. J. Mol. Sci. 2002, 3, 764-776. https://0-doi-org.brum.beds.ac.uk/10.3390/i3070764
Areán CO, Palomino GT, Tsyganenko AA, Garrone E. Quantum Chemical and FTIR Spectroscopic Studies on the Linkage Isomerism of Carbon Monoxide in Alkali-Metal-Exchanged Zeolites: A Review of Current Research. International Journal of Molecular Sciences. 2002; 3(7):764-776. https://0-doi-org.brum.beds.ac.uk/10.3390/i3070764
Chicago/Turabian StyleAreán, C. Otero, G. Turnes Palomino, A. A. Tsyganenko, and E. Garrone. 2002. "Quantum Chemical and FTIR Spectroscopic Studies on the Linkage Isomerism of Carbon Monoxide in Alkali-Metal-Exchanged Zeolites: A Review of Current Research" International Journal of Molecular Sciences 3, no. 7: 764-776. https://0-doi-org.brum.beds.ac.uk/10.3390/i3070764