Introduction
In solid peptides and polypeptides the hydrogen bond plays an important role for forming the second-order structures such as the α-helix, β-sheet,
etc. and higher-order structures [
1,
2,
3]. For this reason, we have studied NMR methodologies for obtaining information about the hydrogen-bonded structure of peptides and polypeptides in the solid state through the observation of solid state NMR chemical shifts. Then, we have elucidated the relationship between the hydrogen-bond lengths and solid state NMR chemical shifts of
13C [
4,
5,
6,
7,
8],
15N [
9,
10] and
17O [
11,
12,
13] nuclei from the experimental and theoretical aspects. From these systematic works, it has been obtained that the observation of the main-chain chemical shifts leads to the determination of the hydrogen-bond length in solid peptides and polypeptides including proteins.
Most recently, the amide proton NMR spectra of glycine-containing peptides and polypeptides in the solid state, of which the hydrogen-bond lengths are distributed in the wide range, have been successfully measured by high frequency 800 MHz NMR [
14] and 300 MHz NMR with the frequency-switched Lee-Goldburg (FSLG) homo-nuclear dipolar decoupling method [
15]. These experimental results have showed that the amide proton chemical shifts move downfield with a decrease in R
N…O (hydrogen-bond length between the amide nitrogen atom and amide carbonyl oxygen atom) or R
H…O (hydrogen-bond length between the amide hydrogen atom and amide carbonyl oxygen atom). It has been preliminarily explained by
ab initio 6-31G**basis set using a GlyGly supermolecule.
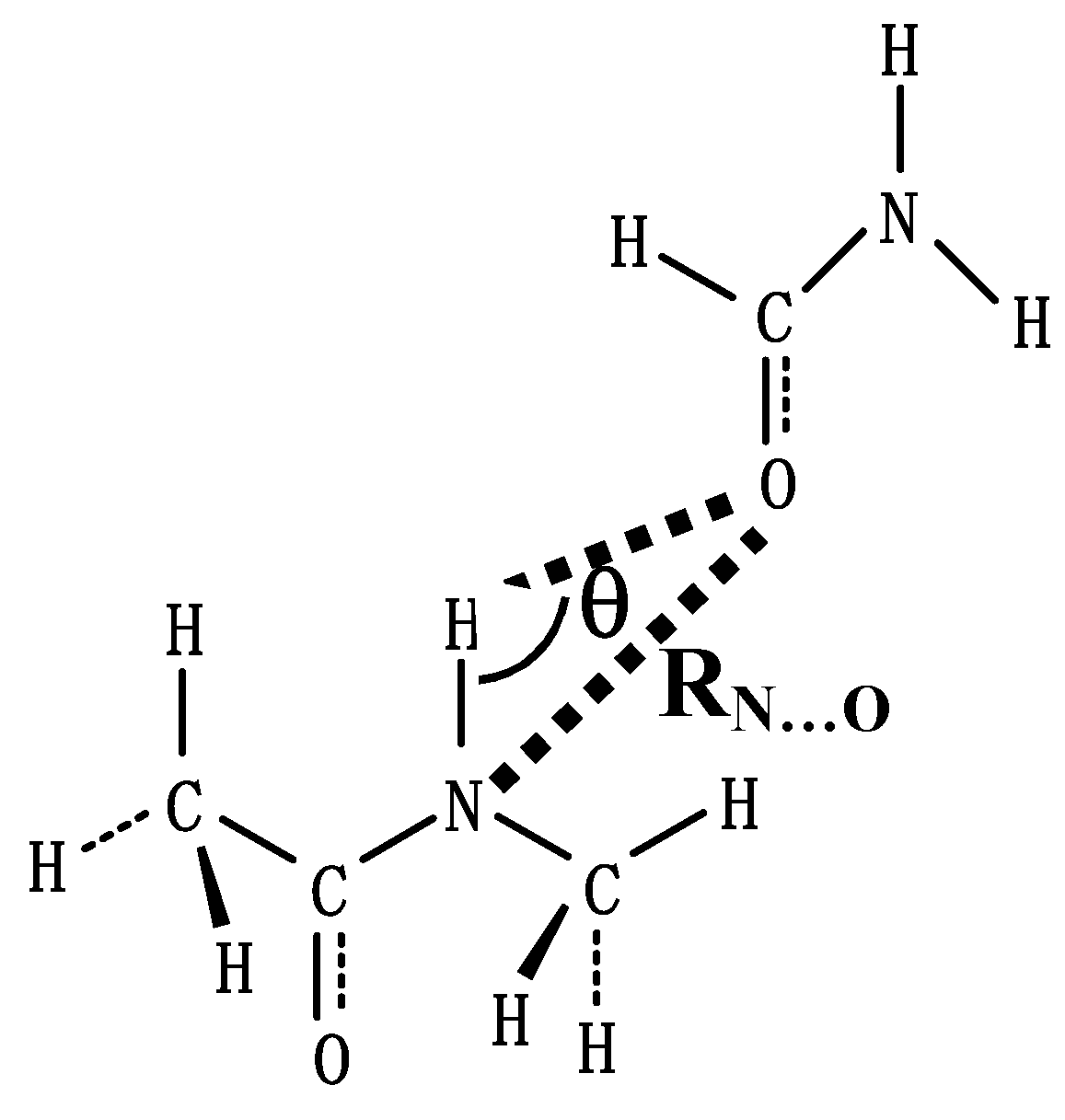
In this work we aim to calculate more sophisticatedly isotropic
1H chemical shifts and
1H chemical shift tensor components of the amide proton in a supermolecule, an N-methylacetamide hydrogen-bonded with a formamide, as functions of hydrogen-bond length R
N…O and hydrogen-bond angles θ with the FPT-GIAO method within the HF/STO 6-31
++G(d,p)
ab initio MO framework [
16,
17,
18] in the Gaussian 98 program [
19], in order to understand deeply behavior for the experimental isotropic
1H chemical shifts of Gly-containing peptides and polypeptides associated with hydrogen bonding.
Results and Discussion
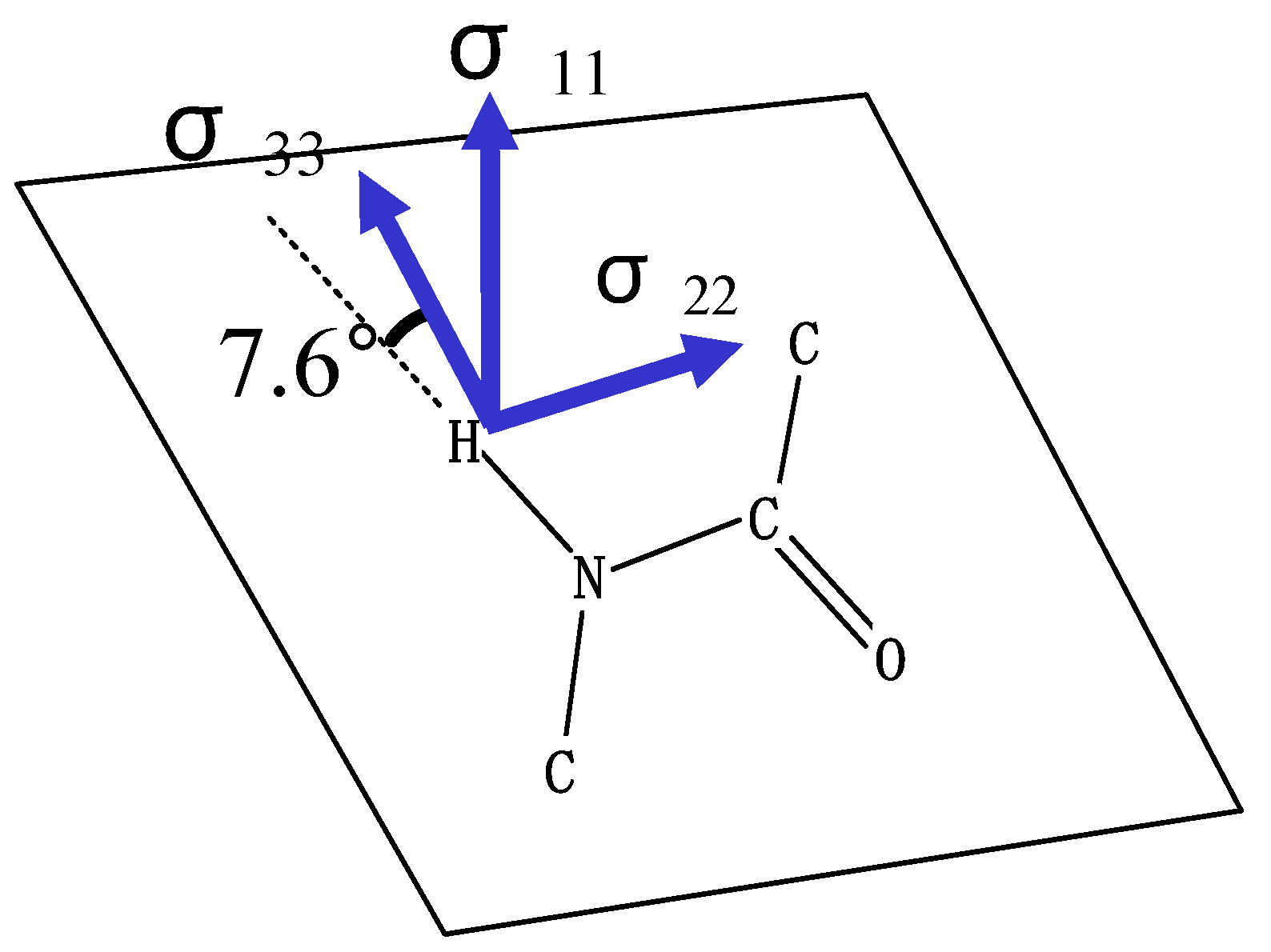
For understanding a feature of the directions of the
1H chemical shift tensor components of the amide proton, the calculated
1H chemical shifts of an N-methylacetamide molecule are listed in
Table 1 and the directions of the chemical shift tensor components are shown together with chemical structure of the molecule in
Fig. 2. The chemical shift tensor components are asymmetric, and the anisotropy( = σ
11 – σ
33) is about 11.3 ppm. The σ
33 is almost directed to the amide N-H bond with the small deviation of 7.6
o from the linear hydrogen-bond (N-H…O=C) for which the hydrogen-bond angle θ is defined to be 0
o, the σ
11 is perpendicular to the amide plane and the σ
22 is perpendicular to the amide N-H bond. At this stage, according to our best knowledge there are no experimental data on
1H chemical shift tensor components of peptides and polypeptides in spite of its importance.
The calculated isotropic
1H chemical shifts of the amide proton of the supermolecule as function of R
N…O and hydrogen-bond angle θ together with the experimental data on various Gly-containing peptides and polypeptides reported previously [
14,
15] are listed in
Table 2. In the calculations the hydrogen-bond lengths and hydrogen-bond angles for various Gly-containing peptides and polypeptides as determined by X-ray diffraction are used.
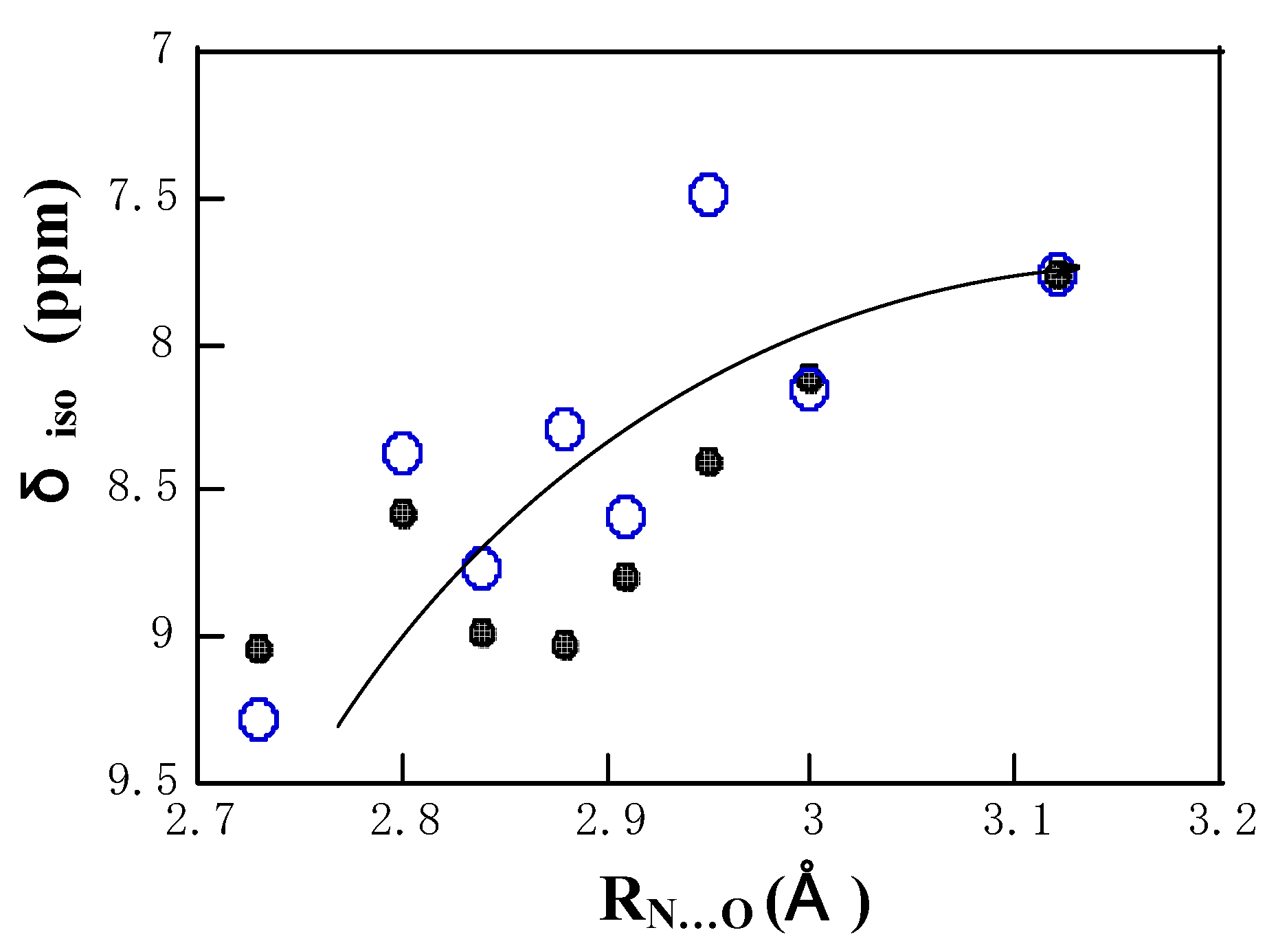
Fig. 3 shows the plots of the calculated and experimental amide
1H chemical shifts against R
N…O. For convenience, the calculated chemical shielding value corresponding to GlyGly.HNO
3 was adjusted to that of the experimental chemical shift value of peptide with the maximum hydrogen-bond length in order to convert from the chemical shielding value to the chemical shift value. The isotropic
1H chemical shifts move downfield with a
Table 1.
1H chemical shieldings (ppm) of the amide proton of N-methyl-acetamide calculated by HF/STO STO 6-31++G(d,p) ab initio MO method
Table 1.
1H chemical shieldings (ppm) of the amide proton of N-methyl-acetamide calculated by HF/STO STO 6-31++G(d,p) ab initio MO method
| σiso | σ11 | σ22 | σ33 |
| 28.1 | 21.1 | 29.8 | 33.4 |
Figure 2.
The directions of the chemical shift tensor components σ11, σ22 and σ33 of the hydrogen-bonded amide proton are shown together with chemical structure of an N-methylacetamide molecule.
Figure 2.
The directions of the chemical shift tensor components σ11, σ22 and σ33 of the hydrogen-bonded amide proton are shown together with chemical structure of an N-methylacetamide molecule.
Figure 3.
The plots of the calculated and experimental isotropic
1H chemical shifts δ
iso of the amide proton as a function of R
N…O, where the calculated chemical shielding value corresponding to GlyGly.HNO
3 was adjusted to that of the experimental chemical shift value of peptide with the maximum hydrogen-bond length in order to convert from the chemical shift shielding value to the chemical shift value. The open circular symbol is for the calculation and the closed circular symbol for the experiment. The data points for peptide and polypeptide samples can be recognized by seeing the hydrogen-bond lengths in
Table 2
Figure 3.
The plots of the calculated and experimental isotropic
1H chemical shifts δ
iso of the amide proton as a function of R
N…O, where the calculated chemical shielding value corresponding to GlyGly.HNO
3 was adjusted to that of the experimental chemical shift value of peptide with the maximum hydrogen-bond length in order to convert from the chemical shift shielding value to the chemical shift value. The open circular symbol is for the calculation and the closed circular symbol for the experiment. The data points for peptide and polypeptide samples can be recognized by seeing the hydrogen-bond lengths in
Table 2
Table 2.
Calculated
1H chemical shifts of the amide proton of a supermolecule, an N-methylacetamide hydrogen-bonded with a formamide, as functions of hydrogen-bond length R
N…O and hydrogen-bond angles by FPT-GIAO method within the framework of HF/STO 6-31
++G(d,p)
ab initio MO method together with the experimental data reported previously [
14,
15].
Table 2.
Calculated 1H chemical shifts of the amide proton of a supermolecule, an N-methylacetamide hydrogen-bonded with a formamide, as functions of hydrogen-bond length RN…O and hydrogen-bond angles by FPT-GIAO method within the framework of HF/STO 6-31++G(d,p) ab initio MO method together with the experimental data reported previously [14,15].
| Sample | Experimental hydrogen-bonded glycine amide proton chemical shifta δiso (ppm) | Calculated hydrogen-bonded glycine amide proton chemical shiftb δiso (ppm) | Hydrogen-bond length RN…O (Å) | Hydrogen-bond angle θ (degree) |
| Poly glycine ( form II ) | 9.04 | 9.29 | 2.73 | 146.2 |
| Tyr - Gly – Gly | 9.03 | 8.29 | 2.88 | 144.0 |
| Pro - Gly – Gly | 8.99 | 8.77 | 2.84 | 151.9 |
| Gly – Gly | 8.59 | — | 2.94 | — |
| Poly glycine ( form I ) | 8.40 | 7.49 | 2.95 | 132.8 |
| Ala - Gly – Gly | 8.12 | 8.15 | 3.00 | 160.2 |
| Val - Gly – Gly | 8.80 | 8.59 | 2.91 | 163.1 |
| Sar - Gly –Gly | 8.57 | 8.37 | 2.80 | 137.5 |
| Gly - Gly・HNO3 | 7.76 | 7.76 | 3.12 | 164.6 |
decrease in R
N…O in the calculations and experiments. From this figure, it is shown that the slope of the curve becomes gradually small with an increase in R
N…O. This means that the effect of the hydrogen bonding on the chemical shift is asymptotically decreased with an increase in R
N…O. The calculations reproduce well the experiments.
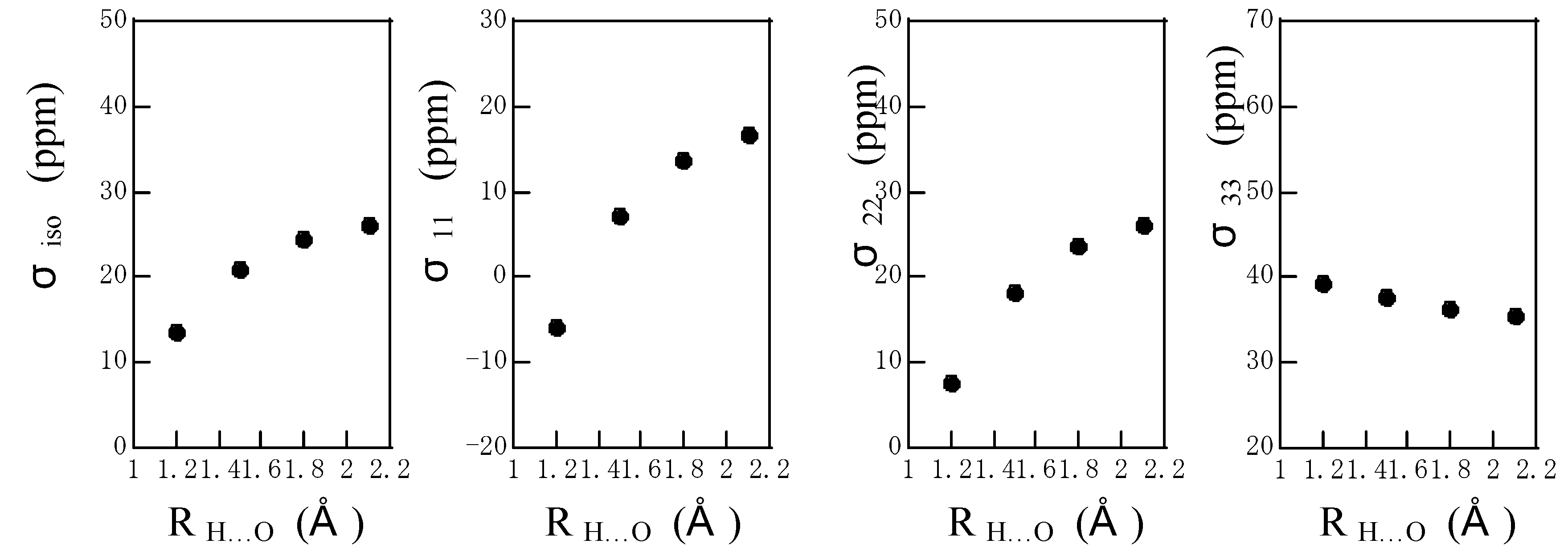
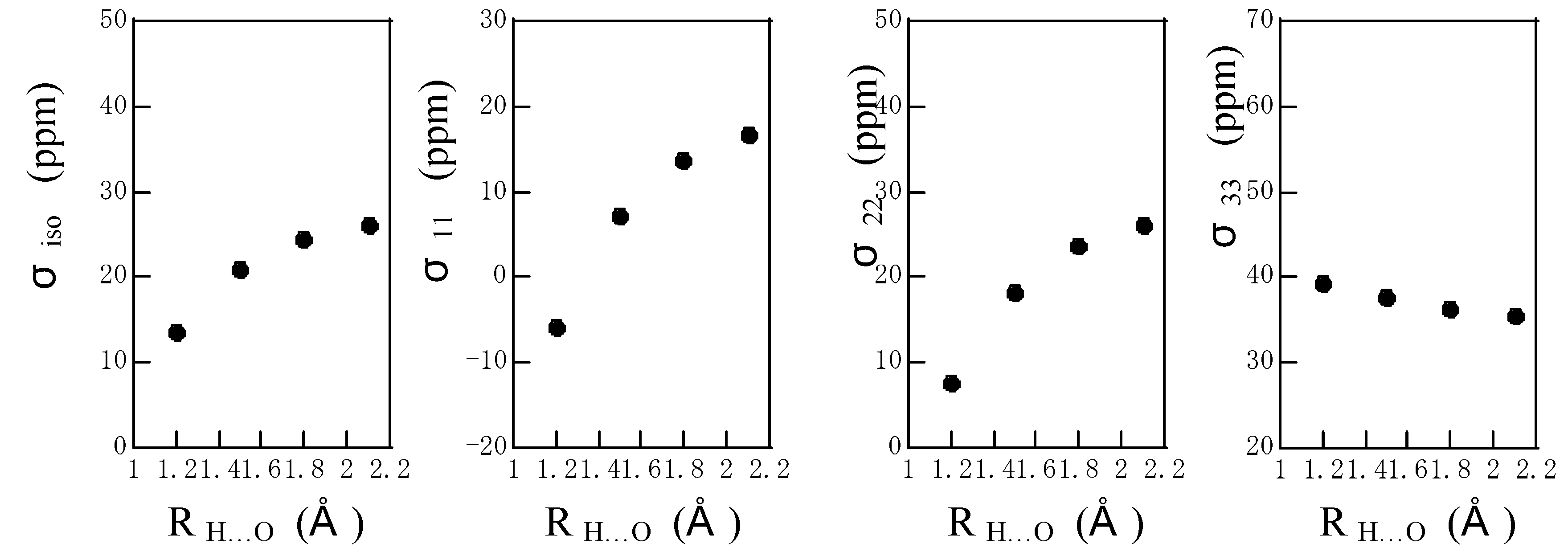
The calculated
1H chemical shielding tensor components of the amide proton are shown as a function of R
N…O in
Fig. 4 together with the isotropic
1H chemical shielding, where the hydrogen-bond angle is fixed to be 180
o. The isotropic chemical shielding moves downfield with a decrease in R
N…O. The σ
11 and σ
22 components move largely downfield with a decrease in R
N…O, but the σ
33 component moves slightly upfield with a decrease in R
N…O. As mentioned above, the σ
33 is almost directed to the amide N-H bond. It can be said that the σ
11 and σ
22 components govern predominantly the downfield shift in isotropic chemical shift. On the other hand, as shown in
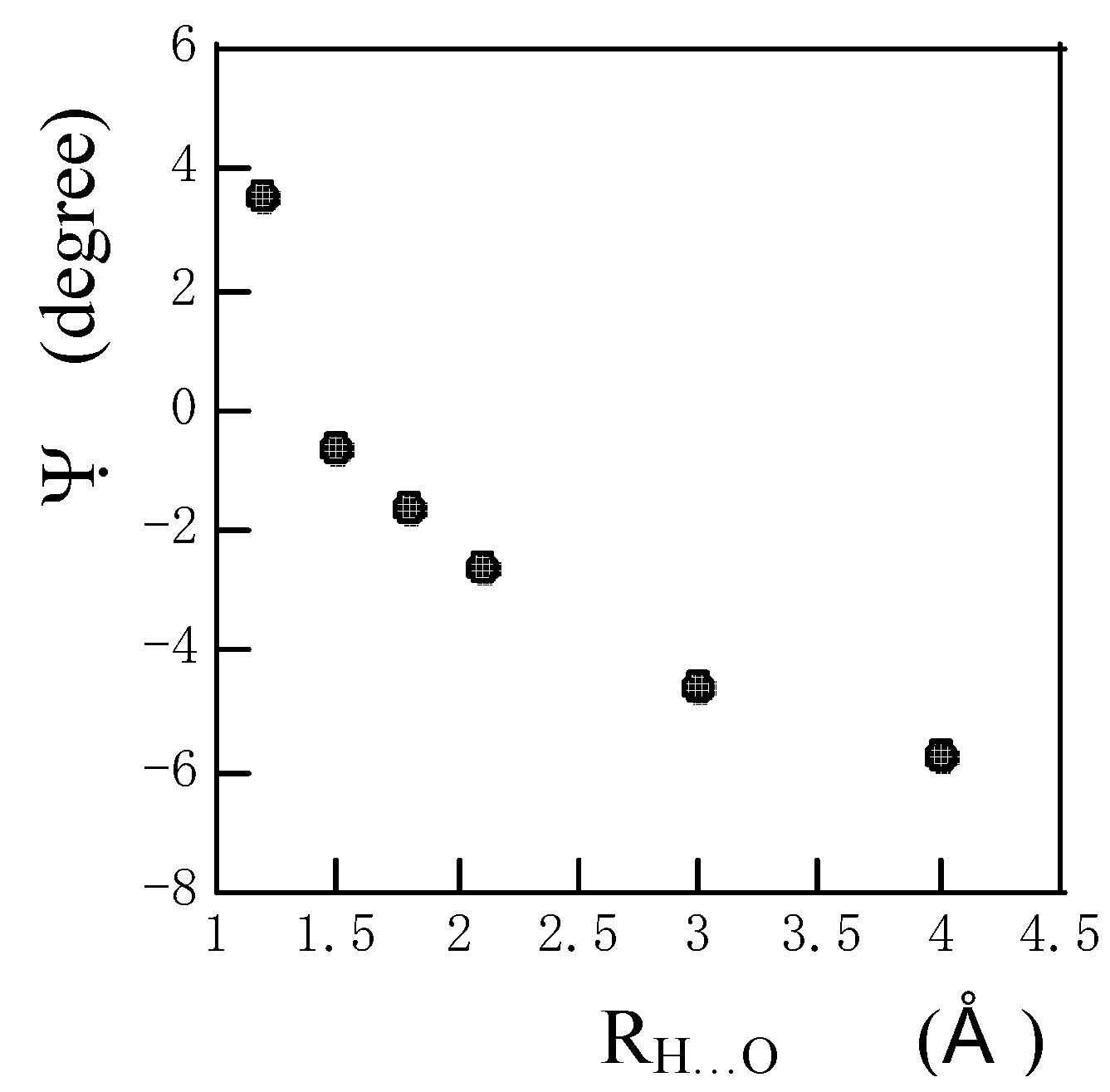
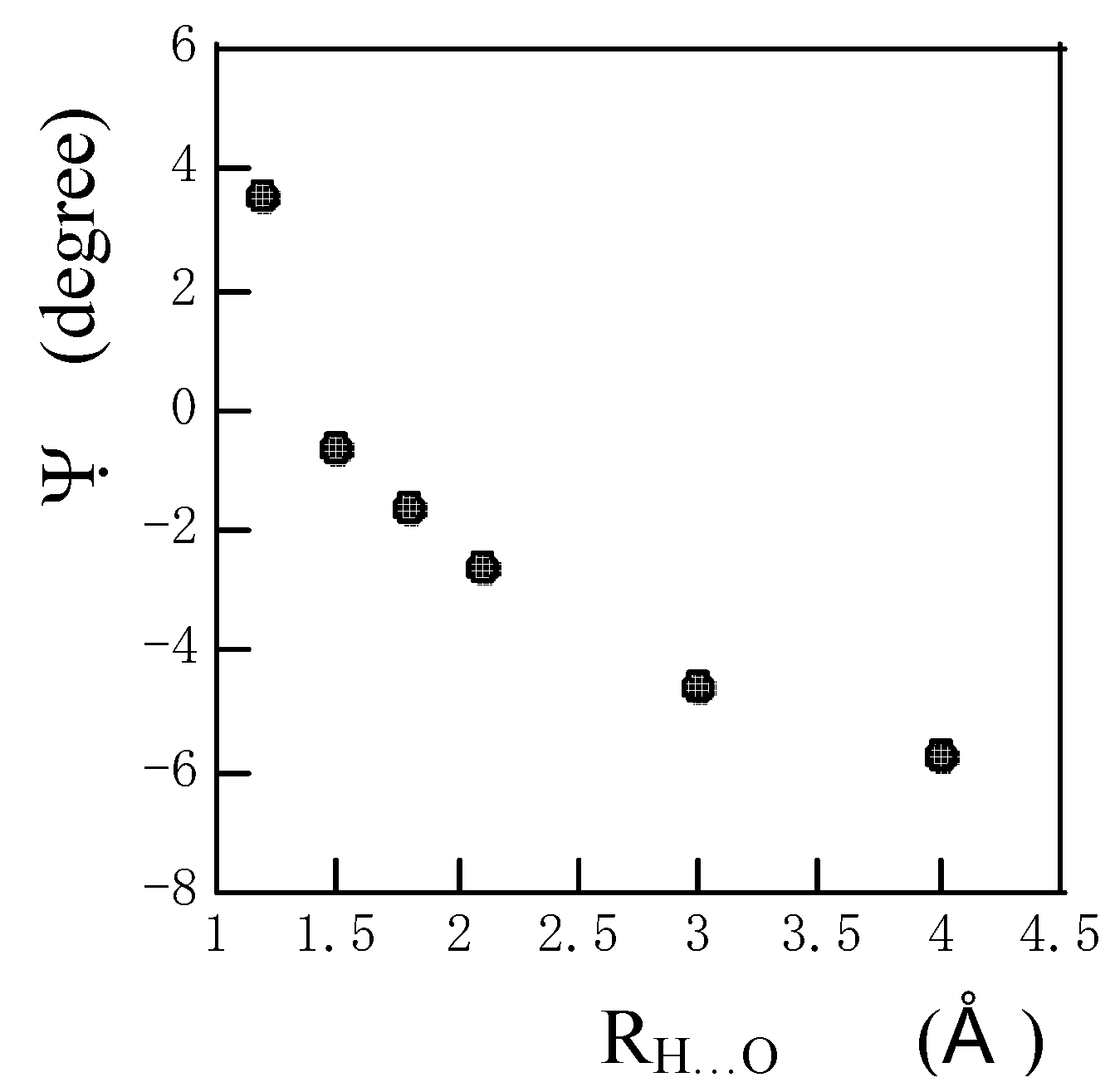
Fig. 5 the angle Ψ between the amide N-H bond and the direction of the σ
33 component approaches 0
o with a decrease in R
N…O until R
N…O = 1.5 A. This shows that the direction of distortion of electronic distribution on the amide proton approaches the N-H bond with a decrease in R
N…O. If R
N…O is further decreased, the angle Ψ deviates with the positive sign from the amide N-H bond again. In the shorter R
N…O range, the direction of the distortion electronic distribution on the amide proton is strongly affected by interactions with neighboring atoms and so the angle Ψ deviates with the positive sign from the amide N-H bond again. As reported previously [
9,
10], the direction of the σ
33 component for the amide nitrogen is along the N-H bond. This direction is the same as the case of the amide proton σ
33 component.
Figure 4.
The plots of the calculated isotropic 1H chemical shielding σiso and 1H chemical shielding tensor components σ11, σ22 and σ33 of the amide proton of an N-methylacetamide hydrogen-bonded with a formamide against RN…O, where the hydrogen-bond angle is fixed to be 180o.
Figure 4.
The plots of the calculated isotropic 1H chemical shielding σiso and 1H chemical shielding tensor components σ11, σ22 and σ33 of the amide proton of an N-methylacetamide hydrogen-bonded with a formamide against RN…O, where the hydrogen-bond angle is fixed to be 180o.
Figure 5.
The plots of the angle Ψ between the amide N-H bond and the direction of the calculated 1H chemical shielding tensor component σ33 of the amide proton of an N-methylacetamide hydrogen-bonded with a formamide against RH…O
Figure 5.
The plots of the angle Ψ between the amide N-H bond and the direction of the calculated 1H chemical shielding tensor component σ33 of the amide proton of an N-methylacetamide hydrogen-bonded with a formamide against RH…O
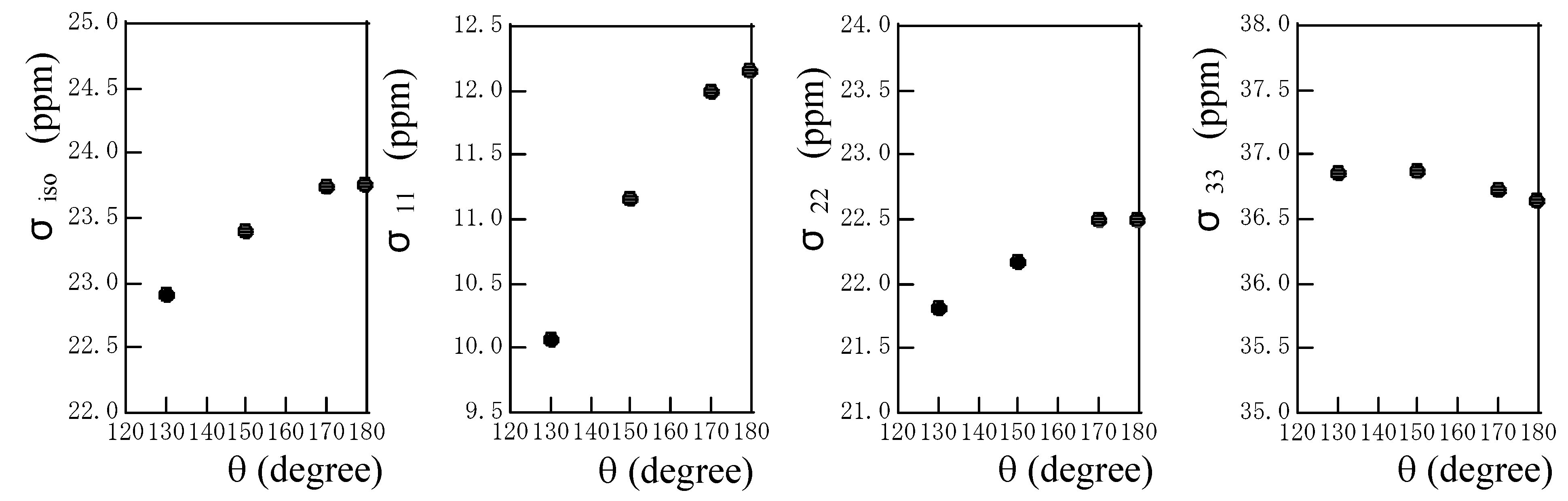
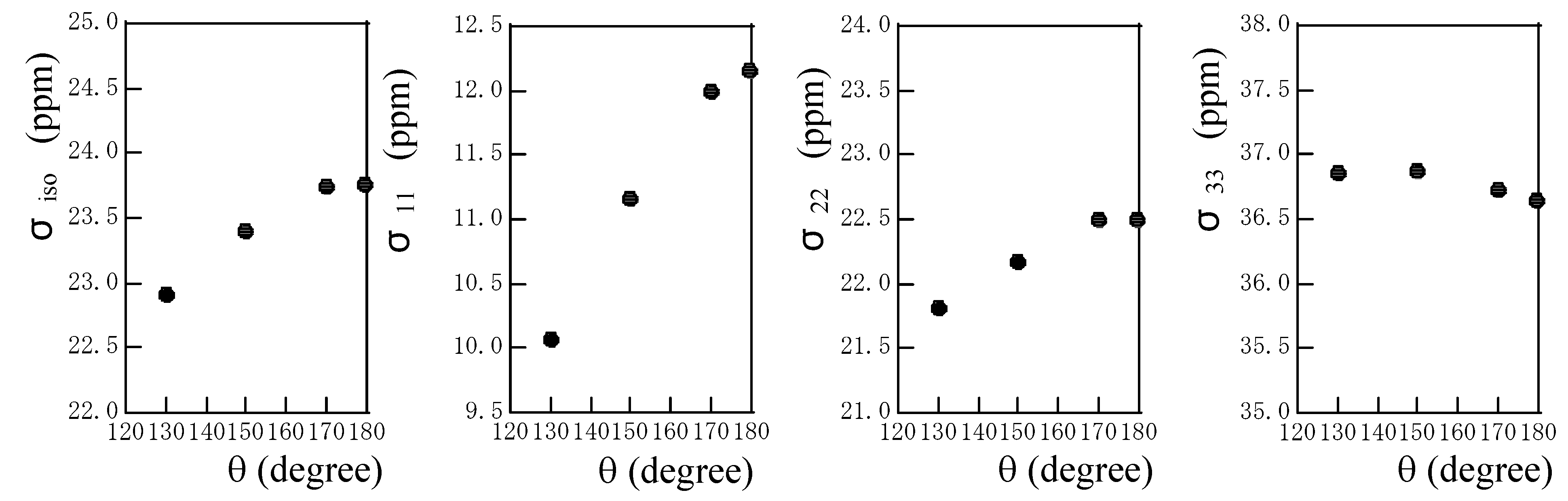
Fig. 6 shows the plots of the calculated isotropic chemical shielding and chemical shielding tensor components against the hydrogen-bond angle θ. The deviation of the linear hydrogen-bond leads to downfield shift for the isotropic chemical shielding. The σ
11 and σ
22 components move largely downfield with a decrease in θ, but the σ
33 component moves slightly upfield with a decrease in R
N…O. It can be said that the σ
11 and σ
22 components govern predominantly the downfield shift in isotropic chemical shift.
At present, there are no experimental data of the amide proton chemical shift tensor components. If these data are obtained, we will be able to obtain deeper insight of hydrogen bonding by comparing the calculations and experiments.
Figure 6.
The plots of the calculated isotropic chemical shielding σiso and chemical shielding tensor components σ11, σ22 and σ33 of the amide proton of an N-methylacetamide hydrogen-bonded with a formamide against the hydrogen-bond angle θ.
Figure 6.
The plots of the calculated isotropic chemical shielding σiso and chemical shielding tensor components σ11, σ22 and σ33 of the amide proton of an N-methylacetamide hydrogen-bonded with a formamide against the hydrogen-bond angle θ.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}