Structure, Stability and Interaction Studies on Schiff Base Analogue Systems

1
Department of Physics, Bharathiar University, Coimbatore-641046, India
2
Department of Physics, Nallamuthu Gounder Mahalingam College, Pollachi-642 001, India
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2004, 5(1), 1-12; https://0-doi-org.brum.beds.ac.uk/10.3390/i5010001
Submission received: 30 August 2004
/
Accepted: 9 December 2004
/
Published: 26 December 2004
Abstract
:Ab initio and density functional theory methods have been applied to study the molecular structure and interaction of water with N-methyl-2-propenylidenimine and N-methyl-2-butenylidenimine molecules. The most possible reactive sites of the above molecules have been identified for the water interactions. The strength of the hydrogen bond is discussed using the atomic charges, which were calculated using the Mulliken population analysis and Natural population analysis schemes at MP2/6-31G* level of theory. The electron density (ρ) and laplacian of electron density (▽2ρ) have been calculated for the possible existence of the hydrogen bonds with CH and CH3 groups of molecules using the “Atoms in molecules” approach. The chemical hardness and chemical potential for these complexes have been calculated at HF/6-31G* level of theory and discussed for the conformational stability of these molecules.
Introduction
Bacteriorhodopsin (bR) is a transmembrane protein and its function as a light-driven proton pump and present in the outer purple membrane of Halobacterium salinarium. The light-absorbing chromosome is a retinal molecule that is covalently bonded via its Schiff base to the ε-amino group of Lys216 [1]. The retinal interaction may include hydrogen bonds with the protonated Schiff base. A resonance Raman study suggests that a negatively charged counter ion located near the Schiff base group is stabilized by water molecules [2]. Solid-state 13C and 15N NMR experimental results are being used to construct a model in which a water molecule is directly hydrogen-bonded to Schiff base [3]. A detailed understanding of Schiff base hydrogen bonding in the various stages of the photocycle will be required for a complete description of bR function. Recent work on synthetic retinal protonated Schiff base lends further support to the idea that Schiff base hydration may play an important functional role [4]. A direct determination of the hydrogen bonding arrangement will require additional experimental work. However, computational chemistry has played an important role in identifying and quantifying hydrogen-bonding geometries and energies of pertinent model systems. The quantum chemical calculations of the whole molecule is computationally expensive and the CPU time required is increasing with approximately the fourth power of the number of atomic orbitals in the system, so the applications are presently restricted to fragments of retinal molecule. Previous theoretical calculations have indicated that water molecules can form two stable hydrogen bonded complexes with a model Schiff base, (E)-N-methyl-2-propenylidenimine (s-trans) [CH2=CH-CH=NH-(CH3)]+, which we call here NMP[5], and (E)-N- methyl-2-cis butenylidenimine, [CH3-NH=CH-CH=CH(CH3)]+ which we call here NMB. The water can bind with these model compounds NMP and NMB along NH and CH sides as a proton acceptor.
Computational details
The second-order Møller-Plesset perturbation theory (MP2) [6] of ab initio method, Becke’s three parameter exact exchange functional (B3) [7] combined with gradient corrected correlation functional of Lee- Yang- Parr (LYP) [8] and Perdew and Wang’s 1991 (PW91) [9] functional of DFT have been employed to optimize the N-methyl-2-propenylidenimine, N-methyl-2-butenylidenimine molecules and with water molecules by implementing 6-31G* basis set. The water interactions have been made along NH, CH and both the sides of the NMP and NMB molecules. The structural optimizations have been performed for all the above said positions of water molecules with NMP and NMB molecules. The Basis set superposition error (BSSE) has been corrected using the Boys and Bernardi’s counter poise method [10]
where EAB is the interaction energy of the complex, EA(AB) and EB(AB) are the energies of monomers in the complex. The chemical hardness (η) and chemical potential (μ) have been calculated using the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies determined by the HF/6-31G* method. The natural population analysis (NPA) and Mulliken population analysis (MPA) studies have been carried out at MP2/6-31G* level of theory. The topological parameters such as electron density and laplacian of electron density have been calculated for CH and CH3 groups with water molecules using the “atoms in molecules” approach to identify the additional hydrogen bonds on the above groups. All the calculations were performed using the Gaussian 98W program [11].
EAB=EAB-(EA(AB)+EB(AB)),
Results and Discussion
NMP-Water Interaction: Geometries and Energetics
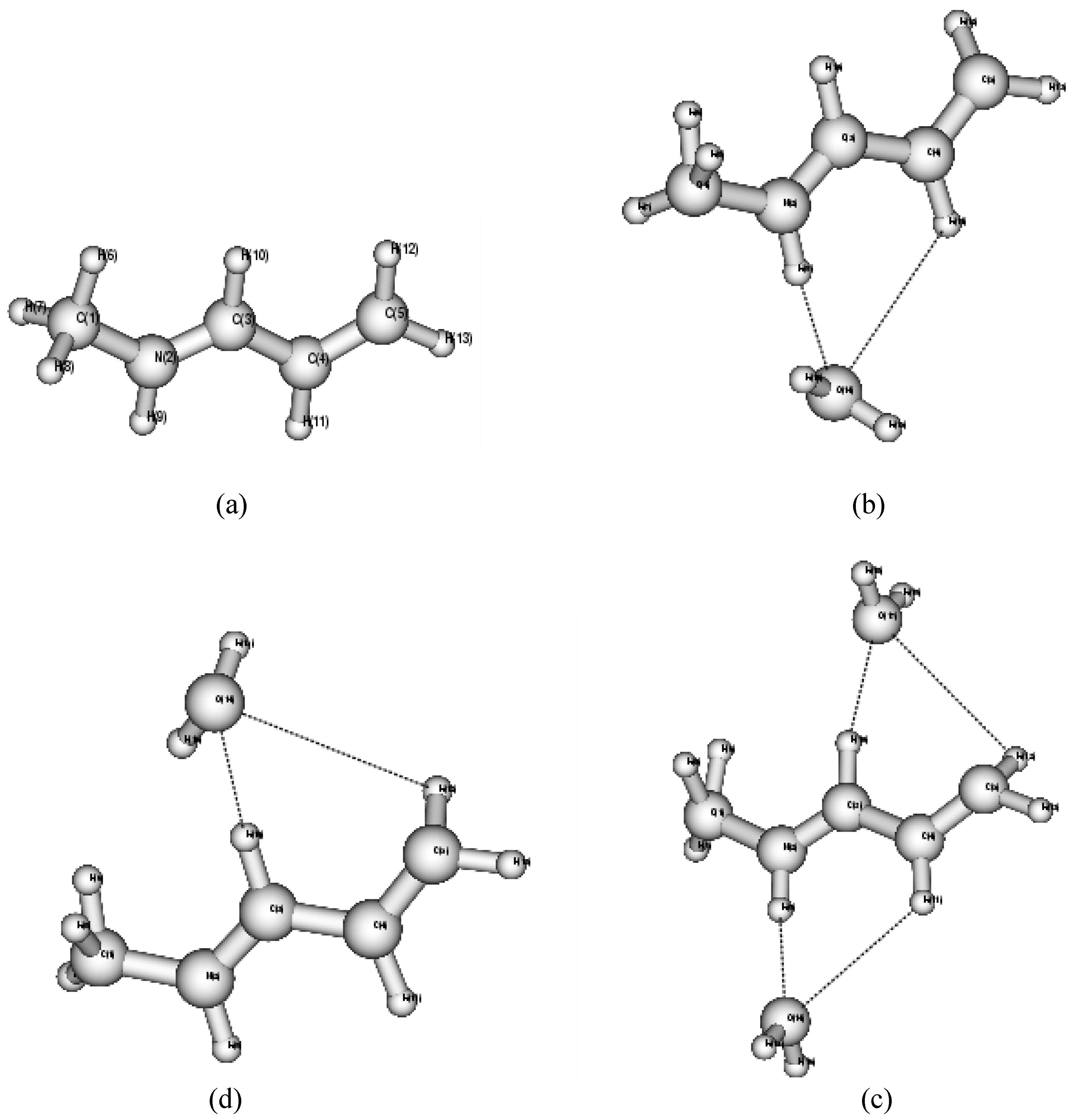
The model Schiff base molecules NMP and NMB and water molecules hydrogen bonded with NMP and NMB in different sites (Fig. 1 and 2) have been optimized at MP2, B3LYP and B3PW91 levels of theory employing 6-31G* basis set and the results are presented in Table 1 and Table 2.
Figure 1.
Molecular structures of (a) NMP (b) NMP-water along NH side (c) NMP-water at CH side (d) NMP-water along both the sides
Figure 1.
Molecular structures of (a) NMP (b) NMP-water along NH side (c) NMP-water at CH side (d) NMP-water along both the sides

Figure 2.
Molecular structures of (a) NMB (b) NMB-water along NH side (c) NMB-water along CH side (d) NMB-water at both the sides.
Figure 2.
Molecular structures of (a) NMB (b) NMB-water along NH side (c) NMB-water along CH side (d) NMB-water at both the sides.


{kind=link}
{kind=link}
Table 1.
Optimized geometrical parameters (bond length in Å, bond angle in degrees), rotational constants RA, RB, RC(in GHz), dipole moment µM (in Debye), total energy E (in Hartree), interaction energy Eint (in k cal/mol), chemical potential μ (in eV) and chemical hardness η (in eV) for NMP and NMP-water complexes.
| Parameters | MP2 | B3LYP | B3PW91 | MP2 | B3LYP | B3PW91 | MM | AM1 |
|---|---|---|---|---|---|---|---|---|
| NMP | NMP-Water along CH side | |||||||
| R(C1-N2) | 1.471 | 1.470 | 1.462 | 1.470 | 1.468 | 1.461 | 1.491 | 1.446 |
| R(N2-C3) | 1.299 | 1.300 | 1.298 | 1.299 | 1.302 | 1.299 | 1.285 | 1.314 |
| R(C3-C4) | 1.435 | 1.433 | 1.431 | 1.436 | 1.434 | 1.432 | 1.447 | 1.446 |
| R(C4-C5) | 1.349 | 1.349 | 1.348 | 1.350 | 1.349 | 1.347 | 1.345 | 1.345 |
| R(N2-H9) | 1.023 | 1.020 | 1.019 | 1.022 | 1.020 | 1.019 | 1.023 | 1.009 |
| R(H10-O14) | … | … | … | 2.015 | 2.040 | 1.987 | … | … |
| R(O14-H15) | … | … | … | 0.972 | 0.970 | 0.968 | … | … |
| θ(Χ1−Ν2−Χ3) | 125.8 | 126.2 | 126.2 | 124.7 | 125.5 | 125.4 | … | … |
| θ(Ν2−Χ3−Χ4) | 123.8 | 124.2 | 124.1 | 123.7 | 123.8 | 123.8 | 122.6 | 124.1 |
| θ(Χ3−Χ4−Χ5) | 119.3 | 120.0 | 119.9 | 118.0 | 118.9 | 118.7 | 121.0 | 121.2 |
| θ(Ν2−Χ1−Η7) | 108.8 | 109.1 | 109.1 | 109.0 | 109.3 | 109.4 | 111.0 | 111.1… |
| θ(Χ3−Η10−Ο14) | … | … | … | 167.5 | 158.9 | 167.5 | … | … |
| θ(Η15−Ο14−Η16) | … | … | … | 104.4 | 104.5 | 104.7 | … | … |
| θ(Χ1−Ν2−Χ3−Χ4) | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | … | … |
| θ(Ν2−Χ3−Χ4−Χ5) | 180.0 | 180.0 | 180.0 | 180.0 | 179.0 | 180.0 | … | … |
| -E | 210.9906 | 211.72692 | 211.64397 | 287.21062 | 288.15841 | 288.0467 | … | … |
| Eint | … | … | … | 14.680 | 14.310 | 13.420 | … | … |
| μΜ | 0.2484 | 0.6997 | 0.7234 | 0.385 | 1.096 | 0.907 | … | … |
| RA | 27.5885 | 27.8687 | 27.9141 | 2.8978 | 3.164 | 2.9314 | … | … |
| RB | 2.2447 | 2.2331 | 2.2443 | 2.1488 | 2.0104 | 2.1483 | … | … |
| RC | 2.1036 | 1.095 | 2.105 | 1.251 | 1.2462 | 1.2569 | … | … |
| η | 6.18* | … | … | 6.23* | … | … | … | … |
| μ | -9.27* | … | … | -8.75* | … | … | … | … |
| NMP-Water along NH side | NMP-2Water | |||||||
| R(C1-N2) | 1.466 | 1.465 | 1.457 | 1.465 | 1.464 | 1.457 | ||
| R(N2-C3) | 1.296 | 1.297 | 1.294 | 1.297 | 1.299 | 1.296 | ||
| R(C3-C4) | 1.439 | 1.438 | 1.435 | 1.440 | 1.439 | 1.437 | ||
| R(C4-C5) | 1.348 | 1.347 | 1.346 | 1.349 | 1.347 | 1.346 | ||
| R(N2-H9) | 1.041 | 1.043 | 1.043 | 1.038 | 1.038 | 1.039 | ||
| R(H9-O14) | 1.772 | 1.761 | 1.741 | 1.795 | 1.785 | 1.7673 | ||
| R(O14-H15) | 0.972 | 0.971 | 0.968 | 0.972 | 0.970 | 0.9679 | ||
| R(H10-O17) | … | … | … | 2.069 | 2.077 | 2.031 | ||
| R(O17-H18) | … | … | … | 0.971 | 0.970 | 0.968 | ||
| θ(Χ1−Ν2−Χ3) | 124.9 | 125.2 | 125.1 | 124.0 | 124.7 | 124.5 | ||
| θ(Ν2−Χ3−Χ4) | 123.0 | 123.7 | 123.6 | 122.8 | 123.2 | 123.2 | ||
| θ(Χ3−Χ4−Χ5) | 119.4 | 120.0 | 119.9 | 118.3 | 119.1 | 118.8 | ||
| θ(Ν2−Χ1−Η7) | 108.8 | 109.2 | 109.2 | 109.0 | 109.4 | 109.4 | ||
| θ(Ν2−Η9−Ο14) | 171.2 | 169.9 | 172.4 | 171.4 | 170.0 | 172.2 | ||
| θ(Η15−Ο14−Η16) | 105.2 | 105.4 | 105.5 | 105.1 | 105.3 | 105.5 | ||
| θ(Χ3−Η10−Ο17) | … | … | … | 164.0 | 158.8 | 166.7 | ||
| θ(Η18−Ο17−Η19) | … | … | … | 104.3 | 104.4 | 104.6 | ||
| θ(Χ1−Ν2−Χ3−Χ4) | 180.0 | 180.0 | 180.0 | 180.0 | 179.9 | 180.0 | ||
| θ(Ν2−Χ3−Χ4−Χ5) | 180.0 | 180.0 | 180.0 | 180.1 | 179.9 | 180.0 | ||
| -E | 287.2197 | 288.16697 | -288.05537 | 363.43722 | 364.59589 | 364.45557 | ||
| Eint | 20.53 | 19.91 | 19.13 | 34.06 | 32.93 | 31.30 | ||
| μΜ | 0.5266 | 1.2245 | 1.2897 | 0.62 | 1.3803 | 1.3676 | ||
| RA | 3.2714 | 3.2906 | 3.3688 | 2.4032 | 2.4413 | 2.3936 | ||
| RB | 2.0295 | 2.0317 | 1.9999 | 1.1159 | 1.1151 | 1.1307 | ||
| RC | 1.2701 | 1.2739 | 1.2726 | 0.7713 | 0.7748 | 0.7773 | ||
| η | 6.21* | … | … | 6.25* | … | … | ||
| μ | -8.68* | … | … | -8.20* | … | … | ||
(*Values are calculated at HF/6-31G* level of theory)
Table 2.
Optimized geometrical parameters (bond length in Ao, bond angle in degrees), rotational constants RA, RB, RC(in GHz), dipole moment µM (in Debye), total energy E (in Hartree), interaction energy Eint (in k cal/mol), chemical potential μ (in eV) and chemical hardness η (in eV) for NMB and NMB-water complexes.
| Parameters | MP2 | B3LYP | B3PW91 | MP2 | B3LYP | B3PW91 |
|---|---|---|---|---|---|---|
| NMB | NMB-Water along CH side | |||||
| R(C1-N2) | 1.470 | 1.468 | 1.460 | 1.470 | 1.468 | 1.460 |
| 1.302 | 1.305 | 1.303 | 1.302 | 1.305 | 1.303 | |
| R(C3-C4) | 1.427 | 1.424 | 1.421 | 1.429 | 1.426 | 1.424 |
| R(C4-C5) | 1.359 | 1.362 | 1.361 | 1.357 | 1.360 | 1.359 |
| R(C5-C6) | 1.489 | 1.487 | 1.482 | 1.790 | 1.489 | 1.483 |
| R(N2-H10) | 1.023 | 1.020 | 1.019 | 1.022 | 1.019 | 1.018 |
| R(C3-H11) | 1.087 | 1.087 | 1.087 | 1.090 | 1.091 | 1.092 |
| R(H11-O17) | … | … | … | 2.074 | 2.056 | 2.057 |
| θ(Χ1−Ν2−Χ3) | 125.8 | 126.2 | 126.2 | 124.8 | 125.3 | 125.4 |
| θ(Ν2−Χ3−Χ4) | 123.1 | 123.8 | 123.7 | 122.6 | 123.1 | 123.0 |
| θ(Χ3−Χ4−Χ5) | 122.8 | 123.4 | 123.3 | 122.2 | 122.9 | 122.9 |
| θ Χ4−Χ5−Χ6 | 129.0 | 129.4 | 129.5 | 128.5 | 129.1 | 129.1 |
| θ(Χ3−Η11−Ο17) | … | … | … | 165.0 | 165.0 | 165.1 |
| θ(Η18−Ο17−Η19 | … | … | … | 104.3 | 104.5 | 104.6 |
| θ(Χ1−Ν2−Χ3−Χ4) | 180.1 | 180.2 | 180.1 | 180.0 | 180.0 | 180.0 |
| θ(Ν2−Χ3−Χ4−Χ5) | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 |
| θ(Χ3−Χ4−Χ5−Χ6) | 0.1 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 |
| -E | 250.16614 | 251.05361 | 250.9573 | 326.3872 | 327.48529 | 327.36 |
| Eint | … | … | … | 15.3 | 14.45 | 13.4 |
| μΜ | 1.5137 | 1.8032 | 1.7934 | 1.0178 | 1.3734 | 1.3692 |
| RA | 10.8426 | 10.9277 | 10.9743 | 2.5135 | 2.5375 | 2.5355 |
| RB | 1.5148 | 1.5011 | 1.5081 | 1.4902 | 1.475 | 1.4805 |
| RC | 1.3516 | 1.3419 | 1.3482 | 0.9508 | 0.9479 | 0.9499 |
| η | 5.98* | … | … | 6.31* | … | … |
| μ | -8.80* | … | … | -8.35* | … | … |
| NMB-Water along NH side | NMB-2Water | |||||
| R(C1-N2) | 1.465 | 1.464 | 1.456 | 1.466 | 1.465 | 1.457 |
| R(N2-C3) | 1.300 | 1.301 | 1.299 | 1.300 | 1.302 | 1.300 |
| R(C3-C4) | 1.432 | 1.429 | 1.426 | 1.434 | 1.431 | 1.429 |
| R(C4-C5) | 1.357 | 1.358 | 1.357 | 1.356 | 1.357 | 1.356 |
| R(C5-C6) | 1.491 | 1.489 | 1.484 | 1.492 | 1.491 | 1.486 |
| R(N2-H10) | 1.040 | 1.039 | 1.040 | 1.031 | 1.036 | 1.037 |
| R(C3-H11 | 1.088 | 1.088 | 1.088 | 1.089 | 1.090 | 1.092 |
| R(H10-O17) | 1.784 | 1.776 | 1.759 | 1.806 | 1.798 | 1.782 |
| R(H11-O20) | … | … | … | 2.104 | 2.090 | 2.093 |
| θ(Χ1−Ν2−Χ3) | 124.8 | 125.2 | 125.2 | 124 | 124.4 | 124.4 |
| θ(Ν2−Χ3−Χ4) | 122.4 | 123.2 | 123.2 | 121.9 | 122.7 | 122.5 |
| θ(Χ3−Χ4−Χ5) | 122.9 | 123.4 | 123.3 | 122.3 | 122.9 | 122.9 |
| θ(Χ4−Χ5−Χ6) | 129 | 129.4 | 129.5 | 128.5 | 129.1 | 129.1 |
| θ(Ν2−Η10−Ο17) | 172.1 | 170.9 | 173.1 | 172.7 | 171.4 | 173.7 |
| θ(Η18−Ο17−Η19) | 105.2 | 105.4 | 105.5 | 105.1 | 105.3 | 105.5 |
| θ(Χ3−Η11−Ο20) | … | … | … | 165.4 | 165.5 | 165.6 |
| θ(Η21−Ο20−Η22) | … | … | … | 104.3 | 104.5 | 104.6 |
| θ(Χ1−Ν2−Χ3−Χ4) | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 |
| θ(Ν2−Χ3−Χ4−Χ5) | 180.1 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 |
| θ(Χ3−Χ4−Χ5−Χ6) | 0 | 0 | 0 | 0 | 0 | 0 |
| -E | 326.39380 | 327.49182 | 327.36690 | 402.6126 | 403.92125 | 403.76735 |
| Eint | 19.61 | 18.76 | 18.00 | 33.90 | 32.07 | 30.29 |
| μΜ | 1.8595 | 2.3063 | 2.3093 | 1.3112 | 1.878 | 1.8962 |
| RA | 3.0968 | 3.1039 | 3.1727 | 1.5716 | 1.5693 | 1.581 |
| RB | 1.1816 | 1.1787 | 1.1679 | 1.0083 | 1.0119 | 1.0052 |
| RC | 0.8681 | 0.8670 | 0.8663 | 0.6226 | 0.6236 | 0.6229 |
| η | 6.01* | … | … | 6.05* | … | … |
| μ | -8.26* | … | … | -7.83* | … | … |
(*Values are calculated at HF/6-31G* level of theory)
In each molecule in the cationic form, the water molecule can hydrogen bonded with the NH group of the Schiff base, which is oriented towards the extra cellular side of the membrane in bR before photon absorption. On the other hand, water molecule can hydrogen bonded with retinal CH group, on the opposite side of the molecule, oriented towards the cytoplasmic side of the membrane. Ab initio calculation for vibrational spectra of similar model Schiff base have already been reported [12]. Nina et. al. [2] have performed ab initio calculations using RHF/6-31G* for these molecules. The calculated geometrical parameters are compared with the semiempirical and molecular mechanics (MM) geometrical parameters of isolated NMP and NMP-water complexes. In the present study, the Post Hartree-Fock and density functional theory methods have been applied for NMP and NMB molecules and with water interactions of the above molecules. We have included more number of water molecules on both the sides for the above molecules. It is interesting to study the variation of structural parameters due to the formation of hydrogen bond with water molecule. The C1-N2 and N2-C3 bond lengths are found to be the same for the water interaction along CH side and there is a small decrease on these two bond lengths for the water interaction along NH side. More negative charges have been transferred to the N atom, which indicates the strong attraction between the atoms. The C3-C4 and N2-H9 bond lengths and C1-N2-H9 bond angle are increased and bond angles C1-N2-C3 and N2-C3-C4 are decreased while the interaction of water molecules along the NH side. The bond angles C1-N2-C3 and C3-C4-C5 are found to be decreased due to the interaction of water along CH side. All other structural parameters are practically unchanged while forming the hydrogen bond either at NH or at CH sides of the cationic form of NMP.
The calculated total energy for NMP and NMP-water complex are given in Table 1. The complex in which water molecule forms the hydrogen bond along the NH donor group of NMP has lower energy compared to the hydrogen bonded along CH group of the same molecule in all the three levels of theory and the relative energies are 5.70, 5.37 and 5.46 kcal/mol, respectively at MP2, B3LYP and B3PW91 levels of theory. Electron-correlated and DFT methods have predicted these energies very reasonably. The hydrogen bond lengths for the former case are shorter than the later case. The simultaneous presence of water molecules in each position is also optimized for the completion of the study. The presence of another water molecule does not make any appreciable change in the structural parameters of the complex. The chemical hardness and chemical potential values are calculated for these molecules at HF level of theory and it is found that NMP-water complex which is energetically more stable, does not have the higher chemical hardness value, and it is found to be increasing while forming the hydrogen bond with water molecule along CH and NH sides of the molecule. According to the principle of maximum hardness [13], the complex, which has the water molecule along NH side, should have maximum hardness value than the CH side of the NMP molecule. At the same time, the chemical hardness value is increasing for the complex molecule and decreasing for the complex, in which two water molecules are present. Therefore, according to the chemical hardness values, the complexes have more stability than the isolated molecule and the order of stability could not be predicted by these values. The interaction energy after BSSE correction shows that the complex having water molecule along NH side has higher interaction energy compared to that of CH side. The atomic charge distributions are calculated using NPA and MPA schemes and are presented in Table 3 and Table 4. The atomic charge of H9 is 0.541 for the NMP-water complex, where the water binds with NH side and the charge on H10 atoms of the complex is 0.380 where the water binds with the CH side. Therefore, more positive charge of H9 atom has strong attraction with the more negative charge of oxygen atom than the attraction between H10 and O14 atoms. This strong attraction reduces the hydrogen bond length and increases the interaction energy. This happened due to the charge transfer from the proton acceptor to the proton donor atoms.
NMB-water Interaction: Geometries and Energetics
In the next case, when the water binds with NH group of NMB molecule, the bond lengths C1-N2, N2-C3 and bond angles C1-N2-C3 and N2-C3-C4 are slightly decreased compared to that of isolated NMB cation. The hydrogen bond is formed almost linearly with the NH group of NMB cation, and the optimized bond angle N2-H10-O17 and hydrogen bond lengths are shown in Table 2. There is no significant change has been observed in the optimized structural parameters of the NMB-water along CH side hydrogen bond of complex compared to that of NMB. The water molecule makes bond angle with the CH group of NMB is less than the bond formed with the NH group of NMB. The calculated interaction energies show that the water molecule binds stronger in NMB-water along NH side than the CH side. Further it can be seen in the Table 1 and Table 2, the application of the DFT techniques result in the prediction of shorter single bonds and longer double bonds compared to HF method for these polyene Schiff base models, which confirms the results of Tajhorshid and Suhai [14] that the DFT calculations have overestimate the extend of the л-electronic delocalization in the polarized polyenes.
The chemical hardness and chemical potential are calculated for these complex molecules at HF/6-31G* level of theory. Similar to NMP-water complex, the NMB-water along CH side has higher chemical hardness value that is not the minimum energy structure. This study shows that the maximum hardness principle is not obeyed for both the complexes. It is understood that the maximum hardness principle is not able to predict the most stable isomer for hydrogen bonded complexes in many occasions and the same conclusion have been arrived for the number of cases of hydrogen bonded systems [15,16]. It is believed that water molecule can also interact with CH and CH2 groups of NMP molecules as in the structure (b), (c), and (d). Similarly, water molecule can also bind with the CH and CH3 groups of NMB molecules as in the structure 2b, 2c, and 2d. To confirm the above statement the topological parameters electron density (ρ) and laplacian of electron density (▽2 ρ) have been calculated using the “Atoms in molecules” approach. The values are given in the Table 5. The electron density values (table 5) indicate that the weak hydrogen bond is formed between the water and CH and CH2 groups of the NMP molecule and same type of hydrogen bond is formed between the water and CH and CH3 groups of NMB molecule. This has been shown in figure also. Normally the electron density values should be in the range of 0.02 to 0.04 a.u and laplacian of electron density should have positive values for the normal hydrogen bonding [17].
| Atom | NMP | NMP-Water along CH | Atom | NMP-Water along NH | NMP-2 Water | ||||
|---|---|---|---|---|---|---|---|---|---|
| NPA | MPA | NPA | MPA | NPA | MPA | NPA | MPA | ||
| C1 | -0.418 | -0.325 | -0.417 | -0.322 | C1 | -0.420 | -0.322 | -0.419 | -319 |
| N2 | -0.569 | -0.663 | -0.578 | -0.674 | N2 | -0.577 | -0.708 | -0.586 | -0.719 |
| C3 | 0.340 | 0.258 | 0.331 | 0.210 | C3 | 0.3324 | 0.237 | 0.317 | 0.194 |
| C4 | -0.412 | -0.257 | -0.410 | -0.265 | C4 | -0.398 | -0.259 | -0.397 | -0.270 |
| C5 | -0.161 | -0.295 | -0.173 | -0.309 | C5 | -0.194 | -0.310 | -0.203 | -0.325 |
| H6 | 0.240 | 0.228 | 0.242 | 0.231 | H6 | 0.235 | 0.219 | 0.237 | 0.221 |
| H7 | 0.256 | 0.250 | 0.250 | 0.240 | H7 | 0.250 | 0.238 | 0.244 | 0.230 |
| H8 | 0.256 | 0.250 | 0.250 | 0.240 | H8 | 0.250 | 0.238 | 0.244 | 0.230 |
| H9 | 0.461 | 0.453 | 0.455 | 0.446 | H9 | 0.498 | 0.541 | 0.492 | 0.535 |
| H10 | 0.250 | 0.300 | 0.282 | 0.380 | H10 | 0.254 | 0.286 | 0.273 | 0.363 |
| H11 | 0.267 | 268 | 0.260 | 0.259 | H11 | 0.274 | 0.279 | 0.267 | 0.270 |
| H12 | 0.233 | 0.254 | 0.242 | 0.264 | H12 | 0.229 | 0.244 | 0.239 | 0.258 |
| H13 | 0.2579 | 0.279 | 0.250 | 0.267 | H13 | 0.252 | 0.266 | 0.244 | 0.254 |
| O14 | … | … | -1.010 | -0.906 | O14 | -1.013 | -0.912 | -1.01 | -0.909 |
| H15 | … | … | 0.514 | 0.469 | H15 | 0.524 | 0.481 | 0.521 | 0.477 |
| H16 | … | … | 0.514 | 0.470 | H16 | 0.524 | 0.481 | 0.521 | 0.477 |
| O17 | … | … | -1.005 | -0.90 | |||||
| H18 | … | … | 0.510 | 0.466 | |||||
| H19 | … | … | 0.510 | 0.466 | |||||
| Atom | NMB | NMB-Water along CH | Atom | NMB-Water along NH | NMB-2 Water | ||||
|---|---|---|---|---|---|---|---|---|---|
| NPA | MPA | NPA | MPA | NPA | MPA | NPA | MPA | ||
| C1 | -0.417 | -0.32 | -0.422 | -0.335 | C1 | -0.419 | -0.318 | -0.424 | -0.333 |
| N2 | -0.578 | -0.672 | -0.581 | -0.681 | N2 | -0.585 | -0.717 | -0.587 | -0.727 |
| C3 | 0.342 | 0.262 | 0.333 | 0.219 | C3 | 0.328 | 0.242 | 0.320 | 0.200 |
| C4 | -0.442 | -0.315 | -0.437 | -0.319 | C4 | -0.429 | -0.321 | -0.424 | -0.323 |
| C5 | 0.059 | -0.053 | 0.046 | -0.060 | C5 | 0.028 | -0.067 | 0.016 | -0.073 |
| C6 | -0.710 | -0.562 | -0.713 | -0.574 | C6 | -0.705 | -0.556 | -0.708 | -0.569 |
| H7 | 0.253 | 0.246 | 0.245 | 0.233 | H7 | 0.248 | 0.234 | 0.239 | 0.222 |
| H8 | 0.237 | 0.224 | 254 | 0.249 | H8 | 0.232 | 0.215 | 0.249 | 0.241 |
| H9 | 0.253 | 0.246 | 0.245 | 0.233 | H9 | 0.247 | 0.235 | 0.239 | 0.222 |
| H10 | 0.458 | 0.449 | 0.451 | 0.441 | H10 | 0.495 | 0.539 | 0.488 | 0.533 |
| H11 | 0.244 | 0.292 | 0.274 | 0.372 | H11 | 0.236 | 0.279 | 0.265 | 0.360 |
| H12 | 0.263 | 0.257 | 0.258 | 0.249 | H12 | 0.272 | 0.271 | 0.267 | 0.274 |
| H13 | 0.259 | 0.270 | 0.254 | 0.262 | H13 | 0.253 | 0.259 | 0.248 | 0.251 |
| H14 | 0.227 | 0.191 | 0.239 | 0.209 | H14 | 0.224 | 0.187 | 0.237 | 0.206 |
| H15 | 0.276 | 0.243 | 0.268 | 0.231 | H15 | 0.270 | 0.235 | 0.262 | 0.223 |
| H16 | 0.276 | 0.243 | 0.268 | 0.231 | H16 | 0.270 | 0.235 | 0.262 | 0.223 |
| 017 | … | … | -0.008 | -0.900 | 017 | -1.011 | -0.910 | -1.001 | -0.907 |
| H18 | … | … | 0.514 | 0.469 | H18 | 0.523 | 0.479 | 0.520 | 0.476 |
| H19 | … | … | 0.514 | 0.469 | H19 | 0.523 | 0.479 | 0.520 | 0.476 |
| O20 | … | … | -1.004 | -0.896 | |||||
| H21 | … | … | 0.512 | 0.466 | |||||
| H22 | … | … | 0.512 | 0.466 | |||||
| Parameters | NMP | Parameters | NMB | ||
|---|---|---|---|---|---|
| ρ | ▽2ρ | ρ | ▽2ρ | ||
| Water along | Water along | ||||
| NH side | NH side | ||||
| O(14)-H(9) | 0.030 | 0.103 | O(17)-H(10) | 0.029 | 0.101 |
| O(14)-H(11) | 0.005 | 0.021 | O(17)-H(12) | 0.004 | 0.187 |
| Water along CH side | Water along CH side | ||||
| O(14)-H(10) | 0.019 | 0.065 | O(17)-H(11) | 0.016 | 0.058 |
| O(14)-H(12) | 0.005 | 0.021 | O(17)-H(8) | 0.007 | 0.027 |
| O(17)-H(14) | 0.005 | 0.022 | |||
| Water at both | |||||
| sides | Water at both | ||||
| O(14)-H(9) | 0.028 | 0.097 | sides | ||
| O(14)-H(11) | 0.004 | 0.020 | O(17)-H(10) | 0.027 | 0.095 |
| O(17)-H(10) | 0.018 | 0.062 | O(17)-H(12) | 0.004 | 0.020 |
| O(17)-H(12) | 0.004 | 0.019 | O(20)-H(8) | 0.006 | 0.026 |
| O(20)-H(11) | 0.015 | 0.055 | |||
| O(20)-H(14) | 0.005 | 0.021 | |||
Conclusion
The MP2/6-31G* levels of theory of ab initio and B3LYP/6-31G*, B3PW91/6-31G* levels of theory of DFT method have been employed to the NMP, NMB, NMP-water and NMB-water complexes to study the hydrogen bond interactions, structural parameters and relative stability of these complexes. The NPA and MPA schemes have been employed to study the charge distribution of these complexes and the strong attraction between the hydrogen bonded atoms along NH side increases the interaction energy more and hence more stability. The calculated chemical hardness values for these complexes could not predict the order of stability, as found in the minimum energy structure. The topological parameter indicates that weak hydrogen bond is formed between water, and CH, CH2, CH3 groups of NMP and NMB molecules.
Acknowledgement
One of the authors (R.K.) expresses his sincere thanks to UGC, New Delhi, for the award of FIP fellowship during the IX plan period and he is thankful to the Management and the Principal, Nallamuthu Gounder Mahalingam College, Pollachi, for allowing him to undergo this program. P.K. is thankful to DST for the financial support in the form of Research Project.
References
- Rothschild, K. J.; Argade, P. V.; Earnest, T. N.; Huang, K.-S.; London, E.; Liao, M.-J.; Bayley, H.; Khorana, H. G.; Herzfeld, J. J. Biol. Chem. 1982, 257, 8592.
- Nina, M.; Roux, B.; Smith, J. C. Biophys. J. 1995, 68, 25.
- De Groot, H.J.M.; Smith, S.O.; Courtin, J.; van der Berg, E.; Winkel, C.; Lugtenburg, J.; Griffin, R.G.; Herzfeld. J. Biochem. 1990, 29, 6873.
- Gat, Y.; Sheves, M. J. Am. Chem. Soc. 1993, 115, 3772. [CrossRef]
- Nina, M.; Smith, J.C.; Roux, B. J. Mol. Struct. (Theochem). 1993, 286, 231. [CrossRef]
- Møller, C.; Plesset, M.S. Phys. Rev. 1934, 46, 618. [CrossRef]
- Becke, A.D. Phys. Rev. A. 1988, 38, 3098. [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Phys. Rev. B. 1988, 37, 785. [CrossRef]
- Perdew, J.P.; Wang, Y. Phys. Rev. B. 1992, 45, 244. [CrossRef]
- Boys, S. F; Bernardi, F. Mol Phys. 1970, 19, 553. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery Jr., J.A.; Stratmann, R.E.; Burant, J.C.; S. Dapprich, J. M.; Millam, A. D.; Daniels, K. N.; Kudin, M. C.; Strain, Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Rega, N.; Salvador, P.; Dannenberg, J.J.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Ortiz, J.V.; Baboul, A.G.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.L.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Andres, J.L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian98, Revision A.11.2; Gaussian, Inc.: Pittsburgh PA, 2001. [Google Scholar]
- Deng, H.; Huang, L.; Groesbeek, M.; Lugtenburg, L.; Callender, R.H. J. Phys. Chem. 1994, 98, 4776. [CrossRef]
- Pearson, R.G. J. Chem. Edu. 1987, 64, 561. [CrossRef]
- Tajkhorshid, E.; Suhai, S. J. Phys. Chem. B 1999, 103, 5581.
- Arulmozhiraja, S.; Kolandaivel, P. Int. J. Quant. Chem. 1996, 64, 221.
- Kanakaraju, R.; Kolandaivel, P.; Gowenlock, B.G. J. Mol. Struct. (Theochem). 2002, 577, 121. [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A quantum Theory; Oxford University Press: Oxford, 1990. [Google Scholar]
© 2004 by MDPI (http://www.mdpi.org). Reproduction for noncommercial purposes permitted.
Share and Cite
MDPI and ACS Style
Kolandaivel, P.; Kanakaraju, R. Structure, Stability and Interaction Studies on Schiff Base Analogue Systems. Int. J. Mol. Sci. 2004, 5, 1-12. https://0-doi-org.brum.beds.ac.uk/10.3390/i5010001
AMA Style
Kolandaivel P, Kanakaraju R. Structure, Stability and Interaction Studies on Schiff Base Analogue Systems. International Journal of Molecular Sciences. 2004; 5(1):1-12. https://0-doi-org.brum.beds.ac.uk/10.3390/i5010001
Chicago/Turabian StyleKolandaivel, P., and R. Kanakaraju. 2004. "Structure, Stability and Interaction Studies on Schiff Base Analogue Systems" International Journal of Molecular Sciences 5, no. 1: 1-12. https://0-doi-org.brum.beds.ac.uk/10.3390/i5010001