Dipole Correlation of the Electronic Structures of theConformations of Water Molecule Evolving Through theNormal Modes of Vibrations Between Angular (C2v) to Linear(D∝h) Shapes

Abstract
:1. Introduction
1.1 Dipole moment as a probe of the electronic structure of water molecule.
2. Method of Computation
2.1 Localization and Computation of hybridization
2.2 Computation of molecular dipole and its partitioning into bond and lone pair moments





- 1)
- μchg , a contribution from net atomic charge densities
- 2)
- μhyb, a contribution from atomic polarization or hybridization resulting from mixing of the 2s and 2p orbitals.




3. Results and Discussion
3.1 The structural effect

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LMO’s | q = 90° | q = 100° | q = 104° | q = 104.1° |
| AO’s | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 |
| O2s | 0.2664 -0.0599 0.8540 | -0.2898 0.8353 0.0000 | -0.2995 0.8262 0.0000 | -0.2996 -0.8261 0.0000 |
| O2px | -0.5465 -0.0000 - 0.0000 | -0.5363 - 0.0000 0.0000 | -0.5328 0.0000 0.0000 | 0.5329 -0.0000 0.0000 |
| O2py | -0.0000 0.9975 0.0700 | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 |
| O2pz | -0.4414 -0.0362 0.5155 | 0.4403 0.5497 0.0000 | 0.4392 0.5634 0.0000 | 0.4391 -0.5636 0.0000 |
| H1s1 | 0.0249 -0.0000 -0.0000 | -0.6591 -0.0000 0.0000 | -0.6584 -0.0000 0.0000 | -0.6584 0.0000 0.0000 |
| H1s2 | 0.6595 0.0000 0.0000 | -0.0074 0.0000 0.0000 | -0.0011 -0.0000 0.0000 | -0.0009 -0.0000 0.0000 |
| LMO’s | q = 105° | q = 110° | q = 115° | q = 120° |
| AO’s | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 |
| O2s | -0.3018 -0.8240 0.0000 | -0.3145 -0.8111 0.0000 | -0.3286 -0.7962 0.0000 | -0.3431 -0.7802 0.0000 |
| O2px | 0.5321 0.0000 0.0000 | 0.5285 0.0000 0.0000 | 0.5249 0.0000 0.0000 | 0.5218 -0.0000 0.0000 |
| O2py | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 |
| O2pz | 0.4388 -0.5666 0.0000 | 0.4362 -0.5849 0.0000 | 0.4326 -0.6050 0.0000 | 0.4279 -0.6256 0.0000 |
| H1s1 | -0.6582 0.0000 0.0000 | -0.6569 0.0000 0.0000 | -0.6551 0.0000 0.0000 | -0.6530 0.0000 0.0000 |
| H1s2 | 0.0005 0.0000 0.0000 | 0.0078 0.0000 0.0000 | 0.0150 0.0000 0.0000 | 0.0219 -0.0000 0.0000 |
| LMO’s | q = 125° | q = 130° | q = 135° | q = 140° |
| AO’s | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 |
| O2s | -0.3594 -0.7611 0.0000 | -0.3773 -0.7387 0.0000 | -0.3974 -0.7115 0.0000 | -0.4190 -0.6803 0.0000 |
| O2px | 0.5189 -0.0000 0.0000 | 0.5161 -0.0000 0.0000 | 0.5136 -0.0000 0.0000 | 0.5112 -0.0000 0.0000 |
| O2py | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 |
| O2pz | 0.4216 -0.6487 0.0000 | 0.4134 -0.6741 0.0000 | 0.4024 -0.7026 0.0000 | 0.3889 -0.7329 0.0000 |
| H1s1 | -0.6504 0.0000 0.0000 | -0.6473 0.0000 0.0000 | -0.6437 0.0000 0.0000 | -0.6398 0.0000 0.0000 |
| H1s2 | 0.0290 -0.0000 0.0000 | 0.0362 -0.0000 0.0000 | 0.0436 -0.0000 0.0000 | 0.0512 0.0000 0.0000 |
| LMO’s | q = 145° | q = 150° | q = 155° | q = 160° |
| AO’s | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 |
| O2s | -0.4431 -0.6423 0.0000 | -0.4706 -0.5934 0.0000 | -0.4997 0.0000 -0.5345 | -0.5307 0.0000 -0.4596 |
| O2px | 0.5086 -0.0000 0.0000 | 0.5065 -0.0000 0.0000 | 0.5042 0.0000 0.0000 | 0.5023 0.0000 0.0000 |
| O2py | 0.0000 0.0000 1.0000 | 0.0000 0.0000 1.0000 | 0.0000 1.0000 0.0000 | 0.0000 1.0000 0.0000 |
| O2pz | 0.3712 -0.7664 0.0000 | 0.3468 -0.8049 0.0000 | 0.3160 0.0000 -0.8452 | 0.2747 0.0000 -0.8881 |
| H1s1 | -0.6353 0.0000 0.0000 | -0.6302 0.0001 0.0000 | -0.6248 0.0000 -0.0000 | -0.6192 0.0000 -0.0000 |
| H1s2 | 0.0594 -0.0000 0.0000 | 0.0676 0.0000 0.0000 | 0.0763 0.0000 0.0000 | 0.0846 0.0000 0.0000 |
| LMO’s | q = 165° | q = 170° | q = 175° | q = 180° |
| AO’s | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 | s(O–H) l.p.O1 l.p.O2 |
| O2s | -0.5608 0.0000 -0.3692 | -0.5876 0.0000 -0.2588 | -0.6060 -0.1328 0.0000 | 0.6126 0.0000 0.0000 |
| O2px | 0.5004 0.0000 0.0000 | 0.4989 0.0000 0.0000 | 0.4981 -0.0000 0.0000 | 0.4977 0.0000 0.0000 |
| O2py | 0.0000 1.0000 0.0000 | 0.0000 1.0000 0.0000 | 0.0000 0.0000 1.0000 | 0.0000 1.0000 0.0000 |
| O2pz | 0.2229 0.0000 -0.9293 | 0.1575 0.0000 -0.9659 | 0.0811 -0.9911 0.0000 | 0.0000 0.0000 1.0000 |
| H1s1 | -0.6138 0.0000 -0.0000 | -0.6092 0.0000 0.0000 | -0.6060 0.0000 0.0000 | -0.1054 0.0000 0.0000 |
| H1s2 | 0.0926 0.0000 0.0000 | 0.0994 0.0000 0.0000 | 0.1038 0.0000 0.0000 | 0.6049 0.0000 0.0000 |
| ÐHOH angle | Hybridization of bond pair | Hybridization of lone pair | ÐHOH angle | Hybridization of bond pair | Hybridization of lone pair |
|---|---|---|---|---|---|
| 90 | sp6.9 | sp0.36 | 135 | sp2.7 | sp0.97 |
| 100 | sp5.73 | sp0.43 | 140 | sp2.4 | sp1.2 |
| 104 | sp5.3 | sp0.46 | 145 | sp2 | sp1.4 |
| 104.1 | sp5.3 | sp0.46 | 150 | sp1.7 | sp1.8 |
| 105 | sp5.2 | sp0.48 | 155 | sp1.4 | sp2.5 |
| 110 | sp4.7 | sp0.52 | 160 | sp1.2 | sp3.7 |
| 115 | sp4.3 | sp0.59 | 165 | sp0.95 | sp6.3 |
| 120 | sp3.9 | sp0.62 | 170 | sp0.77 | sp13.9 |
| 125 | sp3.5 | sp0.71 | 175 | sp0.71 | sp55.6 |
| 130 | sp3.1 | sp0.83 | 180 | sp0.67 | p |
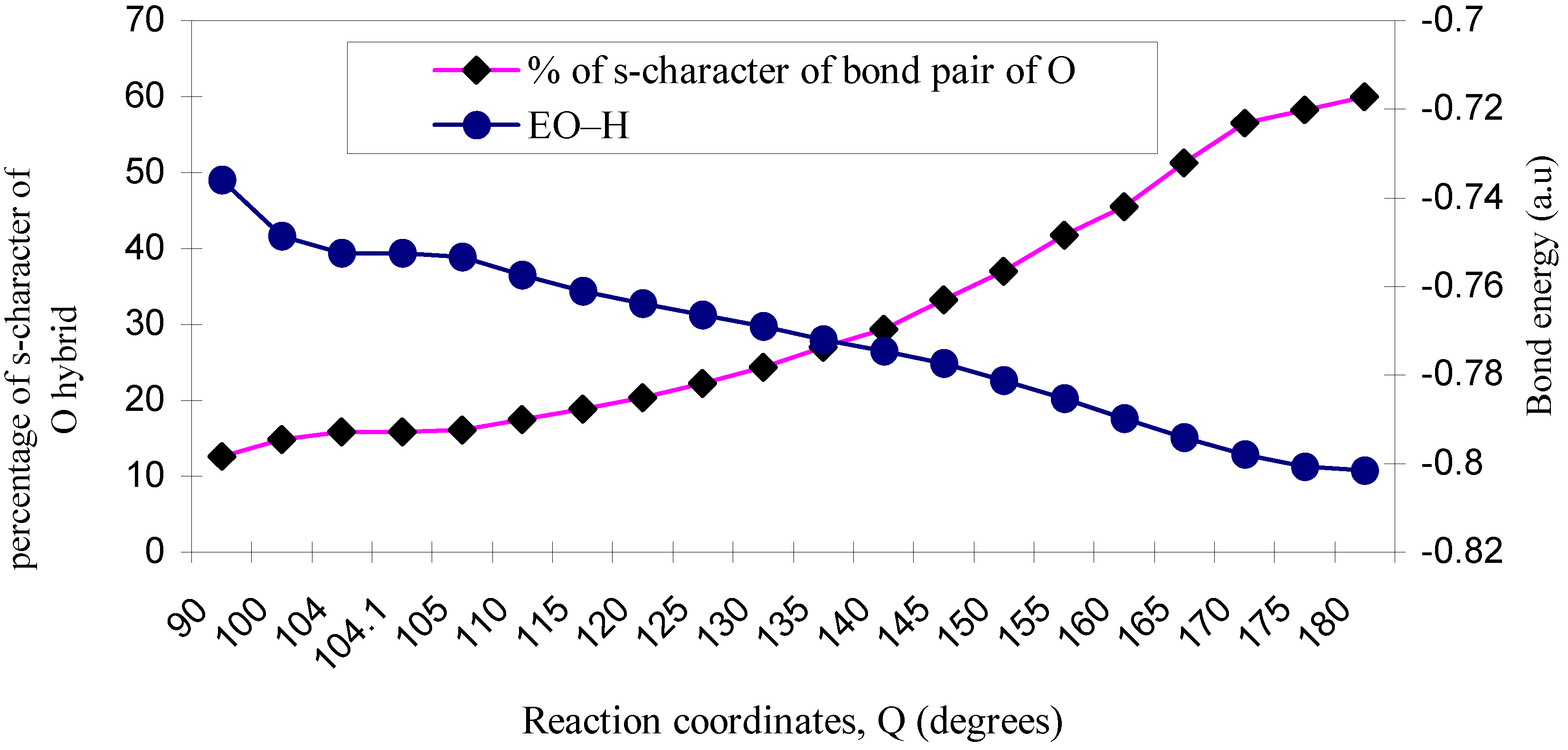
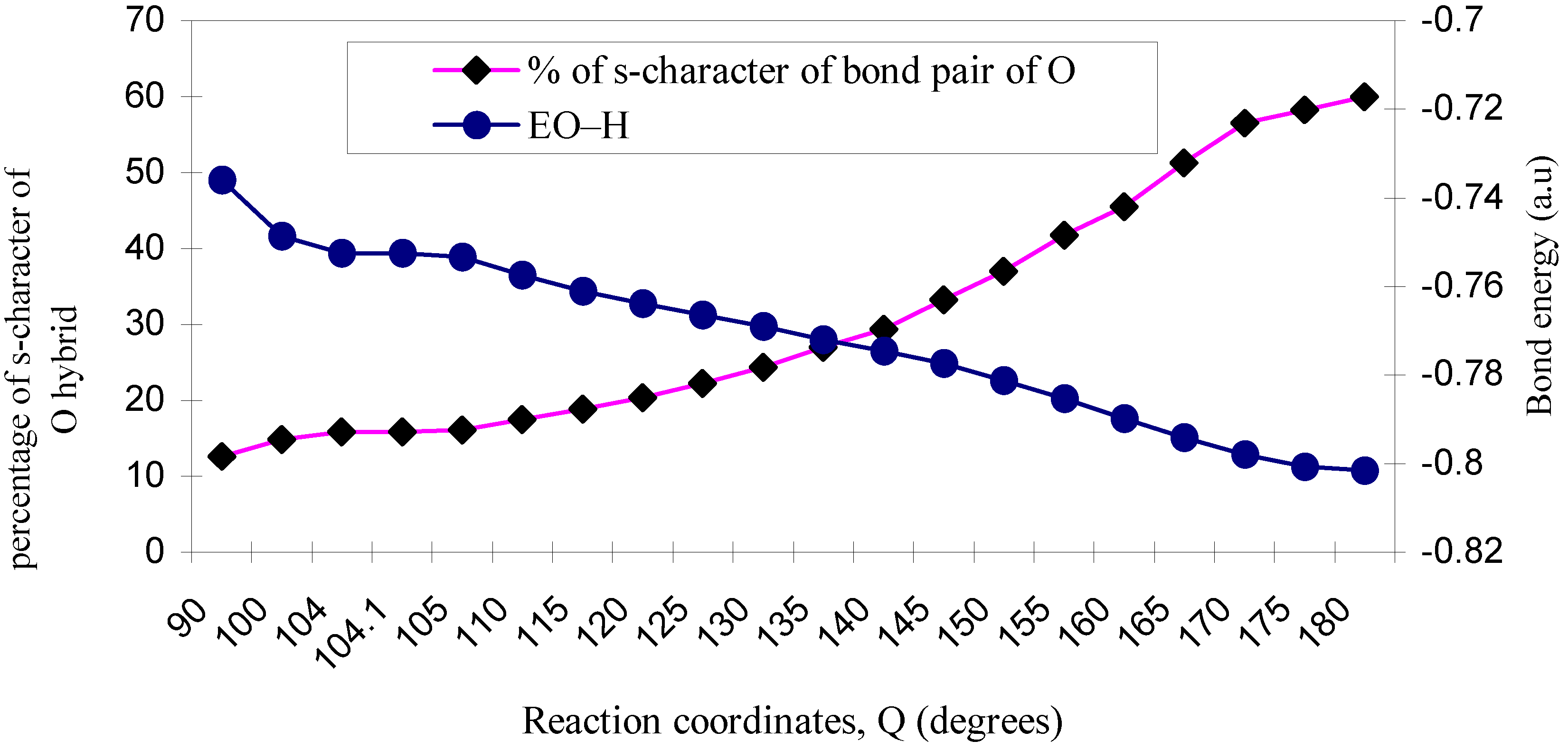
| ÐHOH | O–H bond length | EO–H | % of s-character of bond pair of O |
|---|---|---|---|
| 90 | 1.035 | -0.7361 | 12.66 |
| 100 | 1.030 | -0.7487 | 14.86 |
| 104 | 1.029 | -0.7526 | 15.85 |
| 104.1 | 1.029 | -0.7526 | 15.9 |
| 105 | 1.029 | -0.7534 | 16.1 |
| 110 | 1.027 | -0.7574 | 17.5 |
| 115 | 1.026 | -0.7610 | 18.9 |
| 120 | 1.025 | -0.7638 | 20.41 |
| 125 | 1.024 | -0.7665 | 22.2 |
| 130 | 1.023 | -0.7691 | 24.4 |
| 135 | 1.021 | -0.7720 | 27.03 |
| 140 | 1.020 | -0.7746 | 29.4 |
| 145 | 1.020 | -0.7774 | 33.3 |
| 150 | 1.017 | -0.7813 | 37.04 |
| 155 | 1.016 | -0.7852 | 41.7 |
| 160 | 1.013 | -0.7898 | 45.5 |
| 165 | 1.012 | -0.7940 | 51.2 |
| 170 | 1.011 | -0.7979 | 56.5 |
| 175 | 1.010 | -0.8007 | 58.3 |
| 180 | 1.010 | -0.8015 | 60.0 |
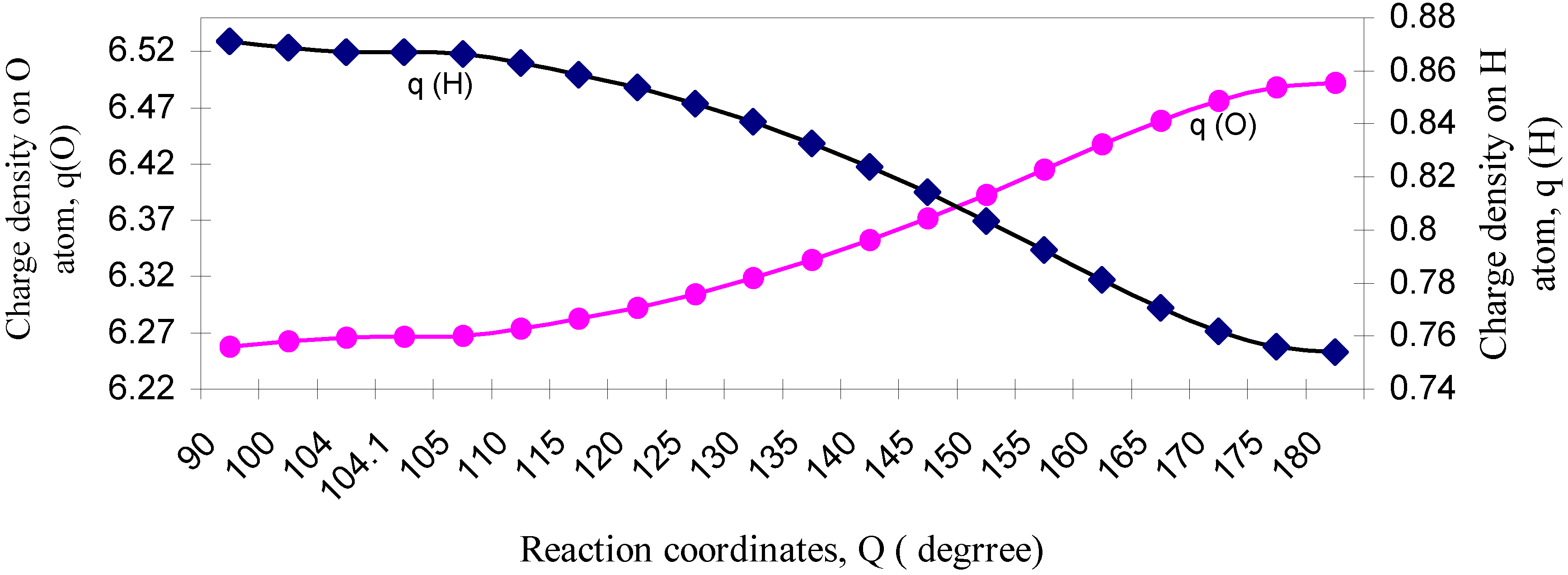
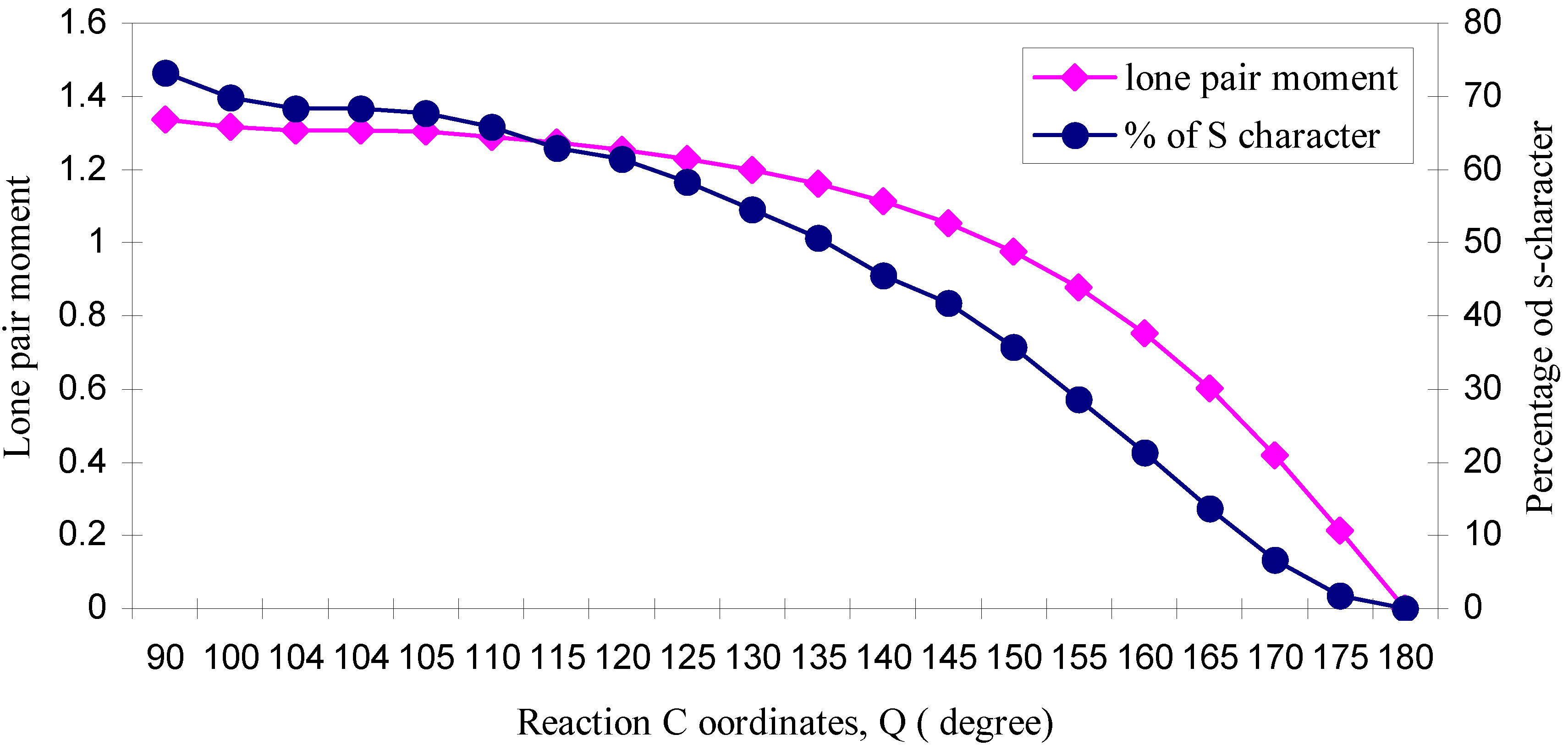
| ÐHOH | q (O) | q (H) | Dipole Moment (D) | Bond Moment (D) | Lone pair Moment (D) | % of s-character of lone pair of O |
|---|---|---|---|---|---|---|
| 90 | 6.2578 | 0.8711 | 2.2429 | 0.9063 | 1.3366 | 73.26 |
| 100 | 6.2623 | 0.8688 | 2.1497 | 0.8342 | 1.3155 | 69.79 |
| 104 | 6.2661 | 0.8669 | 2.1154 | 0.8097 | 1.3057 | 68.3 |
| 104.1 | 6.2662 | 0.8669 | 2.1147 | 0.8092 | 1.3055 | 68.3 |
| 105 | 6.2671 | 0.8664 | 2.1069 | 0.8039 | 1.3030 | 67.74 |
| 110 | 6.2739 | 0.8631 | 2.0646 | 0.7751 | 1.2895 | 65.8 |
| 115 | 6.2825 | 0.8587 | 2.0213 | 0.7483 | 1.2730 | 63.0 |
| 120 | 6.2926 | 0.8537 | 1.9740 | 0.7205 | 1.2535 | 61.5 |
| 125 | 6.3047 | 0.8476 | 1.9216 | 0.6922 | 1.2294 | 58.3 |
| 130 | 6.3186 | 0.8407 | 1.8613 | 0.6619 | 1.1994 | 54.5 |
| 135 | 6.3347 | 0.8326 | 1.7899 | 0.6284 | 1.1615 | 50.7 |
| 140 | 6.3522 | 0.8239 | 1.7046 | 0.5904 | 1.1142 | 45.5 |
| 145 | 6.3715 | 0.8142 | 1.6008 | 0.5474 | 1.0534 | 41.7 |
| 150 | 6.3929 | 0.8035 | 1.4722 | 0.4969 | 0.9753 | 35.7 |
| 155 | 6.4150 | 0.7925 | 1.3153 | 0.4385 | 0.8768 | 28.6 |
| 160 | 6.4376 | 0.7812 | 1.1224 | 0.3698 | 0.7526 | 21.3 |
| 165 | 6.4584 | 0.7708 | 0.8922 | 0.2909 | 0.6013 | 13.7 |
| 170 | 6.4761 | 0.7619 | 0.6207 | 0.2015 | 0.4192 | 6.7 |
| 175 | 6.4879 | 0.7561 | 0.3176 | 0.1033 | 0.2143 | 1.8 |
| 180 | 6.4920 | 0.7540 | 0.0000 | 0.0000 | 0.0000 | 0.00 |
3.2 Hybridization
| ÐHOH angle | EU | EJ | EK | EO |
|---|---|---|---|---|
| 90 | -31.58767 | 16.18246 | -2.18136 | -17.58657 |
| 100 | -31.59862 | 16.20565 | -2.18183 | -17.57480 |
| 104 | -31.61243 | 16.22516 | -2.18344 | -17.57071 |
| 104.1 | -31.61266 | 16.22546 | -2.18347 | -17.57067 |
| 105 | -31.61634 | 16.23043 | -2.18391 | -17.56982 |
| 110 | -31.64297 | 16.26535 | -2.18724 | -17.56486 |
| 115 | -31.67830 | 16.31029 | -2.19179 | -17.55980 |
| 120 | -31.72037 | 16.36259 | -2.19726 | -17.55504 |
| 125 | -31.77149 | 16.42557 | -2.20401 | -17.54993 |
| 130 | -31.83098 | 16.49839 | -2.21196 | -17.54455 |
| 135 | -31.89983 | 16.58259 | -2.22127 | -17.53851 |
| 140 | -31.97510 | 16.67408 | -2.23160 | -17.53262 |
| 145 | -32.05862 | 16.77533 | -2.24331 | -17.52660 |
| 150 | -32.15127 | 16.88867 | -2.25649 | -17.51909 |
| 155 | -32.24723 | 17.00558 | -2.27048 | -17.51213 |
| 160 | -32.34468 | 17.12527 | -2.28491 | -17.50432 |
| 165 | -32.43543 | 17.23637 | -2.29869 | -17.49775 |
| 170 | -32.51231 | 17.33083 | -2.31056 | -17.49204 |
| 175 | -32.56366 | 17.39420 | -2.31853 | -17.48799 |
| 180 | -32.58154 | 17.41610 | -2.32135 | -17.48679 |
| ÐHOH angle | EU | EJ | EK | EH |
|---|---|---|---|---|
| 90 | -0.55637 | 0.28454 | -0.14227 | -0.41410 |
| 100 | -0.55495 | 0.28307 | -0.14154 | -0.41342 |
| 104 | -0.55374 | 0.28185 | -0.14092 | -0.41281 |
| 104.1 | -0.55372 | 0.28183 | -0.14092 | -0.41281 |
| 105 | -0.55342 | 0.28152 | -0.14076 | -0.41266 |
| 110 | -0.55127 | 0.27933 | -0.13967 | -0.41161 |
| 115 | -0.54850 | 0.27654 | -0.13827 | -0.41023 |
| 120 | -0.54528 | 0.27331 | -0.13665 | -0.40862 |
| 125 | -0.54142 | 0.26945 | -0.13472 | -0.40669 |
| 130 | -0.53696 | 0.26503 | -0.13251 | -0.40444 |
| 135 | -0.53182 | 0.25998 | -0.12999 | -0.40183 |
| 140 | -0.52625 | 0.25455 | -0.12728 | -0.39898 |
| 145 | -0.52010 | 0.24864 | -0.12432 | -0.39578 |
| 150 | -0.51323 | 0.24212 | -0.12106 | -0.39217 |
| 155 | -0.50618 | 0.23551 | -0.11776 | -0.38843 |
| 160 | -0.49898 | 0.22886 | -0.11443 | -0.38455 |
| 165 | -0.49233 | 0.22279 | -0.11140 | -0.38094 |
| 170 | -0.48668 | 0.21772 | -0.10886 | -0.37782 |
| 175 | -0.48290 | 0.21435 | -0.10718 | -0.37573 |
| 180 | -0.48160 | 0.21319 | -0.10660 | -0.37501 |
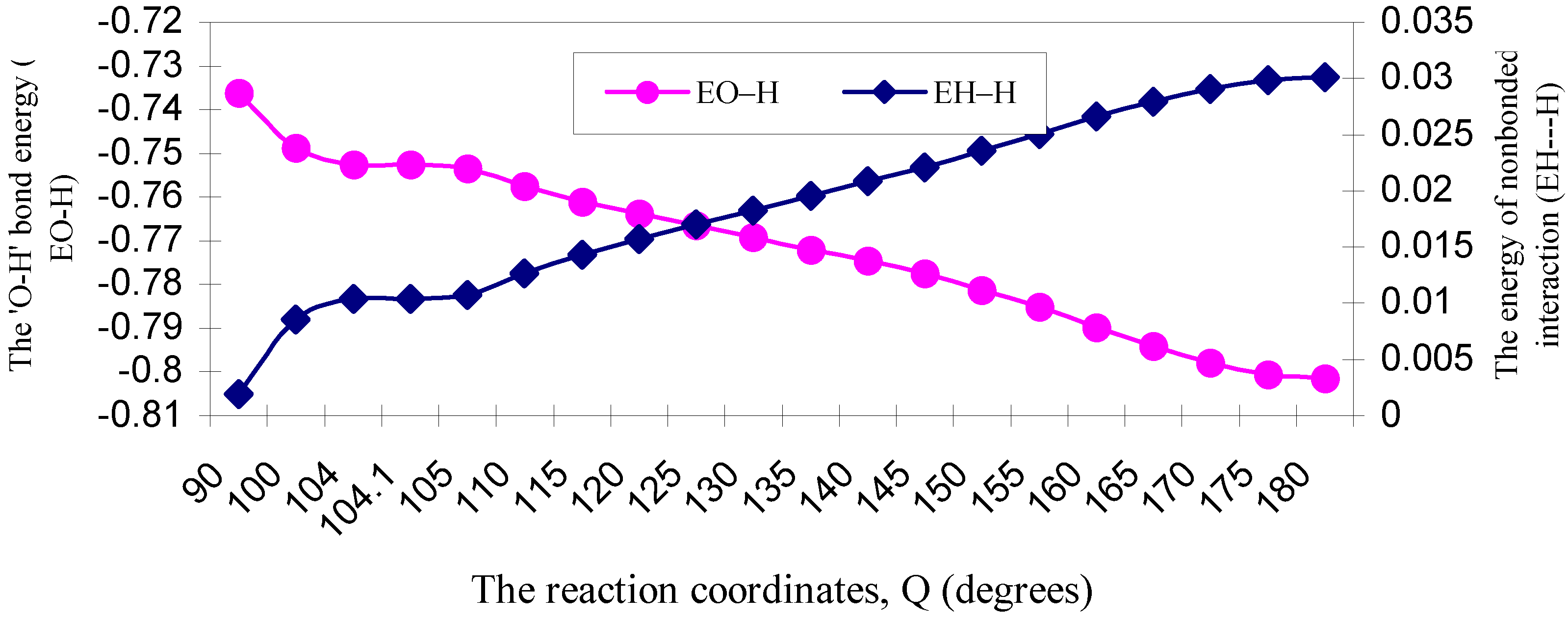
| ÐHOH angle | EJO–H | ENO–H | EVO–H | EKO–H | ERO–H | EO–H |
|---|---|---|---|---|---|---|
| 90 | 2.58453 | 3.06787 | -5.44511 | -0.23210 | -0.71133 | -0.73614 |
| 100 | 2.58869 | 3.08253 | -5.45976 | -0.23371 | -0.72650 | -0.74875 |
| 104 | 2.58656 | 3.08570 | -5.46024 | -0.23385 | -0.73074 | -0.75257 |
| 104.1 | 2.58639 | 3.08552 | -5.45996 | -0.23384 | -0.73074 | -0.75263 |
| 105 | 2.58535 | 3.08552 | -5.45904 | -0.23381 | -0.73140 | -0.75338 |
| 110 | 2.58170 | 3.09153 | -5.46031 | -0.23383 | -0.73649 | -0.75740 |
| 115 | 2.57411 | 3.09454 | -5.45600 | -0.23343 | -0.74027 | -0.76105 |
| 120 | 2.56493 | 3.09756 | -5.45013 | -0.23284 | -0.74333 | -0.76381 |
| 125 | 2.55345 | 3.10059 | -5.44241 | -0.23199 | -0.74617 | -0.76653 |
| 130 | 2.53982 | 3.10362 | -5.43289 | -0.23089 | -0.74879 | -0.76913 |
| 135 | 2.52546 | 3.10969 | -5.42514 | -0.22968 | -0.75234 | -0.77201 |
| 140 | 2.50764 | 3.11275 | -5.41224 | -0.22803 | -0.75475 | -0.77463 |
| 145 | 2.48586 | 3.11275 | -5.39379 | -0.22586 | -0.75636 | -0.77740 |
| 150 | 2.46654 | 3.12193 | -5.38457 | -0.22384 | -0.76139 | -0.78133 |
| 155 | 2.44276 | 3.12500 | -5.36714 | -0.22117 | -0.76461 | -0.78516 |
| 160 | 2.42161 | 3.13426 | -5.35680 | -0.21866 | -0.77024 | -0.78983 |
| 165 | 2.39873 | 3.13735 | -5.34049 | -0.21580 | -0.77381 | -0.79402 |
| 170 | 2.37940 | 3.14046 | -5.32722 | -0.21329 | -0.77723 | -0.79788 |
| 175 | 2.36691 | 3.14357 | -5.31956 | -0.21164 | -0.77996 | -0.80068 |
| 180 | 2.36200 | 3.14357 | -5.31562 | -0.21098 | -0.78052 | -0.80155 |
| ÐHOH angle | EJH–H | ENH–H | EVH–H | EKH–H | ERH–H | EH–H |
|---|---|---|---|---|---|---|
| 90 | 0.26717 | 0.36155 | -0.61344 | -0.00076 | -0.01257 | 0.00195 |
| 100 | 0.24857 | 0.33534 | -0.57219 | -0.00000 | -0.00318 | 0.00854 |
| 104 | 0.24142 | 0.32632 | -0.55695 | -0.00000 | -0.00044 | 0.01035 |
| 104.1 | 0.24129 | 0.32614 | -0.55665 | -0.00000 | -0.00039 | 0.01039 |
| 105 | 0.23968 | 0.32417 | -0.55325 | -0.00000 | 0.00016 | 0.01076 |
| 110 | 0.23131 | 0.31456 | -0.53601 | -0.00006 | 0.00285 | 0.01265 |
| 115 | 0.22305 | 0.30581 | -0.51947 | -0.00023 | 0.00516 | 0.01432 |
| 120 | 0.21521 | 0.29810 | -0.50418 | -0.00048 | 0.00705 | 0.01570 |
| 125 | 0.20761 | 0.29132 | -0.48984 | -0.00082 | 0.00875 | 0.01702 |
| 130 | 0.20024 | 0.28539 | -0.47638 | -0.00125 | 0.01028 | 0.01828 |
| 135 | 0.19321 | 0.28051 | -0.46410 | -0.00176 | 0.01172 | 0.01958 |
| 140 | 0.18630 | 0.27606 | -0.45223 | -0.00235 | 0.01306 | 0.02084 |
| 145 | 0.17939 | 0.27199 | -0.44061 | -0.00308 | 0.01441 | 0.02210 |
| 150 | 0.17304 | 0.26935 | -0.43071 | -0.00389 | 0.01581 | 0.02360 |
| 155 | 0.16675 | 0.26674 | -0.42082 | -0.00483 | 0.01718 | 0.02502 |
| 160 | 0.16114 | 0.26522 | -0.41254 | -0.00580 | 0.01856 | 0.02658 |
| 165 | 0.15600 | 0.26370 | -0.40478 | -0.00679 | 0.01979 | 0.02792 |
| 170 | 0.15188 | 0.26271 | -0.39867 | -0.00768 | 0.02083 | 0.02907 |
| 175 | 0.14926 | 0.26221 | -0.39485 | -0.00826 | 0.02151 | 0.02987 |
| 180 | 0.14832 | 0.26196 | -0.39342 | -0.00848 | 0.02174 | 0.03012 |
3.3 Dipole moment



3. 4 Energy partitioning analysis and the origin of barrier
One-center effect

Two-center effect
4. Conclusion


References
- Feynman, R. P.; Leighton, R. B.; Sands, M. The Feynman Lectures on Physics; Addison-Wesley Publishing Company, 1964. [Google Scholar]
- Kuwajima, K. The Role of the Molten Globule State in Protein Folding: The Search for a Universal View of Folding. Proc. Ind. Natl. Sci. Acad. 2002, 68A, 333–40. [Google Scholar]
- Fecko, C. J.; Eaves, J. D.; Loparo, J. J.; Tokmakoff, A.; Geissler, P. L. Ultrafast Hydrogen- Bond Dynamics in the Infrared Spectroscopy of Water. Science. 2003, 301, 1698–1702. [Google Scholar] [CrossRef]
- Myneni, S.; Luo, Y.; Näslund, L. Å.; Cavalleri, M.; Ojamäc, L.; Ogasawara, H.; Pelmenschikov, A.; Wernet, Ph.; Väterlein, P.; Heske, C.; Hussain, Z.; Pettersson, L. G. M.; Nilsson, A. Spectroscopic Probing of Local Hydrogen-Bonding Structures in Liquid Water. J. Phys. Condens. Matter. 2002, 14, L213–L219. [Google Scholar] [CrossRef]
- Bakker, H. J.; Nienhuys, H.-K. Delocalization of Protons in Liquid Water. Science. 2002, 297, 587–590. [Google Scholar] [CrossRef]
- Lykos, P. Modeling the Hydrogen Bond within Molecular Dynamics. J. Chem. Edu. 2004, 81, 147–149. [Google Scholar] [CrossRef]
- Gordon, T. H.; Hura, G. Water Structure from Scattering Experiments and Simulation. Chem. Rev. 2002, 102, 2651–2670. [Google Scholar] [CrossRef]
- Marchi, R. P.; Eyring, H. Application of Significant Structure Theory to Water. J. Phys. Chem. 1964, 68, 221–228. [Google Scholar] [CrossRef]
- Stevenson, D. P. On the Monomer Concentration in Liquid Water. J. Phys. Chem. 1965, 69, 2145–2152. [Google Scholar] [CrossRef]
- Angell, C. A. Two-State Thermodynamics and Transport Properties for Water as Zeroth- Order Results of a Bond Lattice Model. J. Phys. Chem. 1971, 75, 3698–3705. [Google Scholar] [CrossRef]
- Schulson, E. M. The Structure and Mechanical Behaviour of Ice. J. Minerals, Metals and Materials Soc. 1999, 51, 21–27. [Google Scholar] [CrossRef]
- Laing, M. No Rabbit Ears on Water. J. Chem. Edu. 1987, 64, 124–128. [Google Scholar] [CrossRef]
- Martin, R. B. Localized and Spectroscopic Orbitals: Squirrel Ears on Water. J. Chem. Edu. 1988, 65, 668–670. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond, 3rd Edition. edCornell University Press: Ithaca (NY), 1960. [Google Scholar]
- Lennard-Jones, J. E.; Pople, J. A. The Molecular Orbital Theory of Chemical Valency IV. The Significance of Equivalent Orbitals. Proc. Roy. Soc (London). 1950, A202, 166–180. [Google Scholar] [CrossRef]
- Gillespie, R. J.; Nyholm, R. S. Inorganic Stereochemistry. Quart. Rev. 1957, 11, 339–380. [Google Scholar] [CrossRef]
- Jr, G. R. The Magic of Chemistry. Chem. Edu. 1983, 60, 741–743. [Google Scholar] [CrossRef]
- Sweigart, D.A. Lone pair orbital energies in group Vl and VII hydrides. J. Chem. Edu. 1973, 50, 322. [Google Scholar] [CrossRef]
- Levine, I. N. Quantum Chemistry, 4th Edition. ed; Prentice-Hall International, Inc: Englewood Cliffs, N.J., U.S.A., 1991; p. 474. [Google Scholar]
- Coulson, C. A. Valence, 2nd Edition. ed; Oxford University Press: New York, 1961. [Google Scholar]
- Dewar, M. J. S. The Molecular Orbital Theory of Organic Chemistry; McGraw Hill, New York, 1969. [Google Scholar]
- Ghosh, D.C.; Bhattacharyya, S. Computation of Quantum Mechanical Hybridization and Dipole Correlation of the Electronic Structure of the F3B–NH3 supermolecule. Int. J. Quantum. Chem. 2005, 105, 270–279. [Google Scholar] [CrossRef]
- Lewis, G.N. Valence and the Structure of Atoms and Molecules; The Chemical Catalog Company: New York, 1923. [Google Scholar]
- Bent, H.A. Distribution of Atomic s character in Molecules and Its Chemical Implications. J. Chem. Edu. 1960, 37, 616–624. [Google Scholar] [CrossRef]
- (a) Bent, H.A. Electronegativities From Comparison of Bond Lengths in AH and AH+. J. Chem. Phys. 1960, 33, 1258–1259. [Google Scholar] (b) Bent, H.A. Correlation of Bond Shortening by Electronegative Substituents with orbital hybridization. J. Chem. Phys. 1960, 33, 1259–1260. [Google Scholar]
- Roothaan, C.C.J. New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Gill, P.M.W. Hartree-Fock-Wigner Models for Computational Chemistry. In Proc. 2nd Asian Pacific Conference on Theoretical and Computational Chemistry, Bangkok, May 2-6, 2005; p. 3.
- (a) Glendening, E.D.; Weinhold, F. Natural Resonance Theory: I. General Formalism. J. Comput. Chem 1998, 19, 593–609. [Google Scholar] [CrossRef] (b) Glendening, E.D.; Weinhold, F. Natural Resonance Theory: II. Natural Bond Order and Valency. J. Comput. Chem 1998, 19, 610–627. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K; Weinhold, F. Natural Resonance Theory: III. Chemical Applications. J. Comput. Chem. 1998, 19, 628–646. [Google Scholar] [CrossRef]
- Pipek, J.; Mezey, P.G. A Fast Intrinsic Localization Procedure Applicable for ab initio and Semiempirical Linear Combination of Atomic Orbital Wavefunctions. J. Chem. Phys. 1989, 90, 4916–4926. [Google Scholar] [CrossRef]
- Liu, S.; Perez-Jorda, J.M.; Yang, W. Nonorthogonal Localized Molecular Orbitals in Electronic Structure Theory. J. Chem. Phys. 2000, 112, 1634–1644. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chemistry Edu. Research and Practice in Europe. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Ghosh, D.C.; Jana, J.; Biswas, R. Quantum Chemical Study of the Umbrella Inversion of the Ammonia Molecule. Int. J. Quantum. Chem. 2000, 80, 1–26. [Google Scholar] [CrossRef]
- Ghosh, D.C.; Jana, J.; Bhattacharyya, S. Density Functional and Molecular Orbital Study of Physical Process of Inversion of Nitrogen Trifluoride (NF3) Molecule. Int. J. Quantum. Chem. 2002, 87, 111–134. [Google Scholar] [CrossRef]
- Ghosh, D.C.; Chakraborty, A. A Localized Molecular Orbital Study of the Structure and Bonding of Ozone. Indian J. Chem. 2002, 41A, 1995–2007. [Google Scholar]
- Trindle, C.; Sinanoğlu, O. Semiempirical Method for the Determination of Localized Orbitals in Molecules. J. Chem. Phys. 1968, 49, 65–71. [Google Scholar] [CrossRef]
- Trindle, C.; Sinanoğlu, O. Local Orbitals and Bond Index Characterization of Hybridization. J. Am. Chem. Soc. 1969, 91, 853–858. [Google Scholar] [CrossRef]
- Jensen, P. The Potential Energy Surface for the Electronic Ground State of the Water Molecule Determined from Experimental Data Using a Variational Approach. J. Mol. Spect. 1989, 133, 438–460. [Google Scholar] [CrossRef]
- Tarczay, G.; Császár, A.G.; Klopper, W.; Szalay, V.; Allen, W.D.; Schaefer III, H.F. The Barrier to Linearity of Water. J. Chem. Phys. 1999, 110, 11971–11981. [Google Scholar] [CrossRef]
- Fischer, H.; Kollmar, H. Energy partitioning with the CNDO method. Theoret. Chim. Acta. 1970, 16, 163–174. [Google Scholar] [CrossRef]
- Ghosh, D.C. A Density Functional and Molecular Orbital Study of the Methylamine Conformers. Int. Electronic. J. Mol. Design. 2005, 4, 31–58. [Google Scholar]
- Ghosh, D.C.; Bhattacharyya, S. Molecular Orbital and Density Functional Study of the Formation, Charge Transfer, Bonding and the Conformational Isomerism of the Boron Trifluoride (BF3) and Ammonia (NH3) Donor-Acceptor Complex. Int. J. Mol. Sci. 2004, 5, 239–264. [Google Scholar] [CrossRef]
- (a) Pople, J.A.; Santry, D.P.; Segal, G.A. Approximate Self-Consistent Molecular Orbital Theory.I. Invariant Procedures. J. Chem. Phys. 1965, 43, S129–S135. [Google Scholar] [CrossRef] (b) Pople, J.A.; Santry, D.P.; Segal, G.A. Approximate Self-Consistent Molecular Orbital Theory.II. Calculations with Complete Neglect of Differential Overlap. J. Chem. Phys. 1965, 43, S136–S151. [Google Scholar]
- Pople, J.A.; Beveridge, D.L. Approximate Molecular Orbital Theory; McGraw Hill Book Company: New York, 1970. [Google Scholar]
- Davis, D.W. All Valency Electron Molecular Orbital Calculations I. Dipole Moments, Spin Densities and 19F Shielding Constants in Some Fluorobenzenes and Fluoronitrobenzenes. Mol. Phys. 1967, 13, 465–477. [Google Scholar] [CrossRef]
- Bloor, J. E.; Breen, D. L. Valence-Shell Calculations on Polyatomic Molecules II. CNDO SCF Calculations on Monosubstituted Benzenes. J. Phys. Chem. 1968, 72, 716–722. [Google Scholar] [CrossRef]
- Bloor, J. E.; Breen, D. L. Valence-Shell Calculations on Polyatomic Molecules I. CNDO SCF Calculations on Nitrogen and Oxygen Heterocyclics. J. Am. Chem. Soc. 1967, 89, 6835–6841. [Google Scholar] [CrossRef]
- Bloor, J.E.; Gilson, B.R.; Billingsley, F.P. Valence-Shell Calculations on Polyatomic Molecules III. The Calculation of Dipole Moments and the Structure of Sydnone. Theo. Chim. Acta. 1968, 12, 360–372. [Google Scholar] [CrossRef]
- Hush, N. S.; Yandle, J. R. Chem. Phys. Lett. 1967, 1, 493. [CrossRef]
- Pople, J.A.; Gordon, M. Molecular Orbital Theory of the Electronic Structure of Organic Compounds. I. Substituent Effects and Dipole Moments. J. Am. Chem. Soc. 1967, 89, 4253–4261. [Google Scholar] [CrossRef]
- Sichel, J. M.; Whitehead, M. A. Semi-Empirical All Valence Electrons SCF-MO-CNDO Theory IV. Dipole Moments. Theo. Chim. Acta. 1968, 11, 254–262. [Google Scholar] [CrossRef]
- Roothaan, C. C. J. A Study of Two-Center Integrals Useful in Calculations on Molecular Structure I. J. Chem. Phys. 1951, 19, 1445–1457. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Hall, M.B. Valence Shell Electron Pair Repulsions and the Pauli Exclusion Principle. J. Am. Chem. Soc. 1978, 100, 6333–6338. [Google Scholar] [CrossRef]
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ghosh, D.C.; Chakraborty, A. Dipole Correlation of the Electronic Structures of theConformations of Water Molecule Evolving Through theNormal Modes of Vibrations Between Angular (C2v) to Linear(D∝h) Shapes. Int. J. Mol. Sci. 2006, 7, 71-96. https://0-doi-org.brum.beds.ac.uk/10.3390/i7030071
Ghosh DC, Chakraborty A. Dipole Correlation of the Electronic Structures of theConformations of Water Molecule Evolving Through theNormal Modes of Vibrations Between Angular (C2v) to Linear(D∝h) Shapes. International Journal of Molecular Sciences. 2006; 7(3):71-96. https://0-doi-org.brum.beds.ac.uk/10.3390/i7030071
Chicago/Turabian StyleGhosh, Dulal C., and Arindam Chakraborty. 2006. "Dipole Correlation of the Electronic Structures of theConformations of Water Molecule Evolving Through theNormal Modes of Vibrations Between Angular (C2v) to Linear(D∝h) Shapes" International Journal of Molecular Sciences 7, no. 3: 71-96. https://0-doi-org.brum.beds.ac.uk/10.3390/i7030071