1. Introduction
The human asialoglycoprotein receptor is an integral membrane protein belonging to the C-type lectin family which is located on the surface of the hepatocyte plasmatic membrane.
C-type lectins are grouped into several subclasses: the most abundant ones are the Man-type (capable of linking D-Mannose, D-Glucose or L-Fucose) and the Gal-type ones [that link D-Galactose or D-N-Acetylgalactosamine (NAG)]. The ASGP-R belongs to the Gal-type group and its affinity for NAG has been proved to be significantly higher with respect to galactose.
ASGP-R is responsible for the clearance of desialylated, galactose-terminal glycoproteins from the circulation by receptor-mediated endocytosis. The Glycoproteins, after accomplishing their physiological functions, lose sialic acid molecules at the end of the glucydic chains, becoming galactose-ending or NAG–ending proteins. The Receptor reacts with the carbohydrate moieties at the end of the carbohydrate chain giving rise to receptor-ligands complexes which penetrate the cells by a receptor-mediate endocytosis event. Once the complex reaches the acidic compartment of a mature endosome it breaks down, the receptor is recycled on the cell surface while the ligand is metabolized into the lysosomes.
The functional ASGP-R is an hetero-oligomer made up of two different kinds of subunit referred as H1 and H2. Different domains are recognizable within each subunit: a cytosolic, a transmembrane, and an extracellular moiety that comprises a “
stalk” and the carbohydrate recognition domain (CRD) which includes binding sites for galactose and NAG [
1–
2].
The ASGP-R has been considered for several years a liver specific receptor which could be exploited for targeting endogenous molecules to hepatocytes. More recently C-type lectins with high affinity for glycoconjugates bearing terminal galactose residues have also been identified on the surfaces of peritoneal macrophages and Kupffer cells and appear to mediate recognition of tumor cells. Due to the above characteristics, the interest presented by the ASGP-R for diagnostic and therapeutic purposes is still remarkable [
3].
In this paper several computational methods, based on the receptor structure, were exploited with the aim of understanding the molecular features accounting for the binding selectivity showed by the CRD toward Galactose and NAG with respect to other sugar molecules, such as fucose, mannose and glucose, according to reports taken from the literature.
The theoretical models for complexes were first validated by comparison with the experimental data. The same computational approach was then used to estimate the affinity between CRD and other monosaccharides deriving from natural polymers of interest. In particular monomers deriving from alginates and ulvans were screened “in silico” against the receptor in order to acquire information enabling to increase the potential of their applications in the biomedical field.
Indeed alginates [
4–
7] and ulvans [
8–
9] display unique properties, that strongly encourage their use for biomedical application, such as i) high gel porosity allowing high diffusion rates of macromolecules, ii) possibility to tune porosity with simple coating procedures, iii) dissolution and biodegradation of the system under physiological conditions, iv) mucoadhesive properties.
Commercial alginates consist of biopolymers mostly extracted from three different species of brown algae such as Laminaria hyperborea, Ascophyllum nodosum and Macrocystis pyrifera. In these species the primary polysaccharide is the alginate, as it represents the 40% of the dry weight. In terms of chemical structures alginates are random, anionic, linear polymer consisting of different ratio of guluronic and mannuronic acid units. The physical properties of alginates are accounted by composition and extent of sequences as well as by the molecular weight.
Ulvans represent the major bio-polymeric fraction of the cell wall of
Ulva and
Enteromorpha marine green seaweeds. They are capable of supplying a relevant biomass that is only slightly exploited at present. Additional ways to use such a biomass, besides compost, methane production or paper making, could be based on specific properties of their cell-wall polysaccharides. These macromolecules retain heavy metals and several
Ulva and
Enteromorpha species are used as bioindicators of pollution. In edible Ulvans, cell-wall polysaccharides play a nutritional role as dietary fibre and, from different genera, they demonstrate biological activities or gelling abilities [
10].
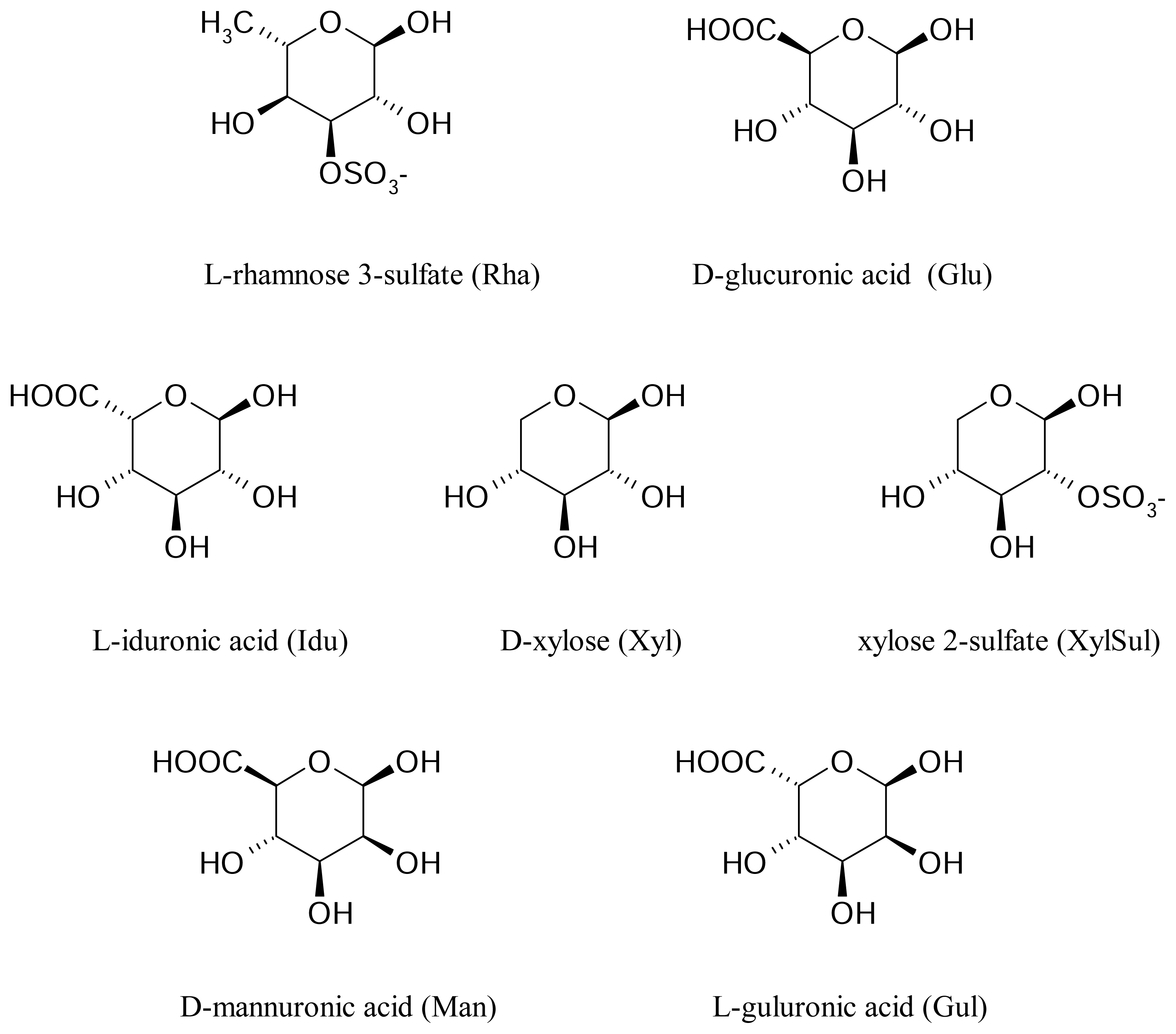
Ulvans mostly consist of different ratio of D-glucuronic acid, D-xylose and two uncommon sugars such as iduronic acid and sulfated L-rhamnose.
2. Results and Discussion
2.1. Molecular modeling and validation step
The thee-dimensional (3D) structure of the H1 subunit of the CRD of human asialoglycoprotein was taken from the Protein Data Bank (PDB) [
11] (ID: 1DV8). Molecular structures of the monosaccharides were built within the InsightII package [
12]. The starting orientation of galactose in the CRD binding site was obtained by replacing the oxygen atom of the two crystallographic Cacoordinated water molecules (11 and 13) with the oxygen atoms of 3 and 4 OH- groups of galactose in order to retain the correct geometry coordination of the calcium ion, since the calcium ion is coordinated to eight oxygen atoms within the binding site [
1]. The other monosaccharides were built on the basis of galactose, taken as the starting template. The putative complexes with CRD were built by InsightII as well and simply relaxed by energy minimization before molecular docking. More accurate optimization did not seem to be required at this level, since, the active site structure of calcium-binding proteins is considerably stabilized by the structural requirements related to the coordination geometry of the calcium ion [
13]. The structure of the CRD in complex with ligands is expected to show quite high similarity with regard to its free form, on the basis of the low RMS values obtained for other C-type lectins, available in the PDB, when comparing their free forms with the ones in complex with ligands. As an example, the RMS values calculated for the backbone atoms of two C-type lectins, are reported below. The crystal structures refer to their unliganded form and the corresponding complex with a monosaccharide ligand. The calculation was made in a subset of 5 Å radius centered on the Ca
2+ ion: RMS = 0.13 was obtained in the comparison of 1PW9 (free form) with 1PWB (complex with glucose); RMS = 0.30 was obtained when comparing 1AFD (free form) and 1AFB (complex with NAG).
2.2. DOCK5 calculations
The DOCK5.2 program [
14] was initially used to search optimal orientations of Galactose, NAG (Gal-type ligands) and fucose, glucose, mannose (Man-type ligands) into the putative high affinity binding site of the H1-CRD, by roughly estimating interaction energies of many different complex configurations, sampled during the docking procedure, and by ranking them according to their scores. On this basis, plausible configurations of protein-ligand complexes were selected. The DOCK energy components are based on the implementation of the force field scores, which are approximately given by molecular mechanics interaction energies, consisting of van der Waals and electrostatic components. Three runs were performed for each ligand by using different seed numbers in order to perform a better sampling and to make sure that the convergence was reached in the pose found for each ligand. In spite the strong approximations contained within the DOCK energy scoring function, the GAL and NAG (Gal-type ligands) were observed, yet at this level, to show a more favorable interaction energy with CRD in comparison to mannose, fucose and glucose (Man-type) ligands. Moreover, NAG was observed to bind the CRD more efficiently than GAL. All the above observations are in good agreement with the well known qualitative data regarding Gal- and Man- type ligands, and the K
i values reported in the literature for GAL and NAG (K
i(GAL)/K
i(NAG) = 60). It has to be pointed out here that only K
i values referring to the rat protein are available, but the affinity toward the human and the rat proteins are expected to be very close to each other, as the two proteins show very high sequence homology, especially in the binding site [
3,
15–
16].
After accomplishing the preliminary step of molecular docking, the models appeared to be valid. Nevertheless, due to the complexity of the analyzed system, where hydrophobic and hydrophilic contributions are known to be involved in the protein-ligand interaction energies, more accurate estimate of interaction energies was attempted by using the DOCK5 GBSA scoring function.
2.3. DOCK5 GBSA calculations
The three poses obtained in the previous docking calculations for each ligand were then re-scored by using the Generalized Born Solvent Area (GB/SA) scoring function [
17].
The GB/SA scores for these systems did not correctly reproduce the experimental data reported in the literature. In fact, NAG, i.e. the ligand that most strongly binds to CRD, shows a less favorable score in comparison with glucose and galactose. Since the GB/SA scoring function showed do not be able of realistically representing the systems of interest, a more accurate validation approach was considered, before affording the prediction step. The DelPhi program was selected for more accurate estimates of the interaction energies in the complexes involving Galactose, NAG (Gal-type ligands) and fucose, glucose, mannose (Man-type ligands).
2.4. Structure analysis
The analysis of the complexes obtained after the DOCK calculations shows that the orientations of the ligands in the binding site allow retaining the bipyramidal pentagonal coordination geometry of the calcium ion. It is mainly due to the oxygen atoms of the 3-OH and 4-OH groups of the sugar. The mean values of the inter-atomic distances between the calcium ion and the oxygen atoms of the ligands (obtained from the three structure collected for each ligand) are reported in
table 1. In the same table is also reported the maximum number of H bonds observed in the three orientations for each ligand.
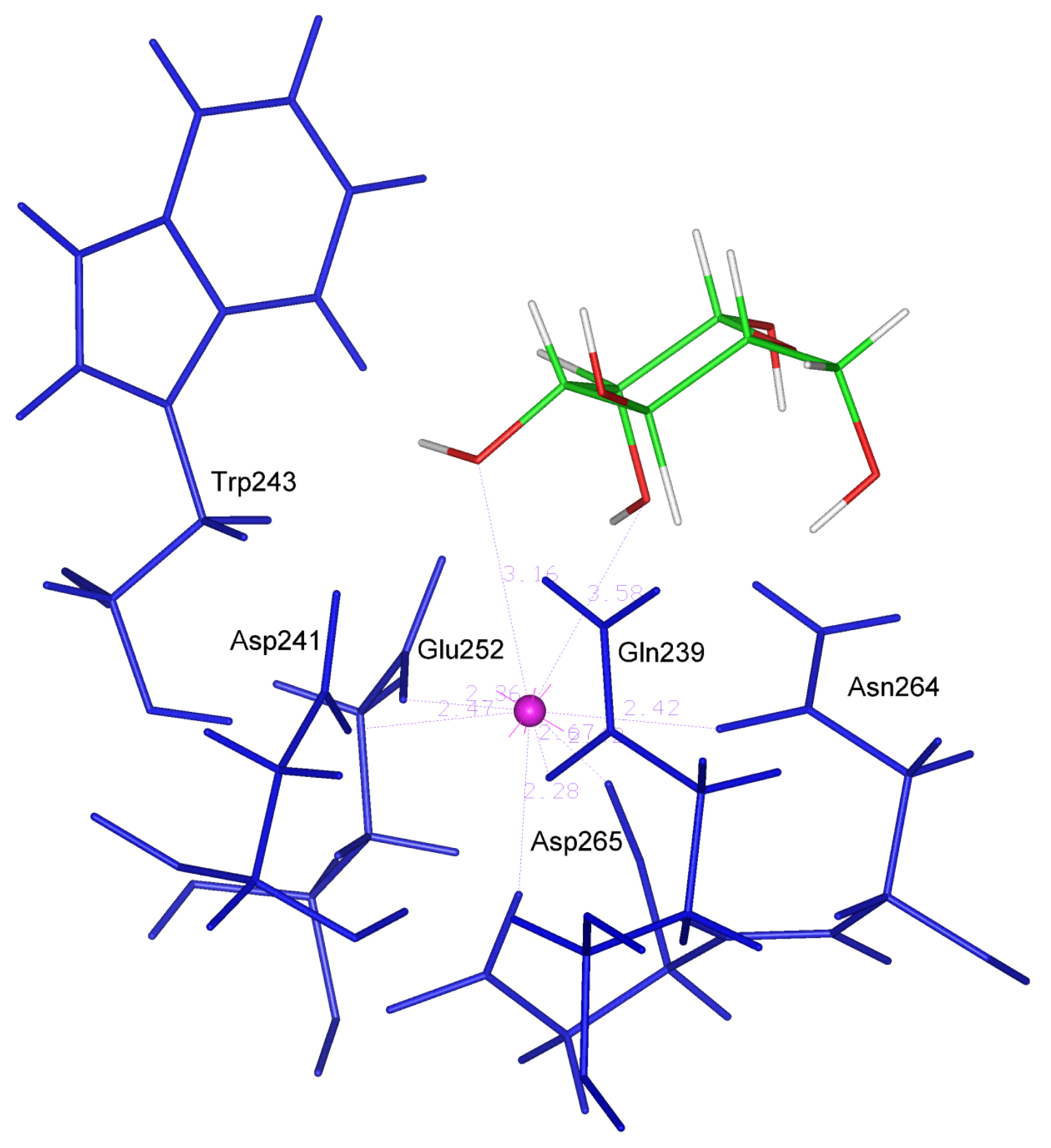
In the case of Gal-type ligands, the 3- and 4-OH groups of the sugar are approximately in the same position of the two water molecules 11 and 13 and they appear to be close enough to the Ca ion, so that they can form the required coordination bonds. In this case, the retention of the Ca coordination geometry allows the pyranose ring to have an optimal orientation, stabilized by good stacking interaction with Trp 243. In fact, the apolar patch formed by the 3, 4, 5, and 6 carbons of galactose and NAG packs against the Trp 243 side chain, according to an interaction mode observed in all the galactose-lectin complexes analyzed to date.
The angle between the least squares plane through the pyranose ring of the ligands and the plane of the indole ring in the Trp243 side chain is 22.29 and 28.25 for galactose and NAG respectively (See
Table 2). This is in agreement with previous findings reported in the literature [
3], where it has been showed that, in several galactose-binding lectins, such an angle ranges within an optimal interval of 17–52 degrees.
Furthermore, galactose and NAG (
figure 1) make four hydrogen bonds with residues Gln 239, Asp 241, Glu 252 and Asn 264, i.e. with the same residues that are involved in the binding of the calcium ion.
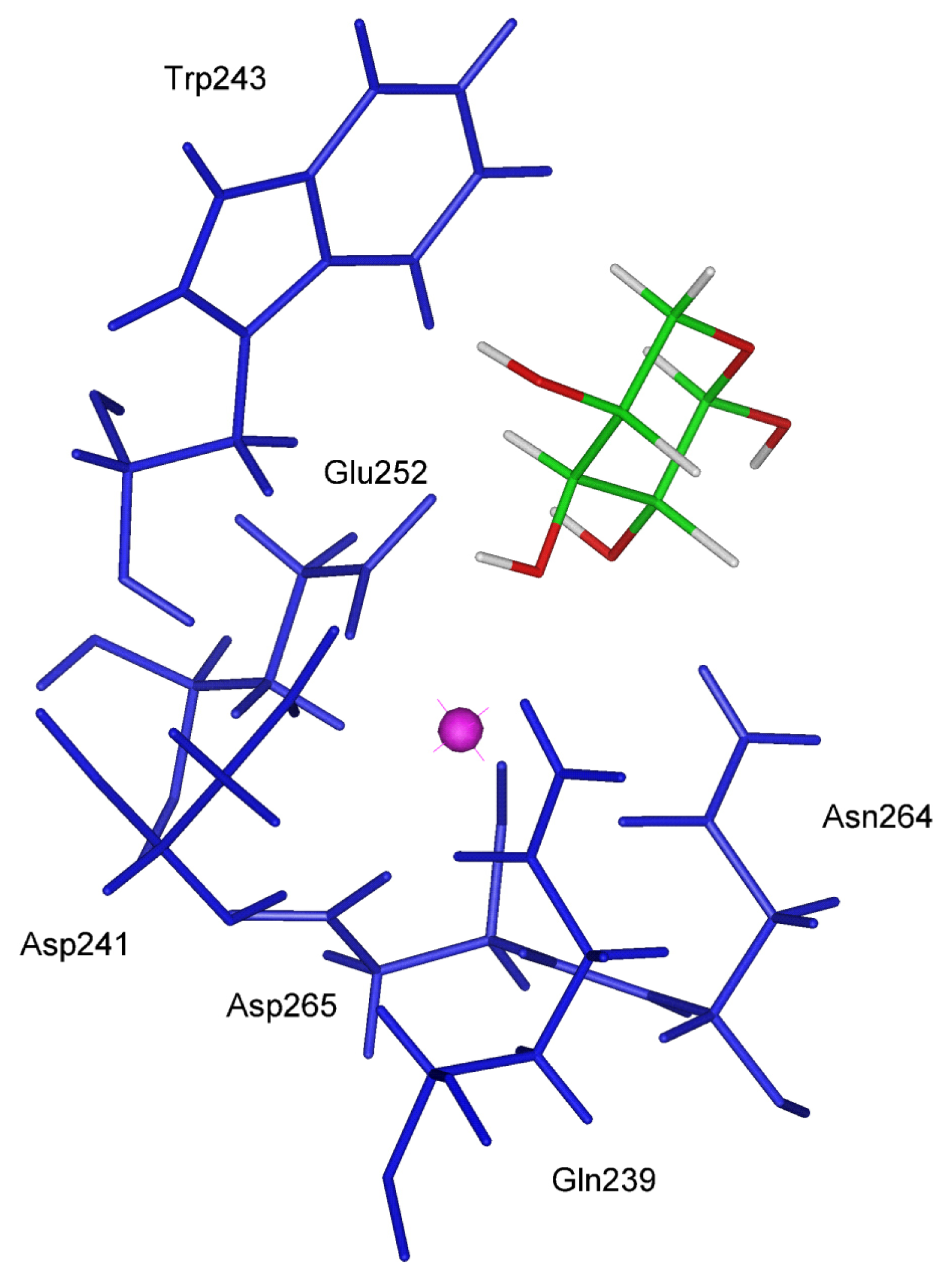
In the CRD-Mannose complex modeled in this work (
figure 2), the location of the sugar ring is different from the one observed for the CRD-Gal complex. The pyranose ring rotates in the binding site so that its oxygen atoms belonging to the hydroxyl groups 2–OH and 3–OH adopt the required distance with calcium. This orientation dictated by the Ca coordination geometry requirement is only stabilized by two H bonds and the hydrophobic interaction with Trp243 has a minor extent as indicated by the value (61.40 degrees) assumed by the critical angle described above.
Glucose is capable of satisfying the Ca coordination geometry requirement with its O3 and O4 oxygen atoms and forming four stabilizing H bonds. Nevertheless the hydrophobic interaction with Trp243 is weaker (angle of 72.96 degrees), like in the case of mannose. L-fucose, even if capable of forming four H bonds with CRD, is unable of satisfying the Ca coordination geometry requirements. In fact only the O2 atom appears to be close enough to Ca to form a coordination bond, moreover it shows a weak hydrophobic interaction with Trp243 (angle of 102.81 degrees).
2.5. DelPhi calculations
The DelPhi program [
18] is designed to calculate the electrostatic potentials in and around an irregular macromolecular structure (e.g., protein) that is embedded in a homogeneous dielectric environment (e.g., water). It enables quite accurate estimate of ligand-protein interaction energies by solving the Poisson-Boltzmann equation with the finite-difference method.
For each systems Linear Poisson-Boltzmann equation was solved first at physiological salt concentration (I = 0.1) then all the runs were repeated at zero salt.
In
table 3 the energy contributions calculated by DelPhi and the corresponding ΔG values (as described in
methods) are shown. The simulations were performed using for each ligand its lowest energy conformation among the three ones suggested by DOCK.
The observed ΔG values seem to correlate quite well with respect to the trend of affinities reported in the literature for the analyzed know ligands. In fact the only ligands showing negative values of ΔG are galactose and NAG (Gal-type ligands), while Man-type ligands, as reported in the literature, do not appear likely to bind ASGP-R and they show positive ΔGs. Moreover, the obtained absolute values suggest that NAG binds ASGP-R more tightly than galactose, even though the difference in ΔGs appears to be underestimated.
2.6. Affinity prediction for other monosaccharides
The studies on complexes involving known ligands, obtained by analyzing interaction energies estimated by means of DelPhi, as well as the observations coming from structural analysis of the binding sites, are all consistent with the data reported in the literature and they are capable of satisfactorily explaining the selectivity toward Gal-type ligands showed by the sugar binding site of the human asialoglycoprotein receptor.
The results of these studies show that the ligands are more likely to bind the CRD when:
- —
at least two oxygen atoms of the sugar (preferably 3-OH and 4-OH) are close enough to the calcium ion so that coordination bonds are made possible.
- —
the position of the ligands in the binding site is stabilized by an high number of H bonds.
- —
the pyranose ring of the sugar is oriented so that the hydrophobic interaction between Trp 243 and the C3, C4, C5 and C6 atoms of the ligands is maximum.
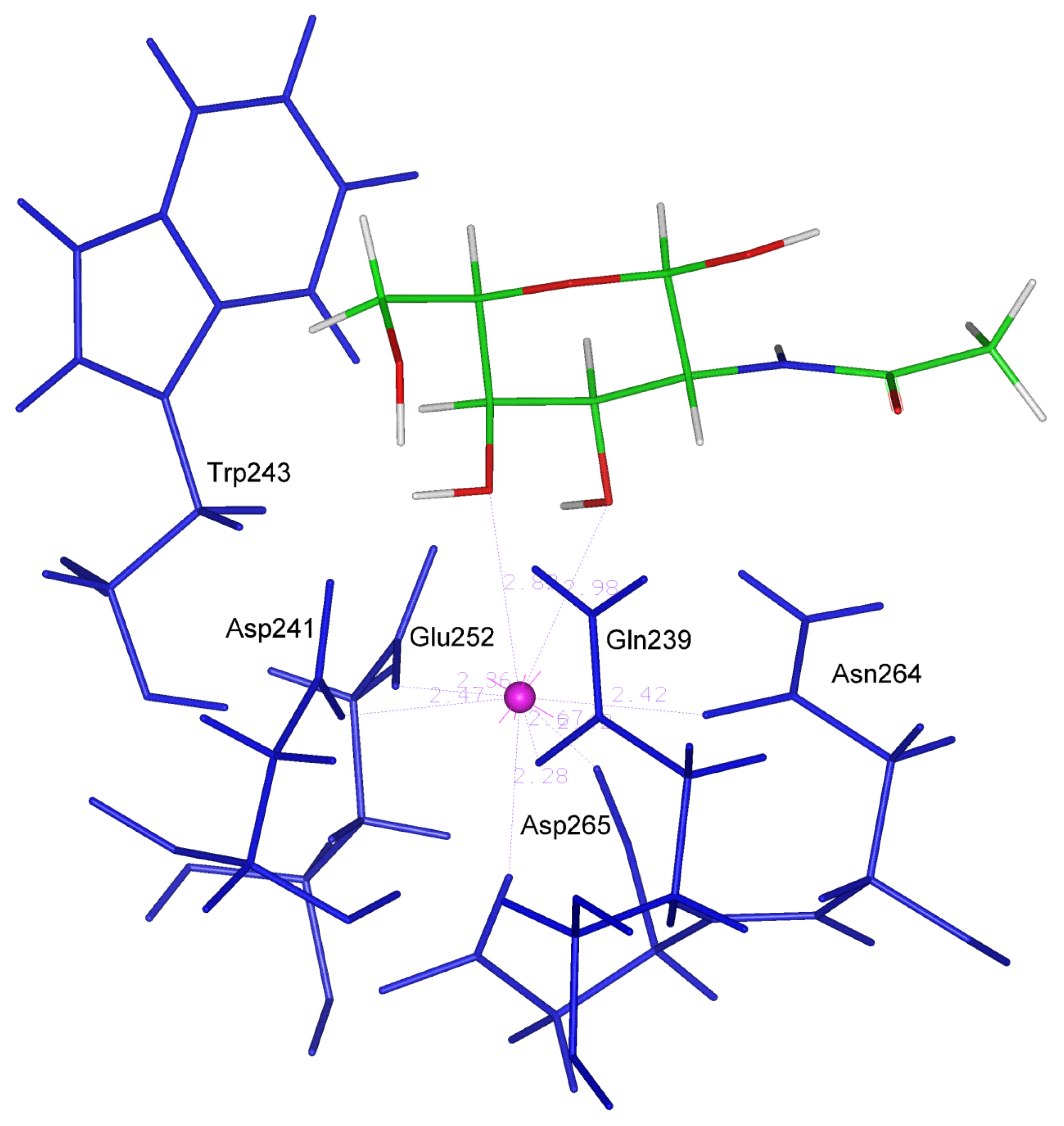
The strategy described above was then applied to the search for favorable interactions occurring between the sugar binding site of the human asialoglycoprotein receptor and other monosaccharides of interest, such as D-mannuronic acid, and L-guluronic acid deriving from alginates and L-rhamnose 3-sulfate, D-glucuronic acid, L-iduronic acid, D-xylose and D-xylose 2-sulfate deriving from ulvanes (
figure 3).
2.6.1. DOCK5 calculations
Optimal orientations of the ligands into the binding site were obtained by means of the DOCK5.2 program. As already done, during the assessment of the theoretical models involving known ligands, three runs were performed for each ligand by using different seed numbers in order to perform an accurate sampling and to make sure that convergence was reached when searching the best pose for each ligand.
Among the above monosaccharides, roughly ranked on the basis of the DOCK energy scores, mannuronic acid, rhamnose 3-sulfate and xylose 2-sulfate showed better interaction with the CRD. It must be pointed out that these ligands (except for xylose) bear a net negative charge: it implies that the score comparison may be done only within the sub-group of charged molecules. More accurate estimates of the binding energies were performed by using the DelPhi program. In analogy to what has been done during the model validation step, the DOCK calculations previously performed mostly served to identify optimal conformations of the ligands in the binding site.
2.6.2. Structural analysis
The analysis of the complexes obtained after the DOCK calculations showed that all the ligands (with the exception of rhamnose 3-sulfate and glucuronic acid) are oriented in the binding site in a way capable of retaining the bipyramidal pentagonal coordination geometry required by the calcium ion. In particular, xylose 2-sulfate engages the oxygen atoms of its 3-OH and 4-OH groups in this interaction by the same way as galactose and NAG do. This monosaccharide is also able to form four H bonds. In
table 4 the values (averaged over the three structure collected for each ligand) of the interatomic distances between the calcium ion and the oxygen atoms of the ligands are reported. In the table the maximum number of H bonds observed in the three orientations for each ligand is also shown.
The averaged angle between the least squares plane through the pyranose ring of the ligands and the plane containing the side chain indole of Trp243 in the CRD is reported in
table 5.
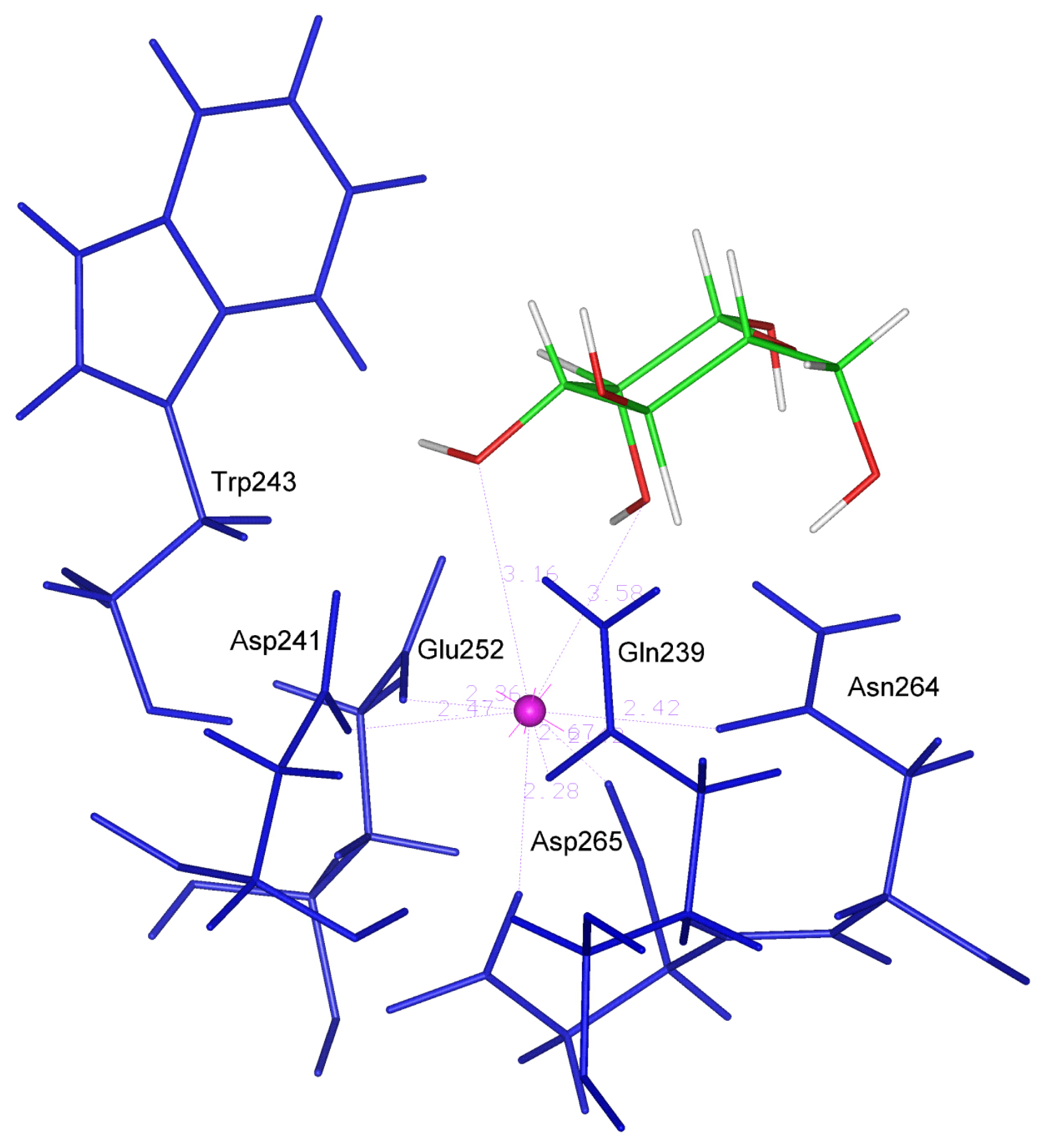
Xylose and Xylose 2-sulfate lack the C6 atom and for this reason the hydrophobic interaction with Trp 243 is expected to be lower than other ligands possessing the C6 atom. On the other hand, only Xyl and XylSul show the same orientation observed in the gal-type ligands, in accordance with the value of the angle above described (51.86 and 39.28 degrees), which turns out to be comprised within the optimal range (In
Figure 4 the best orientation of Xylose in the binding site is shown).
2.6.3. DelPhi calculations
The energy contributions calculated by DelPhi and the ΔG values obtained for the different systems involving the monosaccharides, which are building blocks of alginates and ulvans, are reported in
Table 6.
On the basis of these calculations only xylose shows a negative ΔG value. This finding together with the above observations suggest that xylose has a good probability of interacting with the CRD of the ASGP-R even though the predicted affinity for the single CRD unity is quite low. It can be reasonably expected that saccharides containing several Xylose-ending extremities could interact
in vivo (where the functional ASGP-R is an hetero-oligomer) several fold better with respect to the single CRD. It was already shown that ligands containing multiple galactose-ending moieties bind several fold better than galactose [
19].
3. Conclusions
The selectivity, shown by the CRD toward Gal-type and Man-type ligands, was analyzed by using different computational methods, such as molecular docking and DelPhi calculations. In particular the investigation reported here highlighted some structural features required for a sugar molecule to be able of binding the CRD.
The first part of our work allowed the validation of theoretical models on which prediction of the interactions of interest could be performed. The results show that a carbohydrate ligand is more likely to bind the CRD when i) at least two oxygen atoms of the sugar (preferably 3-OH and 4-OH) are close enough to the Calcium ion so that proper coordination bonds are allowed, ii) the pyranose ring of the sugar is oriented so that the hydrophobic interaction between Trp 243 and the C3, C4, C5 and C6 atoms of the ligands is maximum, iii) the position of the ligands in the binding site is stabilized by an high number of H bonds.
In the subsequent prediction step, the affinity of the CRD for other monosaccharides deriving from alginates and ulvans was estimated by using the previously validated theoretical models. Among the analyzed monosaccharides, one of them, deriving from ulvans, seems to possess proper features that make it a quite good ligand for CRD.
Ulvans with large content of xylose, hence, could be potentially able to interact with ASGP-R even though the affinity of this sugar for the single CRD unit is quite low. It can be reasonably expected that saccharides containing several Xylose-ending extremities could interact in vivo (where the functional ASGP-R is an hetero-oligomer) much better, with respect to the single CRD, as already shown for ligands containing multiple galactose-ending moieties, that bind several fold tighter than galactose.
The above results carry a strong interest in the field of biomedical applications, as the possibility of reliably predicting affinities of sugar molecules for the asialoglycoprotein receptor may help in properly selecting natural carbohydrate polymers well suited for interacting with hepatocytes. A further development of this study will consist of designing suitable experiments for validating the predictive work described here.
4. Methods
4.1. Details on model building
As above mentioned, the starting orientation of galactose in the CRD binding site was obtained by replacing the oxygen atom of the two crystallographic Ca-coordinated water molecules (11 and 13) with the oxygen atoms of 3 and 4 OH- groups of galactose. From the x-ray structure five oxygen atoms (belonging to Gln 239, Asp 241, Glu 252 and Asn 264 residues) appear to be arranged into a pentagonal ring around the metal ion: two of them (belonging to water molecules 11 and 13 ) are located above this plane, one (belonging to Asp265) is located below, so that a roughly bipyramidal pentagonal geometry with a double peak on one side is formed. The Galactose served as the starting template for locating all the other monosaccharides within the CRD binding site. All the saccharides were modeled in their β-D-pyranosidic form, but fucose, rhamnose 3 sulfate, guluronic acid and iduronic acid that were in their L form.
4.2. Dock5 calculations
The DOCK5.2 program allows docking flexible ligands into the active site of a receptor. The program does not require to generate a large number of conformations for the ligand, as the conformational space can be automatically explored by DOCK ‘on the fly’ provided that the geometry of the initial ligand conformation is good enough. The full search is split into orientation and conformation search. The orientation search is suitable for rigid ligands. Flexible ligands are treated as a collection of rigid segments separated by rotatable bonds.
DOCK first generates a negative image of the ligand binding site with a set of overlapping spheres whose centers become the potential locations for the ligand atom rotation. A bounding box is constructed around this sphere cluster with an extra cushion in each direction, which is then gridded to allow the scoring evaluation of docked ligands. In order to rank each potential ligand, a pre-calculated force-field-scoring grid, based on molecular mechanics interaction energies that consist of Van der Waals and electrostatic components, is generated. This energy score is determined both by types and positions of ligand atoms on the energy grids. Subsequently, score optimization allows the conformation and orientation of a molecule to be adjusted to improve the score. The resulting output file for each screening step, based on force field grids, contains the best scoring compounds ranked in order of their scores.
In the DOCK5.2 release, a new scoring function, GB/SA, is available. It has been developed in order to implicitly take into account the effects of water solvation by using the generalized Born approximation.
The absolute values of the scoring function outputs are not directly related to well defined physical properties because of the approximations involved; they are only useful for ranking the ligands with respect to each other. In spite of the many approximations used, it was stated that in several systems the DOCK program offers quite good correlations not only with respect to the orientations of molecules inside the receptor cavity but also with respect to the docking scores [
20]. It has been proved that, in many cases, compounds, which dock better than the others, have potential better binding capacity to the receptor site.
4.3. Delphi calculations
The use of the DelPhi approach made in this work followed the track outlined elsewhere to calculate binding ΔGs in protein complexes [
21].
All molecules were mapped onto a three-dimensional grid. The grid center was located at the center of the complex. Boundary conditions were set to calculate the potential at each boundary point due to every charge in the system, by using the Debye-Hückel approximation (full Coulombic). The boundary between the molecule and solvent is defined by the molecular or solvent-excluded surface, using a 1.4 Å solvent probe. The external and internal dielectric constant were 80 and 2, respectively. All the species were represented with a polar-hydrogen model, and partial atomic charges were from the CHARMM parameter set. Asp, Glu, Lys, and Arg side chains and N- and C-termini were modeled in their ionized state; His and Cys were considered to be neutral. Default atomic radii were used. Further calculations were performed in which the total charge of the binding site was distributed (by using semi-empiric approaches) among Ca++ and the nearest residues (the ones involved in the coordination bonds). This representation (data not shown) did not produce appreciable results, while CHARMM charges produced results in agreement with experimental data.
In the case presented here the simulation must take into account the two water molecules that are coordinated with the Ca++ ions in the receptor and that are displaced out by the ligand upon binding. DelPhi calculations were done on the unliganded protein, the two water molecules, each one of the free ligands and each complex. The system may be schematically represented as
where
A is the receptor (with the two crystallographic water molecules 11 and 13, involved in coordination bonds with the Ca++ ion within the binding site)
B is the ligand
C is the ligand-receptor complex
D refers to each one of the two water molecules displaced by the ligand upon binding
For each A, B, C, D species Linear Poisson-Boltzmann equation was solved first at phisiological salt concentration (I = 0.1) then all the runs were repeated at zero salt. For each ligand-protein system the following energy contributions were computed:
complex: total grid energy (1)
receptor: total grid energy (2)
ligand: total grid energy (3)
water1: total grid energy (4)
water2: total grid energy (5)
complex_nosalt: total grid energy (6)
receptor_nosalt: total grid energy (7)
ligand_nosalt: total grid energy (8)
water1_nosalt: total grid energy (9)
water2_nosalt: total grid energy (10)
complex_nosalt: coulombic energy (11)
receptor_nosalt: coulombic energy (12)
ligand_nosalt: coulombic energy (13)
water1_nosalt: coulombic energy (14)
water2_nosalt: coulombic energy (15)
complex_nosalt: corrected reaction field energy (16)
receptor_nosalt: corrected reaction field energy (17)
ligand_nosalt: corrected reaction field energy (18)
water1_nosalt: corrected reaction field energy (19)
water2_nosalt: corrected reaction field energy (20)
The single contributions were then combined according to the equation reported below, to give rise to the proper calculated ΔG values:
where
ΔG (coulombic) = (11) + (14) + (15) - (12) - (13)
ΔG (reaction field) = (16) + (19) + (20) - (17)-(18)
ΔG (ions) = [(1)-(6)] + [(4)-(9)] + [(5)-(10)] − [(2)-(7)] −[(3)-(8)]




{kind=link}
{kind=link}
{kind=link}
{kind=link}